

The lytic cycle of KSHV is involved not only in the dissemination of the virus but also viral oncogenesis, in which the effect of RTA on the host transcriptome is still unclear. Using genomics approaches, we identified a core set of host genes which are rapidly and directly induced by RTA in the early phase of KSHV lytic reactivation. We found that RTA does not need viral cofactors but requires its host cofactor RBP-Jκ for inducing many of its core RIGs. Importantly, we show a critical role for two of the core RIGs in efficient lytic reactivation and replication, highlighting their significance in the KSHV lytic cycle. We propose that the unbiased identification of RTA-induced host genes can uncover potential therapeutic targets for inhibiting KSHV replication and viral pathogenesis.
KEYWORDS: Kaposi’s sarcoma-associated herpesvirus, RTA, lytic reactivation, primary effusion lymphoma, regulation of gene expression
ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a human oncogenic virus, which maintains the persistent infection of the host by intermittently reactivating from latently infected cells to produce viral progenies. While it is established that the replication and transcription activator (RTA) viral transcription factor is required for the induction of lytic viral genes for KSHV lytic reactivation, it is still unknown to what extent RTA alters the host transcriptome to promote KSHV lytic cycle and viral pathogenesis. To address this question, we performed a comprehensive time course transcriptome analysis during KSHV reactivation in B-cell lymphoma cells and determined RTA-binding sites on both the viral and host genomes, which resulted in the identification of the core RTA-induced host genes (core RIGs). We found that the majority of RTA-binding sites at core RIGs contained the canonical RBP-Jκ-binding DNA motif. Subsequently, we demonstrated the vital role of the Notch signaling transcription factor RBP-Jκ for RTA-driven rapid host gene induction, which is consistent with RBP-Jκ being essential for KSHV lytic reactivation. Importantly, many of the core RIGs encode plasma membrane proteins and key regulators of signaling pathways and cell death; however, their contribution to the lytic cycle is largely unknown. We show that the cell cycle and chromatin regulator geminin and the plasma membrane protein gamma-glutamyltransferase 6, two of the core RIGs, are required for efficient KSHV reactivation and virus production. Our results indicate that host genes that RTA rapidly and directly induces can be pivotal for driving the KSHV lytic cycle.
IMPORTANCE The lytic cycle of KSHV is involved not only in the dissemination of the virus but also viral oncogenesis, in which the effect of RTA on the host transcriptome is still unclear. Using genomics approaches, we identified a core set of host genes which are rapidly and directly induced by RTA in the early phase of KSHV lytic reactivation. We found that RTA does not need viral cofactors but requires its host cofactor RBP-Jκ for inducing many of its core RIGs. Importantly, we show a critical role for two of the core RIGs in efficient lytic reactivation and replication, highlighting their significance in the KSHV lytic cycle. We propose that the unbiased identification of RTA-induced host genes can uncover potential therapeutic targets for inhibiting KSHV replication and viral pathogenesis.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV, also known as human herpesvirus 8) is the only other human herpesvirus besides Epstein-Barr virus which can cause cancer (1). KSHV is responsible for the endothelial neoplasm, Kaposi’s sarcoma (KS), and the two lymphoproliferative diseases, primary effusion lymphoma (PEL), and multicentric Castleman’s disease (2, 3). Similarly to other herpesviruses, KSHV establishes a life-long infection in humans, which KSHV mainly achieves by alternating between its latency and lytic cycle programs in infected B cells (4). While KSHV latency is characterized by the expression of the latent and the repression of lytic genes and virus production, the lytic cycle involves the temporally ordered expression of all immediate early (IE), early (E), and late (L) lytic viral genes, which is accompanied by viral DNA replication and virus production (5). The lytic cycle of KSHV, which can be initiated by either reactivation from latency or following primary infection of certain cell types, can play a role not only in the dissemination of the virus but also in viral oncogenesis (6–9). It has been shown that although the majority of cancer cells are latently infected in KSHV-associated tumors, a partial or full reactivation of the KSHV lytic cycle can often be detected in a subset of cancer cells (6, 10, 11). As a result, lytic genes are expressed, several of which encode viral cytokines, immunomodulatory proteins, and regulatory factors of cell signaling pathways, which can facilitate cellular transformation (3, 12). Also, the full lytic reactivation of KSHV from latently infected cancer cells can result in virus production and infection of neighboring naive cells, which can contribute to the growth of KSHV-associated tumors (3, 6). In fact, previous studies indicated that the treatment of KS patients with the herpesviral DNA replication inhibitor ganciclovir, which can block KSHV lytic replication, could reduce KS development, supporting the notion that the KSHV lytic cycle also contributes to viral tumorigenesis (13–15). Thus, identifying the molecular events driving the lytic cycle is critical to understanding KSHV pathogenesis and finding therapeutic targets for treating KSHV-associated diseases.
It is known that the replication and transcription activator (RTA) encoded by the IE gene ORF50 of KSHV initiates and drives the lytic cycle of the virus (9, 16). RTA has an N-terminal DNA-binding domain and a C-terminal transactivation domain, which are pivotal to RTA’s function of transactivating the promoters of several viral genes (17, 18). RTA can induce its viral target genes by either directly binding to RTA-responsive DNA elements or indirectly via interacting with various host transcription factors (TFs) bound to the gene promoters, which is followed by the recruitment of chromatin-modifying and -remodeling host factors (18–22). Although RTA is a potent transactivator of many KSHV promoters in reporter assays, the interaction of RTA with viral and host factors, as well as the activities of different signaling pathways, in the context of KSHV infection can restrict or modulate the transcriptional activity of RTA (23–30). In fact, it has been demonstrated that the Notch and the NF-κB signaling pathways act antagonistically in RTA-mediated KSHV reactivation. Specifically, RBP-Jκ, which is the major TF and mediator of the Notch signaling, interacts with and can recruit RTA to KSHV promoters whereby RTA can induce viral genes, which is essential for KSHV reactivation (31–34). In contrast, NF-κB can prevent the recruitment of RTA by inhibiting the binding of RBP-Jκ to the viral promoters, which can repress KSHV reactivation (27).
While the role of RTA in the induction of KSHV genes for lytic replication is well established, the direct host target genes of RTA and their role during KSHV lytic phase are still largely unknown (35). One of the major obstacles of studying RTA and its target genes in the lytic cycle of natural KSHV infection is that the default pathway of KSHV infection is the establishment of viral latency in most cell types (36). This is well reflected by the fact that the majority of cells in PEL cell lines derived from KSHV-infected patients carry KSHV in latency, and lytic reactivation can be detected only in a small subset of these cells (37). However, chemical induction of the endogenous RTA in the viral genome or the expression of an RTA transgene in PEL cells can efficiently reactivate KSHV in vitro, allowing the study of RTA and its host target genes in the lytic cycle (38–42). Using RTA-expressing cell lines, a number of Notch signaling-controlled host genes have been identified as RTA targets, which can be linked to different aspects of KSHV pathogenesis (31, 43–45). Recently, RTA has been shown to induce the expression of the Notch receptor ligand JAG1, which can activate Notch signaling-mediated suppression of KSHV reactivation in neighboring KSHV-infected cells, suggesting that RTA-mediated host gene regulation can also be linked to maintenance of viral latency in a KSHV-infected cell population (44). Thus, RTA can affect both latency and the lytic phase of KSHV infection by controlling not only viral genes but also modulating the expression of host genes that are required to sustain persistent KSHV infection of the host. However, despite the essential role of RTA in the KSHV lytic cycle and viral pathogenesis, the RTA host target genes and their role in infected cells are still poorly characterized.
We hypothesized that the host genes that are rapidly and directly upregulated by RTA during the first hours of lytic reactivation could be critical for facilitating the lytic cycle of KSHV. In order to identify the RTA-induced host genes in PEL cells, we performed a comprehensive time course RNA sequencing (RNA-seq) analysis, which was combined with RTA chromatin immunoprecipitation coupled with high-throughput sequencing (RTA ChIP-seq). Subsequently, we demonstrated that geminin (GMNN) and GGT6, two novel RTA-induced host genes, are required for KSHV reactivation and viral production. Thus, our findings support the notion that the host genes, which are rapidly and directly induced by RTA in the early phase of KSHV reactivation, can be essential for driving the KSHV lytic cycle; thus, they can serve as potential therapeutic targets for blocking KSHV replication and viral pathogenesis.
RESULTS
Identification of RTA-binding sites on the KSHV genome.
The essential role of RTA in the induction of KSHV lytic cycle can be partly attributed to the binding of RTA to the promoters of specific viral and host genes resulting in their induction (17). Despite the vast data on RTA function, however, the genome-wide direct target genes induced by transcriptionally active RTA during the early phase of KSHV lytic cycle are still unknown. In order to identify RTA’s rapidly induced target genes, we performed an RTA ChIP-seq analysis to determine the binding sites of RTA on the KSHV and human genomes in PEL cells. For this, we made a TRExBCBL1-3×FLAG-RTA PEL cell line, where the expression of an N-terminally 3×FLAG-tagged RTA transgene can be induced by doxycycline (Dox) treatment of the cells, which can subsequently trigger KSHV lytic reactivation. Figure 1A and B shows that the Dox treatment for 6, 12, and 24 h induced 3×FLAG-RTA expression, which resulted in a gradual induction of the KSHV E (ORF45, K2) and L (K8.1, ORF25) genes. Viral DNA replication begins only after 12 h postinduction (hpi), which is reflected by the expression of L genes only at 24 hpi, as their robust induction depends on viral DNA replication (46).
FIG 1.

Genome-wide mapping of the RTA-binding sites on the viral genome during KSHV lytic reactivation. (A) Immunoblot analysis of viral protein expression in TRExBCBL1-3×FLAG-RTA cells during latency (0 hpi), at 12 hpi, and 24 hpi. 3×FLAG-RTA was detected by anti-FLAG antibody. (B) RT-qPCR analysis of viral gene expression in TRExBCBL1-3×FLAG-RTA at the indicated time points. (C) Snapshot of RTA binding on the KSHV genome (1 to 137168 bp) (GenBank accession no. NC_009333.1) in TRExBCBL1-3×FLAG-RTA cells at 12 hpi, which was identified by RTA (FLAG) ChIP-seq (top graph). The terminal repeats were excluded. The bottom graphs are zoomed-in views of RTA binding and their corresponding inputs in the indicated KSHV genomic regions. The genomic coordinates of the 23 RTA peaks are listed in Table 1. Note that the peak 8 is out of range. The arrow indicates the location of ORF25. (D) RTA (using FLAG antibody) and IgG ChIP assays were performed at 0 hpi and 12 hpi by using TRExBCBL1-3×FLAG-RTA cells, followed by qPCR quantification of the immunoprecipitated DNA at the indicated RTA-binding sites. The ORF25 region and the TRExBCBL1-Vector cell line were used as negative controls. (E) MEME/TOMTOM analysis was applied to identify the most significantly enriched de novo motif within the 50-bp radius of the 23 RTA peak summits on the KSHV genome (top), which matched to the RBP-Jκ-binding motif (bottom). (F) CentriMo analysis of 100-bp radius of the RTA-binding summits shows that the probability of the RBP-Jκ-binding motif is the highest at the summit of the RTA binding.
