

Abstract
Objectives
To identify the prevalence of dystonia in a RNA Polymerase III (POLR3)‐related leukodystrophy patient cohort and to further characterize their dystonic features.
Background
POLR3‐related leukodystrophy is a hypomyelinating leukodystrophy characterized by neurological and non‐neurological features. Dystonia remains a challenging and under‐recognized feature.
Methods
A retrospective chart review was performed in a cohort of 20 patients for whom videos of a standardized neurological examination were available. Patients were recruited at the Montreal Children's Hospital of the McGill University Health Center and the Myelin Disorders Bioregistry Project. Families were consented at the initial assessment and the following data was recorded: age and symptoms at clinical presentation, investigations, causal gene and mutation(s), type and severity of dystonia, and treatment response when needed. Standardized examination videos were reviewed by three independent reviewers and scored using the Global Dystonia Scale.
Results
10 males and 10 females were included in this study; 12/20 had POLR3A mutations, while 8/20 had POLR3B mutations; 19/20 patients had documented dystonia, with 3/19 requiring therapy. There was a good response in two patients to a single agent, and a poor response in one patient to three agents; the majority had mild‐to‐moderate multifocal dystonia without a functional impact.
Conclusions
Dystonia is a common, yet underdiagnosed, slowly progressive manifestation of POLR3‐related leukodystrophy, and in most cases has limited‐to‐no functional impact. When treatment is needed, good response to typically used medication may occur. Further studies are needed to assess evolution of dystonia over time, patients’ functional outcome, and response to therapy (when needed).
Keywords: 4H leukodystrophy, dystonia, POLR3‐related disorder
Introduction
POLR3‐related leukodystrophy is a recently described hypomyelinating leukodystrophy characterized by variable age of onset (early to late childhood) and neurological (i.e., cerebellar, pyramidal, extrapyramidal, and cognitive) and non‐neurological (i.e., dental, ocular, and endocrine) features. Over the last decade, five distinct clinical phenotypes were recognized and are now included under POLR3‐related leukodystrophy. These include: 4H syndrome (hypomyelination, hypodontia, hypogonadotropic hypogonadism)1, 2, 3; ataxia, delayed dentition and hypomyelination (ADDH)4, 5; tremor‐ataxia with central hypomyelination (TACH)6, 7, 8, 9; leukodystrophy with oligodontia (LO)10, 11; and hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC).12, 13 This disease has an autosomal recessive pattern of inheritance with mutations in POLR3A and POLR3B genes,6, 8, 12, 14 shown to be disease‐causing in 2011 and mutations in the POLR1C gene,15 recognized to cause this disorder in 2015.6, 8, 12, 14 Genotype‐phenotype correlations16 have been difficult to establish due to a large number of mutations and an even larger number of combinations of mutations. However, a cross‐sectional clinical study of this disorder suggests that a more severe course is typically seen in patients with POLR3A mutations, when compared to POLR3B. Among the neurological symptoms of this disorder, dystonia, which remains a challenging and under‐recognized feature of this disease, may have an impact on quality of life, especially in advanced disease states. In this study, we report the prevalence and characteristics of dystonia in a cohort of 20 patients with POLR3‐related leukodystrophy who underwent a standardized video protocol.
Methods
Patients
A retrospective cohort study was performed on 20 patients recruited at the Montreal Children's Hospital of the McGill University Health Center, Canada; and the Myelin Disorders Bioregistry Project at The Children's National Medical Center, Washington DC, between March 2012 and October 2014. All patients had molecularly confirmed POLR3‐related leukodystrophy and were initially recruited based on clinical history, examination, and typical MRI findings.
Standard protocol approvals, registrations, and patient consent approvals were obtained from research ethics boards at both institutions, and written informed consent was obtained from parents/legal guardians at the time of the first visit.
Data Collection
Detailed past medical information was obtained through clinical questionnaires completed by referring physicians or the families, supplemented by information from medical records. Pertinent medical information included perinatal, developmental, past medical and family history, age at onset of symptoms, neurological and non‐neurological features, and detailed diagnostic workup (including mutation analysis). Brain MRI studies of all patients were reviewed by the study supervisor (G.B.).