In order to reveal the early target genes of RTA prior to lytic DNA replication, RTA ChIP-seq was performed with FLAG monoclonal antibody in TRExBCBL1-3×FLAG-RTA cells at 12 hpi, when the KSHV genome is still fully chromatinized and the IE and E genes are already induced but KSHV DNA replication is not initiated (Fig. 1C, and data not shown) (39, 46). Thereby, the RTA-binding sites were determined on naturally chromatinized viral DNA in the early phase of lytic reactivation. As a result, the RTA peak calling uncovered 23 RTA-binding sites on the KSHV genome (Table 1). Many of them have been identified by various assays before, validating our RTA ChIP-seq results (17, 47–50). Most recently, the Izumiya lab performed RTA ChIP-seq on the KSHV genome at 24 hpi in an RTA-expressing PEL system similar to ours (51). Importantly, our RTA ChIP-seq results are in agreement with their findings. A snapshot of the RTA ChIP-seq data on the KSHV genome, with the identified peaks and summits depicted underneath, is shown in Fig. 1C, while the coordinates of the RTA peaks are listed in Table 1. We found that the majority of RTA-binding sites are located in genomic regions that mainly contain IE and E genes, which were previously demonstrated to be associated with activating histone marks, such as histone H3 acetylation and H3K4me3 during both latency and lytic reactivation (Fig. 1C) (46, 52). In addition, we could also identify 3 RTA peaks (peaks 11, 19, and 20) in genomic regions (30 to 60 kb and 95 to 115 kb) which contain most of the L genes and are constitutively enriched in repressive histone marks (H3K9me3 and H3K27me3) during KSHV lytic reactivation (46, 52). A close inspection of the lytic genes at these 3 RTA peaks revealed that each of them is adjacent to at least one E gene, suggesting that RTA can also target E genes in a transcriptionally repressive chromatin environment. However, most of the KSHV genomic regions associated with constitutive heterochromatin and containing only L genes were completely devoid of RTA binding. These data indicate that RTA tends to bind to euchromatin-like regions on the KSHV genome. Also, our results are in agreement with previous findings that RTA mainly controls the expression of E genes, although some L genes might also be inducible by RTA in reporter assays, such as the K8.1 and ORF8 promoter regions, where we could also detect RTA binding (50). To further confirm the specificity and reproducibility of the 3×FLAG-RTA ChIP, we performed independent ChIP-qPCR assays with FLAG antibody using uninduced (0 hpi) and Dox-induced (12 hpi) TRExBCBL1-3×FLAG-RTA and Dox-induced TRExBCBL1-Vector cells (negative control) in triplicate. We confirmed that RTA could bind to the identified 3×FLAG-RTA ChIP-seq peak locations within the promoters of the IE gene RTA (peak 13) and the E genes ORF6 (peak 1) and ORF57 (peak 17), but not to the promoter of the L gene ORF25, during KSHV reactivation (Fig. 1D). In contrast, there were negligible FLAG ChIP signals at 0 hpi and in the Dox-induced TRExBCBL1-Vector cells, which were comparable to the IgG negative-control ChIP signals at all tested sites, further confirming the specificity of our FLAG ChIPs (Fig. 1D).
TABLE 1.
Genomic coordinates of the RTA ChIP-seq peaks on the KSHV genome
| RTA peak | Genomic coordinate at: |
||
|---|---|---|---|
| Start | End | Summit | |
| 1 | 2837 | 3411 | 3018 |
| 2 | 4925 | 5378 | 5156 |
| 3 | 7726 | 8746 | 8462 |
| 4 | 13975 | 14546 | 14358 |
| 5 | 17644 | 18957 | 18366 |
| 6 | 20534 | 21883 | 21176 |
| 7 | 22712 | 23168 | 23008 |
| 8 | 23833 | 24228 | 24064 |
| 9 | 26575 | 28554 | 27984 |
| 10 | 28650 | 29064 | 28775 |
| 11 | 51406 | 52035 | 51747 |
| 12 | 68633 | 69680 | 69110 |
| 13 | 69842 | 70426 | 70179 |
| 14 | 74487 | 75231 | 74796 |
| 15 | 75354 | 75918 | 75590 |
| 16 | 78908 | 79180 | 79035 |
| 17 | 81839 | 82288 | 82022 |
| 18 | 91547 | 92207 | 91926 |
| 19 | 96546 | 97116 | 96957 |
| 20 | 100482 | 100698 | 100616 |
| 21 | 120292 | 120566 | 120366 |
| 22 | 123746 | 124108 | 123969 |
| 23 | 127681 | 128120 | 127833 |
RTA can be targeted to the viral promoters either by directly binding to DNA or indirectly through association with other DNA-binding host TFs (17). To determine which host TFs could potentially cooperate with RTA on the viral DNA, we performed a de novo DNA sequence motif scan using 50 bp of DNA sequence upstream and downstream of the 23 RTA-binding peak summits to discover TF motifs that are significantly enriched at the center of RTA peaks. Subsequently, the most significantly enriched de novo DNA motif at the RTA-binding sites was compared to a database of known DNA sequence motifs bound by transcription factors (Fig. 1E). Our analysis revealed that the most significantly enriched DNA motif was matched to that of RBP-Jκ, which in fact could be detected in 18 of the 23 RTA-binding sites (Fig. 1E). Additional analysis of 100 bp of DNA sequence surrounding the RTA summits revealed that the RBP-Jκ motif was the most significantly enriched, which was centered at the summit of the RTA peaks (Fig. 1F). These data suggest that RTA binding is tightly associated with that of RBP-Jκ on the KSHV genome, which can affect the recruitment of RTA to most of the RTA-regulated viral promoters and/or RTA-mediated viral gene expression. This supports previous findings that RBP-Jκ is essential for RTA-mediated KSHV lytic reactivation (34, 53).
Global host gene expression changes during KSHV lytic reactivation.
While viral gene expression has been extensively studied during the lytic phase in various cell culture model systems, we have still limited information about how the lytic cycle of KSHV affects the expression of host genes and to what degree different viral factors, such as RTA, contribute to the reprogramming of the host genome. To address this question, we first determined the global host gene expression changes during KSHV reactivation. To this end, we prepared total RNA from TRExBCBL1-3×FLAG-RTA cells at 0 hpi (latency) and Dox-treated cells at 6, 12, and 24 hpi, which were then subjected to RNA sequencing. Principal-component analysis (PCA) of the RNA-seq data showed that the three biological replicates at each time point clustered together while the data sets per time points were clustered away from one another, indicating a gradual and reproducible shift of host gene expression during KSHV reactivation (Fig. 2A; see also Tables S1 and S2 in the supplemental material). We found that 3,123 host genes showed more than 2-fold expression change (false-discovery rate [FDR], <0.05) during KSHV reactivation relative to latency (Fig. 2B and C and Table S3). As KSHV lytic reactivation progressed, the number of upregulated and downregulated host genes increased similarly, detecting the highest number of gene expression changes at 24 hpi (Fig. 2C and Table S3).
FIG 2.

Global host gene expression changes in PEL cells upon KSHV reactivation. (A) Principal-component analysis (PCA) of the time course RNA-seq data. (B) Hierarchical clustering of 3,123 host genes, which were more than 2-fold differentially expressed (cutoff P value, FDR <0.05) during KSHV reactivation relative to latency (0 hpi). RPKM values are shifted to 0 and scaled to a standard deviation of 1. Blue and yellow represent lower-than-average and higher-than-average gene expression changes, respectively. (C) Venn diagram representation of the 3,123 host genes (described in panel B) showing the numbers of the significantly upregulated and downregulated host genes during KSHV reactivation relative to latency. The percentage of RTA target genes within each group is also shown, which is based on the RTA ChIP-seq data at 12 hpi. Some examples of genes from the middle intersections are shown. (D) Measuring the induction of host genes by RT-qPCR to confirm the RNA-seq results at the indicated time points during lytic reactivation. Significance test was performed between reactivated and latency samples. (E) Example for a host gene upregulated only at 24 hpi. (F) RT-qPCR confirmation of GGT6 induction during KSHV reactivation. GGT6 expression was calculated relative to the expression of 18S rRNA. N.D, not detectable. (G) Examples for genes that are downregulated during lytic reactivation. *, P < 0.05.
Gene ontology analysis of the 257 host genes, which were significantly upregulated more than 2-fold within the first 6 and/or 12 hpi (FDR, <0.05), revealed that there was a significant enrichment of factors involved in the Notch signaling pathway and the positive regulation of RNA polymerase II transcription (Fig. 2C and Table S4). Strikingly, based on the cellular component gene ontology analysis, 26.12% of the upregulated genes encode plasma membrane proteins (e.g., CD244, ephrin-A1 [EFNA1], transforming growth factor beta-3 [TGFB3], sodium-dependent glutamate/aspartate transporter 1A3 [SLC1A3], delta-like 4 [DLL4], and Fc epsilon receptor 2 [FCER2]/CD23) (Fig. 2C and Table S4). The cellular component gene ontology analysis of the 338 genes, which were significantly downregulated more than 2-fold by the first 6 and/or 12 hpi (FDR, <0.05), revealed that many of them code for extracellular matrix proteins and proteins, which reside on the external side of the plasma membrane (e.g., CCR7, CCR5, CCR1, and thrombospondin 1 [THBS1]) (Fig. 2C and Table S4). Of the biological process categories, the downregulated genes were significantly associated with cellular response to tumor necrosis factor, mitogen-activated protein kinase (MAPK) cascade, and the inflammatory response (e.g., CCR5, CCR7, insulin-like growth factor binding protein 4 [IGFBP4], and tumor necrosis factor superfamily member 6B [TNFRSF6B]) (Fig. 2C and Table S4).
Importantly, our RNA-seq data (Fig. 2C and Tables S3 and S4) also included differentially expressed host genes that had been shown before to be RTA-induced genes and were implicated in KSHV pathogenesis, such as FCER2 (CD23), hairy/enhancer of split (HES) related with YRPW protein 1 (HEY1), HES1, and DLL4, validating our RNA-seq analysis (43–45). As an example, some of the most rapidly downregulated host genes were ID2 (helix-loop-helix DNA-binding protein inhibitor), thrombospondin 1 (THBS1), and the zinc finger protein ZIC2. THBS1 is an angiogenesis inhibitor whose expression is regulated by KSHV microRNAs (miRNAs) during latency, while our results show that THSB1 expression is also controlled during lytic reactivation, indicating its critical role in KSHV pathogenesis (54). We also found that ZIC2, which can be degraded by RTA at the protein level and thereby enhance lytic reactivation, is also significantly reduced at the transcriptional level during lytic reactivation, supporting the notion that a reduced level of ZIC2 is essential for facilitating KSHV reactivation (55). Independent quantitative reverse transcription-PCR (RT-qPCR) assays confirmed the rapid induction of 11 host genes, which were among those that were significantly induced by 6 hpi, 12 hpi (Fig. 2D), or only by 24 hpi (Fig. 2E), in accordance with our RNA-seq analysis. In addition, our RNA-seq analysis uncovered host genes whose transcript levels were below detection during latency but rapidly induced during KSHV reactivation, such as the still-uncharacterized gamma-glutamyltransferase 6 (GGT6) (Fig. 2F) (56). We also confirmed that there are host genes which are downregulated at different time points of KSHV lytic reactivation (Fig. 2G). Taken together, our RNA-seq analysis showed dynamic cascade-like host gene expression changes in PEL cells upon KSHV lytic reactivation, which can be mediated by distinct viral factors at different stages of KSHV reactivation. Importantly, our biological pathway analysis revealed numerous differentially expressed host genes that encode regulatory factors of signaling pathways or plasma membrane proteins, which have not yet been implicated in KSHV lytic reactivation.
Identification of host genes directly induced by RTA during KSHV lytic reactivation.
In order to identify RTA-regulated host genes during KSHV lytic reactivation, we further dissected our integrated RTA ChIP-seq and RNA-seq data sets (Fig. 2 and 3). Our RTA peak calling analysis revealed that there were 11,469 RTA-binding sites in the human genome at 12 hpi in TRExBCBL1-3×FLAG-RTA cells (>3-fold enrichment and q <0.005) (Table S5). Our analysis showed that RTA binds at 63% of the upregulated 257 host genes at 6 and/or 12 hpi of KSHV reactivation (Fig. 2C and Tables S3, S5, and S6). Ultimately, we were able to identify 163 human genes by our integrated genomics analysis where RTA binds at 12 hpi and that were significantly upregulated by 6 and/or 12 hpi during KSHV lytic reactivation. We refer to these 163 human genes as the core RTA-induced genes (core RIGs) (Fig. 3A and Table S6). Figure 3B depicts examples of RTA peaks at 4 core RIGs (EFNA1, GGT6, GMNN, and HEY1). RTA binding was further confirmed by independent 3×FLAG-RTA ChIP assays at EFNA1, GGT6, and GMNN genes, while the H19/IGF2 imprint control region was used as a negative control (Neg) for RTA binding (Fig. 3C). The FLAG (RTA) ChIP signal could be detected only in TRExBCBL1-3×FLAG-RTA at 12 hpi but not at 0 hpi or in TRExBCBL1-Vector cells at 12 hpi, where the ChIP signal was comparable to that with the IgG ChIP negative control. These ChIP-qPCR experiments further confirmed the specificity and reproducibility of our FLAG (RTA) ChIPs.
FIG 3.

Identification of the RTA-induced host genes during KSHV reactivation. (A) Heatmap showing the hierarchical clustering of the 163 RTA-induced host genes (core RIGs), which are RTA bound and more than 2-fold upregulated at 6 hpi and/or 12 hpi relative to latency (cutoff P value, FDR < 0.05). RPKM values are shifted to 0 and scaled to a standard deviation of 1. Blue and yellow represent lower-than-average and higher-than-average gene expression changes, respectively. Some genes are shown as examples. (B) Snapshots of the RTA ChIP-seq data showing the RTA binding at four core RIGs. The red arrows mark the RTA-binding sites that were confirmed by RTA ChIP-qPCR described in panel C. (C) RTA (using FLAG antibody) and IgG ChIP-qPCR assays were performed at 0 hpi and 12 hpi by using TRExBCBL1-3×FLAG-RTA to confirm the binding of RTA at their indicated host target sites. Three independent biological replicates were used. The IGF2/H19 host genomic locus (Neg) and the TRExBCBL1-Vector cell line were used as negative controls. (D) Gene ontology analysis of the core RIGs. Listed are the most significantly enriched biological process (top) and cellular component (bottom) terms, which are associated with the core RIGs. Some genes are listed as examples for each gene ontology term. (E) The most significantly enriched de novo motif within the 50-bp radius of the RTA-binding peak summits on the host genome (top) corresponds to the RBP-Jκ-binding motif (bottom) based on MEME/TOMTOM analysis. (F) De novo transcription factor binding motif analysis at the core RIGs identified RBP-Jκ-binding motif.
Gene ontology analysis showed that regulatory factors of the Notch signaling pathway, apoptosis, sphingolipid biosynthesis, and gene transcription were among the most significantly enriched biological processes that were associated with the core RIGs (Fig. 3D). Gene ontology analysis of the enriched cellular components categories showed that 22.08% of the core RIGs encode plasma membrane proteins, suggesting that RTA can alter the composition of the cell surface of cells in which KSHV is undergoing reactivation, which can be critical for the progression of the lytic cycle (Fig. 3D). For example, one of the core RIGs is GGT6, a so-far uncharacterized putative membrane protein, which we found to be essential for KSHV reactivation (see Fig. 9). Also, ligands of the known KSHV entry receptor and angiogenesis regulator EphA2 (e.g., EFNA1, EFNA3, and EFNA4) were among the core RIGs (57, 58). Thus, we propose that many of the core RIGs could play a critical role in KSHV lytic replication, infection, and viral pathogenesis.
FIG 9.

Identification of GGT6 as a pivotal host factor for KSHV lytic reactivation. (A) RT-qPCR measurement of GGT6 expression in shGGT6-treated iSLK cells. (B) RTA expression in shGGT6-treated uninduced and Dox-treated iSLK cells. The quantification of protein expression on the immunoblots is shown as relative fold change. (C) RT-qPCR analysis of latent and reactivated iSLKBAC16 cells treated with different GGT6-specific shRNAs. N.D, not detectable. (D) Cell death analysis of shGGT6-treated reactivated iSLKBAC16 cells at 24 hpi (n.s., not significant). (E) RT-qPCR measurement of the expression of viral genes in the samples described in panel C. (F) Immunoblot analysis of viral protein expression in samples described in panel C. (G and H) iSLKBAC16 cells were treated with shControl or shGGT6 #1 for 48 h, followed by Dox/NaB induction for 72 h, and then viral DNA in the reactivated cells (G) and the virion DNA purified from the supernatant (H) were measured by qPCR. (I) Equal amounts of supernatant from shControl and shGGT6 #1 samples described in panel H were used to infect 293T cells. The KSHV DNA level was measured by qPCR in the infected 293T cells at 24 hpi. Representative immunofluorescence images show 293T cells infected with supernatant derived from shControl- or shGGT6 #1-treated reactivated iSLKBAC16 cells. Green fluorescent protein (GFP) is constitutively expressed from BAC16, which is used for the detection of infected cells. (J) RT-qPCR evaluation of GGT6 expression in shGGT6 #1-treated TRExBCBL1-3×FLAG-RTA cells at 0 hpi (latency) and at 24 hpi (lytic reactivation). (K) TRExBCBL1-3×FLAG-RTA cells were treated with shGGT6 for 2 days followed by Dox induction of 3×FLAG-RTA to trigger lytic viral reactivation for 12 and 24 h. Immunoblot analysis was performed for viral and host proteins indicated on the left. The quantification of protein expression on the immunoblots is shown as relative fold change. N.D, not detectable; *, P < 0.05.
TF-binding motifs associated with the RTA-binding sites on the host genome.
To determine which host TFs could cooperate with RTA on the host genome, we performed a de novo DNA sequence motif scan using the 50-bp radius of the summit of the RTA peaks. In order to compare the TF motifs identified at the RTA-binding sites on the host genome to those on the viral genome, we performed the TF motif analysis in an identical manner. First, we identified the most significantly enriched TF motifs in 100 randomly selected RTA-binding sites at the RTA target host genes (Fig. 3E) and at the core RIGs (Fig. 3F). Strikingly, the identified top TF motifs matched to that of RBP-Jκ, which was virtually identical to what we found at the RTA-binding sites on the KSHV genome. The DNA sequence motif of RBP-Jκ can be found at the RTA-binding sites of 62 core RIGs (e.g., apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3H [APOBEC3H], GGT6, GMNN, EFNA1, EFNA4, FCER2, DLL4, HEY1, and TGFB3), implying that many but not all of the RTA-binding sites involve RBP-Jκ binding (Fig. 3F). In summary, our results signify that RBP-Jκ is vital not only for RTA-mediated KSHV gene regulation but can also be critical in the targeting of RTA to and/or induction of critical host promoters genome-wide, many of which could be essential for efficient lytic KSHV reactivation.
RTA-binding sites on the host genome can function as RTA-responsive enhancers.
To test whether the RTA-binding sites at host genes upregulated during KSHV reactivation can function as RTA-responsive enhancers, we performed an enhancer reporter assay (Fig. 4). To this end, a 406-bp DNA fragment from the 5′ end of GMNN and a 500-bp DNA fragment from the upstream region of the EFNA1 gene, which contain the RTA-binding site, were cloned in both orientations in front of a TATA box of the minimal promoter in a luciferase reporter plasmid. The RTA-binding sites are located in the center of both DNA fragments where we confirmed the binding of RTA with ChIP-qPCR (Fig. 3C). For the enhancer reporter assay, the luciferase reporter plasmids were cotransfected with either 3×FLAG-RTA (wild-type [wt]) or the DNA-binding mutant 3×FLAG-RTA (RTA K152E) into 293T cells (22). As shown in Fig. 4A, the wt RTA and K152E mutant RTA were expressed similarly. The enhancer reporter assays showed that each DNA fragment with the RTA-binding site was able to reproducibly increase luciferase gene expression in the presence of wt RTA, regardless of which orientation they were inserted in front of the minimal promoter (Fig. 4B). In contrast, the DNA-binding RTA mutant exhibited less transcriptional activity than did the wt RTA. These results demonstrated that (i) the tested RTA-binding sites can act as RTA-responsive enhancers of GMNN and EFNA1, and (ii) the DNA-binding activity of RTA also contributes to the RTA-mediated transactivation of GMNN and EFNA1 host genes (Fig. 4B).
FIG 4.