Dystonia Assessment
All participants were assessed by a videotaped examination performed by a pediatric neurologist using a standardized protocol (dystonia coalition adapted for children and degree of disabilities). Videos were scored using the Global Dystonia Scale (GDS).17 Information regarding treatment details were obtained as well. Two independent movement disorder specialists (G.B. and S.C.), together with G.A.Y. (senior neurology resident), reviewed the videotaped examination and rated the severity of dystonia using the GDS. The ratings of one of the three raters tended to be lower than the ratings of the other two. This rater did not observe any dystonia in seven patients for whom the other two raters observed dystonia. Therefore, before averaging ratings across the three raters, all ratings were adjusted using weights formed by the within‐rater averages across all patients and all body parts. The three weights used were 1.22, 1.32, and 0.57. After multiplying each rating by the appropriate weight, an average was taken across raters for each patient and each body part. The resulting values were not whole numbers in accordance with the original scale. Therefore, ranges were used to define severity: none (0), mild (>0 ≤ 2.0), mild‐to‐moderate (>2.0 ≤ 4.0), moderate (>4.0 ≤ 7.0), and severe (>7.0 ≤ 10.0).
Results
Demographic data showed an even distribution of gender among the group with a M:F ratio of 10:10. Among the 20 patients in this study, twelve patients carried mutations in POLR3A, while the remaining eight carried mutations in POLR3B, as indicated in Supporting Table 1. When comparing disease onset, the POLR3A group had a mean age of onset of 21.5 months, which was slightly younger than the POLR3B group's mean age of onset of 28 months. Cerebellar manifestations, such as ataxia and tremor were the presenting features in 10/12 patients with POLR3A mutations while ataxia and motor delay were the presenting features in 5/8 patients with POLR3B confirmed mutations. Other reported presenting manifestations among the group were: spasticity (6/20), behavioral disturbances (6/20), and learning difficulties (4/20). All patients enrolled in the study had the typical POLR3‐related leukodystrophy MRI findings, including diffuse hypomyelination with relative T2 hypointense signal in the dentate nuclei, anterolateral nuclei of the thalami, globi pallidi, posterior limbs of the internal capsules (in some), and optic radiations with a thin corpus callosum and cerebellar atrophy (Fig. 1).
Figure 1.
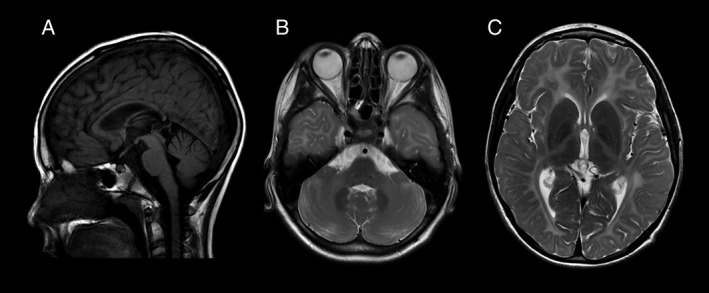
MRI characteristics of POLR3‐related leukodystrophy.
MRI of the brain of patient 17 at the age of 10 years and 7 months. On the sagittal T1 FLAIR, we note a thin corpus callosum and mild superior vermis atrophy (A). Diffuse hypomyelination of the cerebellum and cerebrum is seen on axial T2 weighted images (B, C) with relative preservation of the dentate nuclei (B), optic radiations (C), anterolateral nuclei of the thalamus (C) and globi pallidi (C).
The total dystonia score ranged from 0 to 82, with a mean of 16.66 (SD 18.39). The severity per total dystonia scale is indicated in Fig. 2. Only one of the 20 patients had severe dystonia, while most patients scored in the mild and mild‐to‐moderate range (10/20 patients; Table 1). When considering the dystonia distribution among body parts, the majority of patients had multifocal dystonia, with the distal arms and legs being the most commonly affected, while the trunk was the least frequently affected (in only one patient who had severe dystonia; Table 2). Video 1 illustrates a patient with moderate multifocal dystonia.
Figure 2.
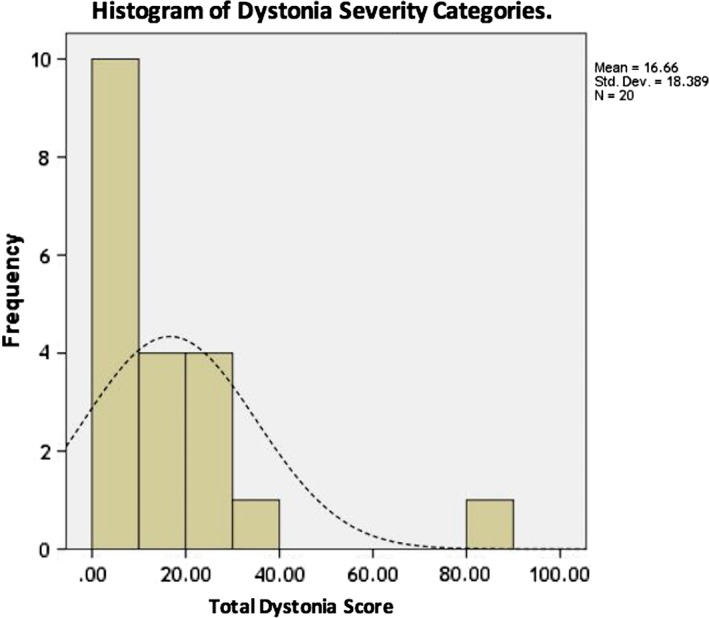
Histogram of Dystonia Severity scores categories.