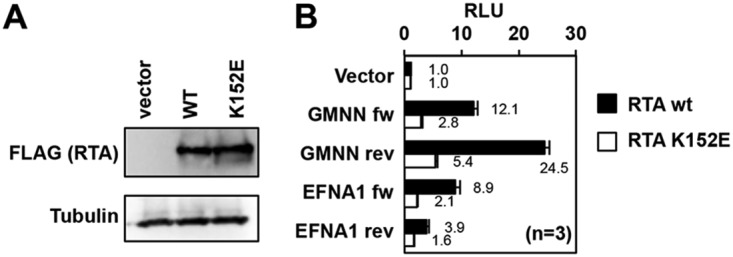
RTA-binding sites at core RIGs act as RTA-responsive enhancers. (A) Immunoblot analysis of the expression of the wild type 3×FLAG-RTA and the DNA-binding mutant 3×FLAG-RTA (RTA K152E) in transfected 293T cells. (B) 293T cells were cotransfected with the luciferase reporter plasmid and either the wild-type or the DNA-binding mutant 3×FLAG-RTA. At 48 h posttransfection, a luciferase reporter assay was performed. The enhancer activities of the DNA fragments containing the RTA-binding sites, which were derived from GMNN and EFNA1 core RIGs, were tested in both orientations (forward [fw] and reverse [rev]). RLU, relative light units.
Testing RTA-mediated host gene induction in different cell types.
To investigate whether RTA can also induce its host target genes in cell types other than BCBL1, we tested the effect of RTA on the expression of several core RIGs in different cell lines with or without KSHV (Fig. 5). First, we confirmed that the increased level of GMNN RNA transcript also resulted in increased GMNN protein production during KSHV lytic reactivation in the TRExBCBL1-3×FLAG-RTA cell line (Fig. 5A). The upregulation of GMNN could also be readily detected in the Dox-inducible RTA-expressing KSHV+ renal epithelial carcinoma cell line iSLKBAC16, in which the lytic reactivation of the recombinant KSHV clone BAC16 was triggered by inducing the expression of the RTA transgene with either Dox alone or Dox and histone deacetylase inhibitor (NaB) together (Fig. 5B). In addition, we confirmed the induction of eight of the core RIGs (APOBEC3H, GGT6, DLL4, HEY1, HES1, TGFB3, EFNA1, and GMNN) in iSLKBAC16 cells at 24 hpi (Fig. 5C and D), as well as in two Dox-inducible RTA-expressing KSHV-free cell lines, such as the B-cell lymphoma cell line TRExBJAB-3×FLAG-RTA and the renal epithelial carcinoma cell line iSLK (Fig. 5E to H). In the KSHV-free cell lines, RTA expression was induced for 6, 12, and 24 h, followed by immunoblot or RT-qPCR analysis of the expression of the core RIGs. We found that RTA could rapidly induce each host gene in the absence of any other viral factors in both KSHV-free cell lines, except for TGFB3 and EFNA1, which could be induced by RTA only in iSLK cells (Fig. 5H). These results indicated that RTA could also induce the expression of its host target genes in multiple different cell types without any other KSHV factors, supporting our notion that RTA is directly responsible for the upregulation of core RIGs during KSHV lytic reactivation. However, our data also indicate that the host cell type can also affect the target gene repertoire of RTA, which likely depends on the differential host chromatin environment and transcriptional cofactors in the various cell types.
FIG 5.

Testing RTA-driven induction of core RIGs in various KSHV-infected and KSHV-free cell types. Immunoblot analysis of KSHV+ TRExBCBL1-3×FLAG-RTA (A) and iSLKBAC16 (B) cell lines at the indicated postinduction time points. The quantification of GMNN expression on the immunoblots is shown as fold change relative to 0 hpi. (C and D) RT-qPCR analysis of the expression of the indicated core RIGs in iSLKBAC16 cells. The induction of core RIGs were calculated as relative to the expression of 18S rRNA (C) or as the fold change relative to 0 hpi (D). ND, not detectable. (E and F) Immunoblot analysis of RTA and GMNN expression in two different KSHV-free cell lines in which the expression of the transgene RTA was induced by Dox for the indicated periods of time. The quantification of GMNN expression on the immunoblots is shown as fold change relative to 0 hpi. (G) Gene expression of the indicated core RIGs was measured by RT-qPCR in two different Dox-inducible RTA-expressing KSHV-free cell lines at the indicated hours postinduction of RTA. (H) RT-qPCR measurement of TGFB3 and EFNA1 induction relative to 0 hpi in iSLK cells. *, P < 0.05.
Role of RBP-Jκ in the induction of RTA’s host target genes.
The significant enrichment of RBP-Jκ’s DNA sequence-binding motif at the core RIGs prompted us to investigate if RBP-Jκ is also required for the induction of the host genes. We tested the requirement for RBP-Jκ both during KSHV reactivation and in KSHV-free cells (Fig. 6 and 7). For this purpose, we used isogenic KSHV+ (iSLKBAC16) and KSHV-free cell lines (iSLK) to be able to directly compare RBP-Jκ requirement for the induction of core RIGs in the presence versus in the absence of other viral factors and to avoid the potential effect of different genetic background of cell lines on the function of RBP-Jκ. We found that while the viability of cells was comparable between shRBP-Jκ and shControl samples, shRBP-Jκ greatly reduced lytic KSHV protein production in reactivated iSLKBAC16 cells, indicating inefficient lytic reactivation (Fig. 6A to C). This is in line with previous studies showing that RBP-Jκ is essential for the RTA-mediated induction of lytic viral genes (31–34, 53). Next, we selected 8 core RIGs which had an RBP-Jκ-binding motif at their RTA-binding sites and found that all except APOBEC3H needed RBP-Jκ for their efficient RTA-mediated induction during KSHV lytic reactivation (Fig. 6D).
FIG 6.

Effect of the shRNA depletion of RBP-Jκ on RTA-induced viral and host gene expression during KSHV reactivation. (A) Confirmation of the shRNA knockdown of RBP-Jκ by RT-qPCR. (B) Immunoblot analysis of viral gene expression in RBP-Jκ-treated reactivated iSLKBAC16 cells. The quantification of protein expression on the immunoblots is shown as fold change relative (Rel.) to shControl (shCtrl) at 0 hpi for RBP-Jκ or relative to shControl at 24 hpi in the case of viral proteins. LANA, latency-associated nuclear antigen. (C) Cell death analysis in shRNA-treated reactivated iSLKBAC16 cells (n.s., not significant). (D) RT-qPCR analysis of the RTA-mediated induction of core RIGs in the absence of RBP-Jκ (*, P < 0.05).
FIG 7.

RBP-Jκ is required for RTA-induced expression of core RIGs in KSHV-free cells. (A) Reduced expression of RBP-Jκ in shRBP-Jκ-treated iSLK cells was confirmed by RT-qPCR. (B) Immunoblot analysis of shRBP-Jκ-treated iSLK cells for the indicated proteins. The quantification of protein expression on the immunoblots is shown as relative fold change. (C) RT-qPCR quantification of the RTA-induced expression of core RIGs in the absence of RBP-Jκ in iSLK cells (*, P < 0.05).
We also tested if the RTA-driven induction of the core RIGs also depends on RBP-Jκ in the KSHV-free iSLK cell line (Fig. 7). Importantly, the expression of Dox-induced RTA transgene was comparable between shControl- and shRBP-Jκ-treated iSLK cells (Fig. 7A and B), and yet, the RTA-mediated induction of core RIGs was abolished in the absence of RBP-Jκ (Fig. 7B and C). We note that despite the presence of RBP-Jκ’s DNA-binding motif at the RTA-binding site of the APOBEC3H gene, its RTA-mediated induction does not need RBP-Jκ expression neither in iSLKBAC16 nor in iSLK cells (Fig. 6D and 7C). Based on our results, we predict that the majority of core RIGs require the cooperation of RTA with RBP-Jκ at the RTA-binding sites for their RTA-induced expression, as is the case at several RTA-induced KSHV genes. However, there are also core RIGs associated with RBP-Jκ-binding sites whose induction may not require RBP-Jκ but rather are repressed by RBP-Jκ, such as in the case of APOBEC3H. RBP-Jκ is also known to be able to act as a transcriptional repressor, which can affect the function of RTA in gene induction (59). Thus, further studies are warranted to investigate to what extent RBP-Jκ cooperates with RTA genome-wide, as well as what other RTA cofactors exist and how they influence RTA-mediated host gene regulation in the cell type- and site-specific chromatin environment.
Evaluating the requisite of the core RIGs GMNN and GGT6 for KSHV lytic reactivation.
Because RTA is responsible for the switch from latency to the lytic cycle in KSHV-infected cells, we hypothesized that the host genes induced directly and rapidly by RTA during KSHV reactivation could also be important for the initiation and/or progression of the viral lytic cycle. To test this idea, we inhibited the expression of two core RIGs, GMNN or GGT6, by shRNAs during KSHV reactivation and analyzed their effects on KSHV lytic gene induction, viral DNA replication, and virus production (Fig. 8 and 9). Geminin (GMNN) is known to be involved in cell cycle regulation and in chromatin remodeling that drives gene expression programs in cell fate decisions (60). GGT6 is a member of the gamma-glutamyltransferase gene family, whose members are integral plasma membrane proteins with extracellular enzymatic activity, which can cleave gamma-glutamyl peptide bonds and are involved in glutathione homeostasis (56). Neither GMNN nor GGT6 has been studied for its role in the regulation of KSHV lytic reactivation.
FIG 8.

Effect of geminin on KSHV lytic reactivation. (A and B) Immunoblot analysis of RTA and GMNN expression in shControl- and shGMNN-treated iSLK cells and the expression of KSHV proteins in shRNA-treated iSLKBAC16 cells at 0 hpi and 48 hpi. (C) Cell death analysis in shRNA-treated reactivated iSLKBAC16 cells at 24 hpi. (D) RT-qPCR analysis of viral gene expression in shControl- and shGMNN #1-treated iSLKBAC16 cells. (E) The copy number of KSHV genome in the shGMNN #1- and shControl-treated iSLKBAC16 cells was determined at the indicated time points of KSHV reactivation relative to 0 hpi. (F) Immunoblot analysis of the expression of viral proteins in shControl- and shGMNN-treated TRExBCBL1-3×FLAG-RTA cells at 0 hpi (latency), 12 hpi, 24 hpi, and 48 hpi. The quantification of protein expression on the immunoblots is shown as relative fold change. (G) Testing the expression of viral genes by RT-qPCR in shGMNN-treated TRExBCBL1-3×FLAG-RTA cells compared to shControl-treated cells at 0 hpi and 24 hpi. (H) The relative KSHV DNA load in shControl- and shGMNN-treated TRExBCBL1-3×FLAG-RTA cells at different time points of KSHV reactivation compared to 0 hpi (latency). (I) The same amount of supernatants from shControl and shGMNN samples at 72 hpi described in panel H was used to infect 293T cells, and the viral DNA level was measured in the infected cells by qPCR at 24 hpi. n.s., nonsignificant; *, P < 0.05.
We used two different GMNN-specific shRNAs to inhibit GMNN expression and found that shGMNN did not affect RTA expression, as shown both in iSLK and iSLKBAC16 cells (Fig. 8A and B). However, while shGMNN did not affect the viability of cells (Fig. 8C), it did greatly reduce the expression of viral genes downstream to RTA and KSHV lytic replication in iSLKBAC16 (Fig. 8B and D, and E). Similarly, shGMNN reduced the induction of lytic viral proteins in TRExBCBL1-3×FLAG-RTA cells during KSHV reactivation, where KSHV reactivation was triggered by treating the cells with Dox for 12, 24, or 48 h (Fig. 8F). We found that despite the comparable expression of the Dox-induced 3×FLAG-RTA in shControl and shGMNN cells, the expression of early KSHV protein ORF45 was modestly reduced as early as 12 hpi, while that of the late KSHV proteins ORF26 and K8.1 was greatly reduced at 24 and 48 hpi during KSHV reactivation in shGMNN-treated cells (Fig. 8F). RT-qPCR analysis of the expression of additional KSHV lytic genes also showed that while only a subset of early genes was affected in shGMNN-treated cells, the induction of all of the tested late genes was significantly reduced (Fig. 8G). Furthermore, we found that shGMNN diminished KSHV DNA replication, resulting in reduced virus production and infection (Fig. 8H and I). In summary, we propose that GMNN supports efficient viral gene expression and KSHV DNA replication through an as-yet-unknown mechanism.
GGT6 is one of the core RIGs whose expression level is below the detection limit during latency, but it is rapidly induced during KSHV reactivation (Fig. 2F and Table S6). To test the effect of GGT6 on KSHV lytic reactivation, we inhibited GGT6 expression by using different GGT6-specific shRNAs (Fig. 9). Importantly, while shGGT6 did not affect the expression of Dox-induced RTA transgene (Fig. 9A and B) and had no significant effect on cell viability (Fig. 9C and D), shGGT6 markedly reduced the induction of lytic viral genes (Fig. 9E and F), viral DNA replication (Fig. 9G), and virus production in iSLKBAC16 cells (Fig. 9H and I). In agreement with this observation, shGGT6 also greatly reduced the induction of all lytic genes that were tested in TRExBCBL1-3×FLAG-RTA cells as early as 12 hpi prior to lytic viral DNA replication, indicating a role for GGT6 in the early phase of KSHV reactivation (Fig. 9J and K). In contrast, neither the latent viral factor LANA nor the polycomb host protein RING1B, which are involved in the inhibition of lytic gene expression, were affected by shGGT6, indicating a specific role for GGT6 in the regulation of lytic gene expression (Fig. 9K). Taken together, these results indicate that the newly discovered core RIGs GMNN and GGT6 are critical host factors that are required for the RTA-mediated efficient lytic reactivation of KSHV.
DISCUSSION
It is well established that the viral transcription activator RTA is essential for the lytic reactivation of KSHV from latency; however, it is still an open question of which host genes are directly induced by RTA and what roles they play in the regulation of KSHV lytic cycle (17, 35). To fill this knowledge gap, we performed an unbiased and comprehensive genomics analysis to identify the RTA-induced target genes in the early phase of KSHV reactivation in PEL cells and test if they are required for KSHV lytic reactivation. Importantly, we focused on the first 12 h of lytic reactivation, which reflects an early stage of KSHV reactivation prior to viral DNA replication in PEL cells. In this phase of KSHV reactivation, the viral genome is still fully chromatinized, but specific histone modification changes already take place on the viral chromatin, accompanied by the induction of the IE and E genes (46). We hypothesized that not just the viral target genes of RTA but also those host target genes which are directly induced by RTA at this early stage of lytic reactivation can be crucial for driving the lytic gene expression program leading to the replication of viral DNA and virus production.
Our RTA ChIP-seq analysis identified 23 and 11,469 RTA-binding sites on the viral and host genomes, respectively. Many of the RTA-binding sites contained the Notch signaling transcription factor RBP-Jκ-binding motif under the RTA peak. We showed that the shRNA knockdown of RBP-Jκ abolished not only the induction of lytic viral genes but also most of the tested core RIGs. Thus, our data support the notion that RBP-Jκ is pivotal for RTA not only to transactivate its viral genes but also many of its host target genes (17, 35). A comparison of RTA binding with the histone modification map of KSHV published previously shows that RTA binds to viral genomic regions, which are either constitutively enriched in the activating histone marks H3 acetylation and H3K4me3 (e.g., 118- to 128-kb latency locus) or show a diminishing level of repressive histone mark H3K27me3 accompanied by increasing amount of H3 acetylation and H3K4me3 during KSHV reactivation (46). In contrast, RTA binding was excluded from most of the viral genomic regions with constitutive heterochromatin enriched in H3K9me3 and H3K27me3 (e.g., 30 to 60 kb and 100 to 115 kb), which contain mainly L genes (46). These data support the notion that RTA mainly induces E genes, which RTA can accomplish by recruiting histone acetyltransferases CBP/p300 and chromatin remodeling factors to its target promoters to modulate their chromatin (20, 21). Importantly, we also detected RTA binding at some L genes, such as ORF8 (RTA peak 3), ORF29 (RTA peak 11), and K8.1 (RTA peak 15), which is in accordance with the observations that RTA can mediate the transactivation of their promoters (34, 50). It is worthy of note that RTA peak 11 is also in close proximity to the E genes ORF30 and ORF31, two members of the L gene transcription preinitiation complex, suggesting that RTA may also be involved in their induction (61–63). Thus, RTA could also promote L gene expression by inducing the expression of viral genes required for the induction of L genes. Most recently, the Izumiya group reported an elegant analysis about the role of RTA binding in the regulation of the 3D structure of the KSHV genome and its connection to viral gene expression during KSHV lytic reactivation (51). They demonstrated that RTA could induce not only the viral genes directly downstream of the RTA-binding sites but also distal viral genes through RTA-mediated chromatin looping within the KSHV episome. Thus, the limited number of RTA bindings on the KSHV genome could virtually regulate the expression of all lytic genes by modulating the three-dimensional (3D) structure of the viral episome during viral reactivation. It remains an open question whether any of the RTA bindings on the viral genome can interact with those on the host genome forming host-virus interchromosomal loops, and if so, what roles they play in KSHV lytic reactivation.
While the RTA-mediated regulation of KSHV genes has been extensively studied, a comprehensive identification of RTA host target genes and their role in KSHV reactivation are still lacking. To begin to fill this knowledge gap, we performed an integrative genomics analysis in reactivated PEL cells. We could identify 163 RTA-bound host genes (core RIGs) that were induced by 12 hpi, despite the fact that we found 11,469 RTA-binding sites on the host genome. This indicates that there are a number of as-yet-unknown factors which can determine whether or not RTA binding at a host gene results in increased gene expression. We note that there are many more induced genes at 24 hpi than at 6 to 12 hpi, many of which could also be regulated by RTA. Further studies will be required to determine which host genes are directly induced by RTA or rather RTA-independent mechanisms at later time points of KSHV reactivation. In addition, the modifications of cellular chromatin, DNA methylation of the host genome, the presence of different host and viral TFs, and the depletion of RNA polymerase II on the host genome during KSHV lytic reactivation can all influence RTA binding on the host genome, whether or not RTA can induce its host target genes, and when it can induce them in the course of KSHV reactivation (17, 23, 26, 28, 35, 42, 64–67). Importantly, we found that several of the core RIGs are involved in the Notch signaling pathway (e.g., HEY1, HES1, and DLL4), which has been shown to play a role in KSHV pathogenesis (44, 45, 68). However, most of the identified core RIGs have not yet been identified as RTA target genes or tested for their role in KSHV lytic reactivation. We showed that the core RIGs (e.g., GMNN and EFNA1) could also be induced by RTA in KSHV-free cells, and their identified RTA-bound genomic regions can function as enhancers, which can be transactivated by RTA in the absence of other viral proteins. These results highlight the direct role of RTA in the induction of the core RIGs, many of which could be required for driving the lytic cycle of KSHV.
For DNA viruses, the regulation of the cell cycle is critical for promoting their replication (69). RTA overexpression has previously been shown to be capable of inducing cell cycle arrest in G0/G1 phase in KSHV-free cells, suggesting that RTA can be involved in cell cycle regulation in KSHV-infected cells undergoing lytic reactivation (70). Here, we discovered that one of the novel core RIGs was geminin (GMNN), which is involved not only in the regulation of cell cycle but can also control cell fate decision during development by driving specific gene expression programs. In fact, GMNN can function both as an inhibitor of DNA replication origin licensing and as a transcription factor, which can interact with other TFs and chromatin regulatory proteins to control cell proliferation and differentiation in multiple cell contexts (60). We found that the shRNA inhibition of GMNN reduced KSHV DNA replication and lytic viral gene expression, resulting in diminished virus production. Importantly, GMNN expression is also increased in human cytomegalovirus-infected cells, which has been linked to the dysregulation of the host cell cycle by the virus (71). In addition, geminin has been shown to play a role in the regulation of Epstein-Barr virus DNA replication, indicating that geminin can affect herpesvirus replication by modulating either the host cell cycle or viral DNA replication (72). Whether or not the replication defect of KSHV in the absence of geminin can be attributed to geminin being involved in cell cycle regulation or directly in the regulation of chromatin of viral and/or host genes critical for KSHV lytic reactivation remains unknown.
Interestingly, our genomics analysis revealed that many of the core RIGs encode plasma membrane proteins, suggesting that RTA can rapidly alter the protein composition of cellular membranes during KSHV reactivation in PEL cells. Many of these core RIGs are associated with the regulation of immune responses, cell adhesion, and cell-to-cell signaling (e.g., FCER2/CD23, IL1RAP, CD244, GPRC5A, PCDH1, EFNA1, EFNA3, and EFNA4) (see Fig. 3D and Table S4); thus, RTA-driven changes can affect many cellular processes, likely impacting KSHV pathogenesis. Importantly, the increased expression of cell adhesion molecules on the surface of reactivated PEL cells can facilitate their interaction with other cell types and formation of transient virological synapses, allowing direct cell-to-cell transmission of replicating KSHV, as shown for spreading of HIV from macrophages to CD4+ T cells (73, 74). In fact, cell-mediated transmission has been demonstrated to be critical for KSHV infection of B cells (75). Thus, we are speculating that RTA-induced host genes can facilitate not only KSHV replication but also its cell-to-cell transmission, which can provide protection for KSHV from being recognized by the immune system during infection.
To test if the plasma membrane-associated core RIGs are required for KSHV lytic reactivation, we investigated the function of GGT6, a novel RTA target host gene. GGT6 is a largely uncharacterized protein which is predicted to be a transmembrane protein in the plasma membrane that belongs to the gamma-glutamyltransferase gene family involved in glutathione homeostasis and protection of cells from oxidative damage of reactive oxygen species (ROS) (56, 76). In the clinic, the elevated expression of GGT in tumors is correlated with drug resistance. Our RNA-seq analysis showed that GGT6 was not expressed in latently infected PEL cells but was rapidly induced during reactivation, while the expression of other GGT members did not change significantly, likely providing steady-state protection for PEL cells. It is known that KSHV reactivation is associated with increased ROS, such as H2O2 production, which enhances lytic reactivation through activating the MAPK signaling pathway (77). However, if H2O2 is produced in excess, it leads to cell death. Therefore, we tested if GGT6 is required for the survival of reactivated KSHV-infected cells and whether the elevated expression of GGT6 results in increased GGT enzymatic activity in cells. We did not observe any significant cell death in shGGT6-treated reactivated iSLKBAC16 cells, excluding the possibility that reduced lytic KSHV reactivation in shGGT6 cells was due to cell death. We also assayed for change in GGT enzymatic activity during lytic reactivation in the presence or absence of shGGT6 treatment (data not shown), but we could not detect any effects. These results are in line with the prediction that GGT6 is enzymatically inactive, due to GGT6 lacking the key threonine residue in its putative enzymatic domain (78). Importantly, regardless of the mode of lytic reactivation, the depletion of GGT6 greatly reduced the induction of lytic genes and KSHV production. These results indicate that GGT6 is required for KSHV lytic reactivation and virus production, but further studies are needed to determine how GGT6 controls KSHV reactivation.
In summary, our study illustrates the power of unbiased integrative genomics studies in KSHV-infected cells for the identification of new candidate host target genes of RTA, which can be tested for their role in the viral life cycle in follow-up mechanistic studies. We hypothesize that many of the identified RTA-inducible host genes could be essential for the progression of KSHV lytic cycle, and therefore, they could serve as targets for therapeutic intervention in KSHV lytic reactivation.
MATERIALS AND METHODS
Cell lines.
293T (ATCC) cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (P/S). The origin and growing conditions of the iSLK cells are described elsewhere (79). The iSLK cell line carrying the KSHV clone BAC16 (iSLKBAC16) was cultured in DMEM medium with 10% FBS, P/S, 250 μg/ml G418, 1 μg/ml puromycin, and 1 mg/ml hygromycin (80). We generated the TRExBCBL1-3×FLAG-RTA and TRExBJAB-3×FLAG-RTA cell lines by electroporating the pOG44 and pcDNA5FRT/TO-3×FLAG-RTA plasmids into the TRExBCBL1 and TRExBJAB cell lines (generously provided by Jae U. Jung from the University of Southern California) and used 200 μg/ml hygromycin to establish stable cell lines. These cell lines and TRExBCBL1-Vector cells were cultured in RPMI medium containing 10% Tet System Approved FBS (TaKaRa), P/S, and hygromycin. The expression of the N-terminally 3×FLAG-tagged RTA was induced by adding 1 μg/ml doxycycline (Dox) to the medium. KSHV reactivation in the iSLKBAC16 cell line was induced with 1 μg/ml Dox and 1 mM sodium butyrate.
Antibodies.
The following antibodies were used for the ChIPs and/or immunoblots: anti-FLAG (Sigma F1804), normal mouse IgG (catalog no. sc-2025; Santa Cruz), anti-RTA (from Yoshihiro Izumiya, University of California, Davis), anti-ORF6 (from Gary S. Hayward, Johns Hopkins University), anti-vIRF1 (from Jae U. Jung, University of Southern California), anti-ORF45 (catalog no. sc-53883; Santa Cruz), anti-ORF26 (catalog no. NBP1-47357; Novus Biologicals), anti-K8.1 (catalog no. sc-65446; Santa Cruz), anti-K8 (catalog no. sc-57889; Santa Cruz), anti-geminin (catalog no. ab195047; Abcam), anti-RBP-Jκ (catalog no. 5313; Cell Signaling), anti-RING1B (catalog no. ab3832; Abcam), and anti-LANA (catalog no. 13-210-100; Advanced Biotechnologies).
Lentiviral shRNA knockdown.
The RBP-Jκ-, GMNN-, and GGT6-specific short hairpin RNAs (shRNAs) were expressed from pLKO.1 lentiviral vector. The target sequences are shown in Table 2. shRBP-Jκ has been validated and used in previous studies (81). The shRNA lentiviruses were produced as described previously (82). TRExBCBL1-3×FLAG-RTA and iSLKBAC16 cells were infected with shRNA lentiviruses in the presence of 8 μg/ml polybrene. Two days after lentivirus infection, the iSLKBAC16 cells were split 1:3 to 1:4 and induced with 1 μg/ml doxycycline and 1 mM sodium butyrate on the following day for the indicated time periods. Similarly, 2 days after lentivirus infection, TRExBCBL1-3×FLAG-RTA cells were treated with 1 μg/ml Dox to induce KSHV reactivation for the indicated time periods.
TABLE 2.
Oligonucleotide sequences
| Name | Oligonucleotide sequence (5′ to 3′) | Methoda |
|---|---|---|
| shGMNN #1 | TGCCAACTCTGGAATCAAA | shRNA kd |
| shGMNN #2 | GCAGAGTACATAACTACATAA | shRNA kd |
| shGGT6 #1 | GTGGCCAAGTCTACCACTAGT | shRNA kd |
| shGGT6 #2 | CCTTCTCACCTCCTCGCTCAA | shRNA kd |
| shRBP-Jκ | CCCTAACGAATCAAACACAAA | shRNA kd |
| GMNN LucF | ACTGAAGCTTATTTGGCTCATGGAAAGAGGT | Cloning |
| GMNN LucR | TATCAAGCTTCCCTACGCAATGTTCTTCAAA | Cloning |
| EFNA1 LucF | ACTGAAGCTTTCAGACTGGGCCAGCTCCAG | Cloning |
| EFNA1 LucR | TATCAAGCTTCCTAGATGCCGGGAATAAAG | Cloning |
| AIMP2 F | TCAAGTGCTTTGGAGAACAGAA | RT-qPCR |
| AIMP2 R | CATCGTCTGGATGCTGAATTT | RT-qPCR |
| APOBEC3H F | CCAAGTCACCTGTTACCTCAC | RT-qPCR |
| APOBEC3H R | AAGATGCCCAGGTTCAGATG | RT-qPCR |
| ATF3 F | TCAAGGAAGAGCTGAGGTTTG | RT-qPCR |
| ATF3 R | CATCTTCTTCAGGGGCTACCT | RT-qPCR |
| DLL4 F | CCCTGGCAATGTACTTGTGAT | RT-qPCR |
| DLL4 R | GTGGTGGGTGCAGTAGTTGAG | RT-qPCR |
| EFNA1 F | CACACCGTCTTCTGGAACAGT | RT-qPCR |
| EFNA1 R | TCATAGTGCGGACAGATGATG | RT-qPCR |
| GGT6 F | CTGGGGATGCTCTACTGAGTCT | RT-qPCR |
| GGT6 R | AGGTAGCTGCTCTGCTGGTTC | RT-qPCR |
| GMNN F | CAAAAGGAAACATCGGAATGA | RT-qPCR |
| GMNN R | AATGACTCCTGGGTGACTCCT | RT-qPCR |
| HES1 F | TCTGGAAATGACAGTGAAGCA | RT-qPCR |
| HES1 R | GTCACCTCGTTCATGCACTC | RT-qPCR |
| HEY1 F | AGGTTCCATGTCCCCAACTAC | RT-qPCR |
| HEY1 R | TGTTATTGATCCGGTCTCGTC | RT-qPCR |
| ID2 F | CACGGATATCAGCATCCTGTC | RT-qPCR |
| ID2 R | CACACAGTGCTTTGCTGTCAT | RT-qPCR |
| MAFB F | CTCAGCACTCCGTGTAGCTC | RT-qPCR |
| MAFB R | TCCTCGAGGTGTGTCTTCTGT | RT-qPCR |
| TBX3 F | GCACCTGGAGGCTAAAGAACT | RT-qPCR |
| TBX3 R | TATCCAGCCCAGAACATCTCA | RT-qPCR |
| TGFB3 F | GATCGAGCTCTTCCAGATCCT | RT-qPCR |
| TGFB3 R | GACATCAAAGGACAGCCACTC | RT-qPCR |
| TSC22D3 F | GCTCTCCATCCTGCTCTTCTT | RT-qPCR |
| TSC22D3 R | GCCTGTTCGATCTTGTTGTCT | RT-qPCR |
| TSLP F | AGCCACATTGCCTTACTGAAA | RT-qPCR |
| TSLP R | CCTTAGTTTTCATGGCGAACA | RT-qPCR |
| ZCCHC12 F | GGAGACCAGAAGGCGGAATC | RT-qPCR |
| ZCCHC12 R | TCAGCTGCACAGAATTCAAGTT | RT-qPCR |
| ORF6 F | TAAAATACGCGTGTGGGAAAG | ChIP-qPCR |
| ORF6 R | CAGAGTCCCCTTTCCTTGTTT | ChIP-qPCR |
| ORF57 F | AGCGAAGTCACGGTAACACTT | ChIP-qPCR |
| ORF57 R | CCACTGGTACCACAAACGAAA | ChIP-qPCR |
| ORF25 F | ACAGTTTATGGCACGCATAGTG | ChIP-qPCR |
| ORF25 R | GGTTCTCTGAATCTCGTCGTGT | ChIP-qPCR |
| RTA F | TGAGGTCTATTTCCCACGACA | ChIP-qPCR |
| RTA R | ACAGCTCCGACGATGAGTATG | ChIP-qPCR |
| EFNA1 F | ACACGCGGCTGTGATACTACT | ChIP-qPCR |
| EFNA1 R | CGAACAGGCTCTTGAATATGG | ChIP-qPCR |
| GGT6 F | CTGCCCCATTAGGACACAAG | ChIP-qPCR |
| GGT6 R | AAGGCTGAACTCCTTCTCGAC | ChIP-qPCR |
| GMNN F | TGCTAAGTGGCTTTCAGCTTT | ChIP-qPCR |
| GMNN R | AAACATTGCTTCCATCACGTC | ChIP-qPCR |
kd, knockdown.
Cell viability assays, KSHV infection, and quantification of viral DNA.
Cell viability was quantified in three independent experiments by using standard Trypan blue exclusion test, according to the manufacturer’s instructions (Thermo Fisher). Cell death was measured in the shRNA-treated iSLKBAC16 cells at 24 hpi and was calculated as the mean values of cell death numbers relative to the total cell numbers in the shRNA-treated samples. KSHV infection and the quantification of viral DNA were performed as described previously (82).
Plasmids, transfections, and luciferase assay.
GMNN-pGL4.27 and EFNA1-pGL4.27 plasmids were generated by PCR amplifying a short DNA fragment containing the RTA-binding sites of the GMNN gene (406 bp) and the EFNA1 gene (500 bp) using the primers listed in Table 2. The PCR fragments were cloned in both orientations into the HindIII site of pGL4.27 luciferase vector in front of the minimal promoter. The full-length wild-type and the K152E mutant RTA were expressed from pCDH-CMV-MCS-EF1-puro vector. 293T cells were cotransfected with pGL4.27 luciferase plasmids and wild-type or K152E mutant RTA using PEI transfection reagent (Polysciences) (22). At 48 h posttransfection, the luciferase assay was performed using a luciferase assay kit from Promega, following the manufacturer’s instructions. Each luciferase experiment was performed at least three times, and three biological samples per treatments were performed.
RNA purification and RT-qPCR.
RNA purification and quantification of gene expression by RT-qPCR were performed as described previously (82). We note that 1 μg of total RNA was used for cDNA synthesis, and 10-fold-diluted cDNA was subjected to quantitative PCR (qPCR) analysis for measuring the expression of the host target genes. Gene expression changes were calculated either by the 2−ΔΔCT method to obtain fold change data or using the 2−ΔCT method, in which target gene expression was determined relative to the level of 18S rRNA. The 2−ΔCT method was only used where the expression of target genes was not detectable at 0 hpi. For significance testing, a two-tailed Student t test was performed. The qPCR was performed in CFX96 real-time PCR machine using SYBR Green supermix (Bio-Rad). The RT-qPCR primers for the KSHV genes have been reported elsewhere (83). The RT-qPCR primer pairs for the host genes are listed in a 5′ to 3′ orientation in Table 2.
ChIP assay.
The ChIPs were performed as previously described for the ChIP-seq assay (65). Briefly, 5 × 107 of TRExBCBL1-Vector and TRExBCBL1-3×FLAG-RTA cells were used for chromatin preparation. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and 0.125 M (final concentration) glycine was added subsequently for quenching the cross-linker. Cells were resuspended in swelling buffer (25 mM HEPES-KOH [pH 7.5], 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1 mM dithiothreitol [DTT], protease inhibitor cocktail) and incubated for 10 min at 4°C. After centrifugation, the pellets were resuspended in sonication buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, protease inhibitors) and sonicated by a Diagonade sonicator, according to the manufacturer’s guidelines. After centrifugation, the sonicated chromatin was diluted with dilution buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% Na-deoxycholate, 0.1% SDS, protease inhibitors) for the ChIP assays. For each ChIP-qPCR experiment, 10 μg of chromatin was incubated overnight with 0.1 μg of FLAG or IgG antibody preconjugated to magnetic beads. For ChIP-seq experiments, pools of 4 individual ChIPs with 20 μg of chromatin and 2 μg of FLAG antibody were used. The ChIPs were washed twice with each of the following buffers at 4°C: dilution buffer (described above), low-salt buffer (20 mM Tris-HCl [pH 8.1], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, protease inhibitors), high-salt buffer (20 mM Tris-HCl [pH 8.1], 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, protease inhibitors), and LiCl buffer (10 mM Tris [pH 8.1], 250 mM LiCl, 1 mM EDTA, 1% Na-deoxycholate, 1% NP-40). Finally, the ChIPs were also washed with Tris-EDTA (TE) buffer once. ChIPs and input chromatin were RNase-A treated in TE buffer for 30 min, followed by proteinase K treatment, while also reverse cross-linking at 60°C overnight. DNA was purified by phenol-chloroform-isoamyl alcohol extraction. Three independent ChIPs were performed and measured in duplicate by qPCR. The ChIP data are the averages of the results of three independent ChIP experiments, which are shown as the percentage of the immunoprecipitated DNA compared to input DNA. For each ChIP primer pair, standard curves were established. The primer sequences are shown in 5′ to 3′ orientation in Table 2. For a negative control, we used the CTCF/cohesin-binding site in the erythroid-specific DNase I hypersensitive site (HS1) of the β-globin gene regulatory region, whose ChIP-qPCR primers have been published previously (82).
Integrated RNA-seq and ChIP-seq analysis.
Multiplexed libraries were sequenced with 75-bp reads on Illumina sequencers. Total RNA was purified from TRExBCBL1-3×FLAG-RTA cells at 0, 6, 12, and 24 hpi. Strand-specific RNA-seq libraries were prepared from 3 biological replicates at each time point by using a KAPA Stranded RNA-seq kit, according to the manufacturer’s instructions. Reads from the time course RNA-seq data were aligned to the hg38 human genome with STAR aligner (2.4.1d version), using default settings in the FLOW 6.0 (6.0.17.1212 build) software analysis package (Partek, Inc., St. Louis, MO). The RefSeq Transcripts 83 (2017-11-01 version) human transcript genomic database was applied. Gene- and transcript-level reads per kilobase per million (RPKM) values were calculated from the aligned reads in the Genomics Suite (7.17.1103 build) software analysis package with standard settings (Partek Inc.). The gene-level RPKM values were set to a minimum of 0.001 RPKM prior to the differential gene expression analysis. We used 1-way analysis of variance (ANOVA) model for significance testing in the differential gene expression analysis. The list of genes, which were more than 2-fold differentially expressed, was generated by using step-up Benjamini-Hochberg false-discovery rate correction (FDR) of <0.05 in the Partek Genomics Suite platform. The principal-component analysis and the hierarchical clustering of RNA-seq data were performed within the Partek Genomics Suite platform.
ChIP-seq libraries from the RTA (FLAG) ChIP and input DNA were constructed using the NuGen kit, according to the manufacturer’s instructions. Reads from RTA (FLAG) ChIP-seq and input were mapped to both the KSHV genome (GenBank accession no. NC_009333.1) with the Isaac2 aligner software and the human genome (hg38) with the Bowtie 2 software using the Partek FLOW 6.0 platform, with default settings. Bigwig-formatted ChIP-seq data set files were created from the aligned reads with Galaxy BamCompare tool with 50-bp-bin-length default settings. Subsequently, the RTA ChIP-aligned reads were normalized against the corresponding genomic input, and the RTA peaks were called using the MACS2 software. The RTA peak calling on the KSHV genome was performed with 95-bp extension size, bypassed the shifting model, and retained maximum 30 duplicates with adjusted default parameters. The RTA peak calling on the human genome was performed with 147-bp extension size and retained a maximum 2 duplicates adjusted with default parameters. The curated final RTA peaks with at least 3-fold-enrichment cutoff and stringent q value of <0.005 were retained for further analysis. Using the Partek Genomics Suite, host genes were called RTA bound if there was at least one curated RTA peak in the corresponding gene body or within a 10-kb distance of their transcription start site (TSS). The 10-kb radius of the TSS was selected because this genomic region is known to possess the majority of gene regulatory sites involved in gene transcription control (84, 85). Gene ontology analysis was performed using David Gene ontology bioinformatics tool. The top gene ontology terms with at least 5 genes were listed and ranked based on their EASE score threshold. The supplemental tables contain further detailed information, such as RNA-seq data sets with differential gene expression data, RTA ChIP-seq peak coordinates, and gene ontology analysis.
Data availability.
The genomics data sets are available under GEO accession numbers GSE123897 and GSE123898 in the NCBI GEO Database.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jae U. Jung (University of Southern California) for providing TRExBCBL1, TRExBCBL1-Vector, TRExBJAB, iSLK, and iSLKBAC16 cell lines, as well as the vIRF1 antibody. We are grateful to Gary S. Hayward (Johns Hopkins University) for providing the ORF6 antibody and to Yoshihiro Izumiya (University of California, Davis) for the RTA antibody. We also thank Shannon Wallet, Michael Kladde, and Rolf Renne (University of Florida) for the helpful discussions and support.
B.P. was supported by NIH grants R03DE028029 and R01AI132554, as well as the UF Research Opportunity Seed Fund. Z.T. was supported by NIH grants R01AI132554 and R03DE025562, a UFHCC A1 Accelerator Grant, and the UF Research Opportunity Seed Fund. S.J.J. was supported by NIH training grant T90DE21990. N.M. was supported in part by the UF Emerging Scholars program.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01978-18.
REFERENCES
- 1.Jha HC, Banerjee S, Robertson ES. 2016. The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5:E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purushothaman P, Uppal T, Verma SC. 2015. Molecular biology of KSHV lytic reactivation. Viruses 7:116–153. doi: 10.3390/v7010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavallin LE, Goldschmidt-Clermont P, Mesri EA. 2014. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposi’s sarcoma associated with HIV/AIDS. PLoS Pathog 10:e1004154. doi: 10.1371/journal.ppat.1004154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duus KM, Lentchitsky V, Wagenaar T, Grose C, Webster-Cyriaque J. 2004. Wild-type Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J Virol 78:4074–4084. doi: 10.1128/JVI.78.8.4074-4084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol 77:9738–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang HH, Ganem D. 2013. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13:429–440. doi: 10.1016/j.chom.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hosseinipour MC, Sweet KM, Xiong J, Namarika D, Mwafongo A, Nyirenda M, Chiwoko L, Kamwendo D, Hoffman I, Lee J, Phiri S, Vahrson W, Damania B, Dittmer DP. 2014. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. mBio 5:e01633-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giffin L, Damania B. 2014. KSHV: pathways to tumorigenesis and persistent infection. Adv Virus Res 88:111–159. doi: 10.1016/B978-0-12-800098-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glesby MJ, Hoover DR, Weng S, Graham NM, Phair JP, Detels R, Ho M, Saah AJ. 1996. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: data from the Multicenter AIDS Cohort Study. J Infect Dis 173:1477–1480. [DOI] [PubMed] [Google Scholar]
- 14.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA, Roche Ganciclovir Study Group . 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med 340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 15.Mocroft A, Youle M, Gazzard B, Morcinek J, Halai R, Phillips AN. 1996. Anti-herpesvirus treatment and risk of Kaposi’s sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 10:1101–1105. [PubMed] [Google Scholar]
- 16.Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. 2005. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol 79:3479–3487. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front Microbiol 3:30. doi: 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. 2001. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J Virol 75:6786–6799. doi: 10.1128/JVI.75.15.6786-6799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guito J, Lukac DM. 2015. KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 7:72–109. doi: 10.3390/v7010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gwack Y, Baek HJ, Nakamura H, Lee SH, Meisterernst M, Roeder RG, Jung JU. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol Cell Biol 23:2055–2067. doi: 10.1128/MCB.23.6.2055-2067.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gwack Y, Byun H, Hwang S, Lim C, Choe J. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J Virol 75:1909–1917. doi: 10.1128/JVI.75.4.1909-1917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Wang J, Wood C, Xu D, Zhang L. 2005. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 replication and transcription activator regulates viral and cellular genes via interferon-stimulated response elements. J Virol 79:5640–5652. doi: 10.1128/JVI.79.9.5640-5652.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM. 2007. Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J Virol 81:8451–8467. doi: 10.1128/JVI.00265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guito J, Gavina A, Palmeri D, Lukac DM. 2014. The cellular peptidyl-prolyl cis/trans isomerase Pin1 regulates reactivation of Kaposi’s sarcoma-associated herpesvirus from latency. J Virol 88:547–558. doi: 10.1128/JVI.02877-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gwack Y, Nakamura H, Lee SH, Souvlis J, Yustein JT, Gygi S, Kung HJ, Jung JU. 2003. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol Cell Biol 23:8282–8294. doi: 10.1128/MCB.23.22.8282-8294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Z, Liu Y, Liang D, Wang Z, Robertson ES, Lan K. 2010. Cellular corepressor TLE2 inhibits replication-and-transcription-activator-mediated transactivation and lytic reactivation of Kaposi’s sarcoma-associated herpesvirus. J Virol 84:2047–2062. doi: 10.1128/JVI.01984-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. 2009. NF-κB serves as a cellular sensor of Kaposi’s sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jκ coactivator. J Virol 83:4435–4446. doi: 10.1128/JVI.01999-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izumiya Y, Lin SF, Ellison T, Chen LY, Izumiya C, Luciw P, Kung HJ. 2003. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J Virol 77:1441–1451. doi: 10.1128/JVI.77.2.1441-1451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko YC, Tsai WH, Wang PW, Wu IL, Lin SY, Chen YL, Chen JY, Lin SF. 2012. Suppressive regulation of KSHV RTA with O-GlcNAcylation. J Biomed Sci 19:12. doi: 10.1186/1423-0127-19-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao W, Tang Y, Lin SF, Kung HJ, Giam CZ. 2003. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J Virol 77:3809–3815. doi: 10.1128/JVI.77.6.3809-3815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll KD, Bu W, Palmeri D, Spadavecchia S, Lynch SJ, Marras SA, Tyagi S, Lukac DM. 2006. Kaposi’s sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP- Jκ/CSL to activate the Notch pathway. J Virol 80:9697–9709. doi: 10.1128/JVI.00746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev 16:1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Persson LM, Wilson AC. 2010. Wide-scale use of Notch signaling factor CSL/RBP-Jκ in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol 84:1334–1347. doi: 10.1128/JVI.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scholz BA, Harth-Hertle ML, Malterer G, Haas J, Ellwart J, Schulz TF, Kempkes B. 2013. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog 9:e1003336. doi: 10.1371/journal.ppat.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aneja KK, Yuan Y. 2017. Reactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus: an update. Front Microbiol 8:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J Virol 77:6474–6481. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katano H, Sato Y, Kurata T, Mori S, Sata T. 2000. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 269:335–344. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 38.Li Q, He M, Zhou F, Ye F, Gao SJ. 2014. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle. J Virol 88:6355–6367. doi: 10.1128/JVI.00219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 41.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol 72:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin HJ, DeCotiis J, Giron M, Palmeri D, Lukac DM. 2014. Histone deacetylase classes I and II regulate Kaposi’s sarcoma-associated herpesvirus reactivation. J Virol 88:1281–1292. doi: 10.1128/JVI.02665-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang H, Gwack Y, Kingston D, Souvlis J, Liang X, Means RE, Cesarman E, Hutt-Fletcher L, Jung JU. 2005. Activation of CD21 and CD23 gene expression by Kaposi’s sarcoma-associated herpesvirus RTA. J Virol 79:4651–4663. doi: 10.1128/JVI.79.8.4651-4663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Hu H, He Z, Liang D, Sun R, Lan K. 2016. Fine-tuning of the Kaposi’s sarcoma-associated herpesvirus life cycle in neighboring cells through the RTA-JAG1-Notch pathway. PLoS Pathog 12:e1005900. doi: 10.1371/journal.ppat.1005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yada K, Do E, Sakakibara S, Ohsaki E, Ito E, Watanabe S, Ueda K. 2006. KSHV RTA induces a transcriptional repressor, HEY1 that represses RTA promoter. Biochem Biophys Res Commun 345:410–418. doi: 10.1016/j.bbrc.2006.04.092. [DOI] [PubMed] [Google Scholar]
- 46.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6:e1001013. doi: 10.1371/journal.ppat.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bu W, Palmeri D, Krishnan R, Marin R, Aris VM, Soteropoulos P, Lukac DM. 2008. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J Virol 82:10709–10723. doi: 10.1128/JVI.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Ye F, Xie J, Kuhne K, Gao SJ. 2009. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386:290–302. doi: 10.1016/j.virol.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. 2009. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 387:76–88. doi: 10.1016/j.virol.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziegelbauer J, Grundhoff A, Ganem D. 2006. Exploring the DNA binding interactions of the Kaposi’s sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J Virol 80:2958–2967. doi: 10.1128/JVI.80.6.2958-2967.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell M, Watanabe T, Nakano K, Davis RR, Lyu Y, Tepper CG, Durbin-Johnson B, Fujimuro M, Izumiya Y. 2018. KSHV episomes reveal dynamic chromatin loop formation with domain-specific gene regulation. Nat Commun 9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6:e1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang Y, Ganem D. 2003. Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc Natl Acad Sci U S A 100:8490–8495. doi: 10.1073/pnas.1432843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, Renne R. 2007. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog 3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyu Y, Nakano K, Davis RR, Tepper CG, Campbell M, Izumiya Y. 2017. ZIC2 is essential for maintenance of latency and is a target of an immediate-early protein during KSHV lytic reactivation. J Virol 91:e00980-17. doi: 10.1128/JVI.00980-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heisterkamp N, Groffen J, Warburton D, Sneddon TP. 2008. The human gamma-glutamyltransferase gene family. Hum Genet 123:321–332. doi: 10.1007/s00439-008-0487-7. [DOI] [PubMed] [Google Scholar]
- 57.Chakraborty S, Veettil MV, Bottero V, Chandran B. 2012. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci U S A 109:E1163. doi: 10.1073/pnas.1119592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F. 2012. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat Med 18:961–966. doi: 10.1038/nm.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson JE, Macdonald RJ. 2011. Notch-independent functions of CSL. Curr Top Dev Biol 97:55–74. doi: 10.1016/B978-0-12-385975-4.00009-7. [DOI] [PubMed] [Google Scholar]
- 60.Patmanidi AL, Champeris Tsaniras S, Karamitros D, Kyrousi C, Lygerou Z, Taraviras S. 2017. Concise review: geminin-a tale of two tails: DNA replication and transcriptional/epigenetic regulation in stem cells. Stem Cells 35:299–310. doi: 10.1002/stem.2529. [DOI] [PubMed] [Google Scholar]
- 61.Brulois K, Wong LY, Lee HR, Sivadas P, Ensser A, Feng P, Gao SJ, Toth Z, Jung JU. 2015. The association of Kaposi’s sarcoma-associated herpesvirus ORF31 with ORF34 and ORF24 is critical for late gene expression. J Virol 89:6148–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis ZH, Hesser CR, Park J, Glaunsinger BA. 2016. Interaction between ORF24 and ORF34 in the Kaposi’s sarcoma-associated herpesvirus late gene transcription factor complex is essential for viral late gene expression. J Virol 90:599–604. doi: 10.1128/JVI.02157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jäger S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen CP, Lyu Y, Chuang F, Nakano K, Izumiya C, Jin D, Campbell M, Izumiya Y. 2017. Kaposi’s sarcoma-associated herpesvirus hijacks RNA polymerase II to create a viral transcriptional factory. J Virol 91:e02491-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K. 2017. Cooperative binding of transcription factors orchestrates reprogramming. Cell 168:442–459.e20. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Journo G, Tushinsky C, Shterngas A, Avital N, Eran Y, Karpuj MV, Frenkel-Morgenstern M, Shamay M. 2018. Modulation of cellular CpG DNA methylation by Kaposi’s sarcoma-associated herpesvirus. J Virol 92:e00008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xi X, Persson LM, O'Brien MW, Mohr I, Wilson AC. 2012. Cooperation between viral interferon regulatory factor 4 and RTA to activate a subset of Kaposi’s sarcoma-associated herpesvirus lytic promoters. J Virol 86:1021–1033. doi: 10.1128/JVI.00694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emuss V, Lagos D, Pizzey A, Gratrix F, Henderson SR, Boshoff C. 2009. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog 5:e1000616. doi: 10.1371/journal.ppat.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fehr AR, Yu D. 2013. Control the host cell cycle: viral regulation of the anaphase-promoting complex. J Virol 87:8818–8825. doi: 10.1128/JVI.00088-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar P, Wood C. 2013. Kaposi’s sarcoma-associated herpesvirus transactivator Rta induces cell cycle arrest in G0/G1 phase by stabilizing and promoting nuclear localization of p27kip. J Virol 87:13226–13238. doi: 10.1128/JVI.02540-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biswas N, Sanchez V, Spector DH. 2003. Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication. J Virol 77:2369–2376. doi: 10.1128/JVI.77.4.2369-2376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, Dutta A. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287–296. [DOI] [PubMed] [Google Scholar]
- 73.Groot F, Welsch S, Sattentau QJ. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660–4663. doi: 10.1182/blood-2007-12-130070. [DOI] [PubMed] [Google Scholar]
- 74.Torres-Flores JM, Arias CF. 2015. Tight junctions go viral! Viruses 7:5145–5154. doi: 10.3390/v7092865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Myoung J, Ganem D. 2011. Infection of lymphoblastoid cell lines by Kaposi’s sarcoma-associated herpesvirus: critical role of cell-associated virus. J Virol 85:9767–9777. doi: 10.1128/JVI.05136-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanigan MH. 2014. Gamma-glutamyl transpeptidase: redox regulation and drug resistance. Adv Cancer Res 122:103–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, Gao SJ. 2011. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog 7:e1002054. doi: 10.1371/journal.ppat.1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West MB, Wickham S, Parks EE, Sherry DM, Hanigan MH. 2013. Human GGT2 does not autocleave into a functional enzyme: a cautionary tale for interpretation of microarray data on redox signaling. Antioxid Redox Signal 19:1877–1888. doi: 10.1089/ars.2012.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao WR, Hsieh RH, Hsu KW, Wu MZ, Tseng MJ, Mai RT, Wu Lee YH, Yeh TS. 2007. The CBF1-independent Notch1 signal pathway activates human c-myc expression partially via transcription factor YY1. Carcinogenesis 28:1867–1876. doi: 10.1093/carcin/bgm092. [DOI] [PubMed] [Google Scholar]
- 82.Toth Z, Smindak RJ, Papp B. 2017. Inhibition of the lytic cycle of Kaposi’s sarcoma-associated herpesvirus by cohesin factors following de novo infection. Virology 512:25–33. doi: 10.1016/j.virol.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, Kung HJ, Jung JU. 2013. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog 9:e1003813. doi: 10.1371/journal.ppat.1003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, Lancet D, Cohen D. 2017. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017:1–17. doi: 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.MacIsaac KD, Lo KA, Gordon W, Motola S, Mazor T, Fraenkel E. 2010. A quantitative model of transcriptional regulation reveals the influence of binding location on expression. PLoS Comput Biol 6:e1000773. doi: 10.1371/journal.pcbi.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomics data sets are available under GEO accession numbers GSE123897 and GSE123898 in the NCBI GEO Database.