The horizontal line shows the total dystonia rating score, none (0); mild (>0 ≤ 20); mild to moderate (>20 ≤ 40); moderate (>40 ≤ 70) and severe (>70 ≤ 100), while the vertical line represents the number of patients in each category. The dotted line represents the normal distribution curve.
Table 1.
Dystonia severity classification per different genetic mutation
| Genetic mutation Rating | POLR3A | POLR3B | Total | |||
|---|---|---|---|---|---|---|
| f | % | f | % | f | % | |
| No dystonia | 0 | 0.0 | 1 | 12.5 | 1 | 5.0 |
| Mild | 4 | 33.3 | 1 | 12.5 | 5 | 25.0 |
| Mild to Moderate | 3 | 25.0 | 2 | 25.0 | 5 | 25.0 |
| Moderate | 4 | 33.3 | 4 | 50.0 | 8 | 40.0 |
| Severe | 1 | 8.3 | 0 | 0.0 | 1 | 5.0 |
Abbreviations: (f), frequency of affected patients; (%), percentage among the group.
Table 2.
Prevalence and severity of dystonia for each body part among the study cohort
| Body part | Mild | Mild‐to‐moderate | Moderate | Severe | ||||
|---|---|---|---|---|---|---|---|---|
| f | % | f | % | f | % | f | % | |
| Upper face | 4 | 20 | 1 | 5 | 0 | 0 | 0 | 0 |
| Lower face | 1 | 5 | 0 | 0 | 1 | 5 | 0 | 0 |
| Jaw & tongue | 4 | 20 | 0 | 0 | 1 | 5 | 0 | 0 |
| Larynx | 2 | 10 | 5 | 25 | 0 | 0 | 0 | |
| Neck | 1 | 5 | 2 | 10 | 2 | 10 | 1 | 5 |
| R Proximal arm | 1 | 5 | 0 | 0 | 0 | 0 | 1 | 5 |
| L Proximal arm | 3 | 15 | 0 | 0 | 0 | 0 | 1 | 5 |
| R Distal hand | 6 | 30 | 7 | 35 | 5 | 25 | 1 | 5 |
| L Distal hand | 6 | 30 | 5 | 25 | 7 | 35 | 1 | 5 |
| R Upper leg | 1 | 5 | 0 | 0 | 0 | 1 | 5 | |
| L Upper leg | 0 | 0 | 1 | 5 | 0 | 0 | 1 | 5 |
| R Distal leg | 7 | 35 | 5 | 25 | 5 | 25 | 1 | 5 |
| L Distal leg | 7 | 35 | 3 | 15 | 5 | 25 | 1 | 5 |
| Trunk | 0 | 0 | 1 | 5 | 0 | 0 | 0 | 0 |
Abbreviations: (f), frequency of affected patients; (%), percentage among the group.
Discussion
Dystonia is a movement disorder defined as involuntary muscle contractions of agonist and antagonist muscles causing slow, repetitive movements or abnormal postures or both.18 These movements may be painful, and occasionally have a superimposed tremor component to it. The clinical diagnosis of dystonia is usually challenging, especially in the presence of other comorbid neurologic signs or symptoms. We believe it is an under recognized neurologic feature in POLR3‐related leukodystrophy due to the presence of other confounding comorbid neurologic features such as cerebellar and pyramidal signs. To our knowledge, this is the first study to evaluate dystonia in this disorder systematically.
The pathophysiologic considerations of dystonia remain controversial, principally in relation to which part of the central nervous system is primarily affected (i.e., basal ganglia alone or basal ganglia with other structures such as the thalamus, brainstem, parietal lobe, or cerebellum).19 Recently, several neurophysiological studies have identified functional abnormalities in the sensorimotor network in the basal ganglia and the cerebellothalamic pathway.20 We hypothesize that the underlying etiology of the dystonic phenotype in POLR3‐related leukodystrophy is likely related to diffuse hypomyelination and neuronal loss, which involves the basal ganglia connections, resulting in a network disorder.
While dystonia in the pediatric age group is typically generalized, the majority of our cohort manifested multifocal dystonia. Indeed, all dystonia patients had bilateral and symmetric limb involvement affecting upper and lower limbs in variable degrees, but never an isolated neck, laryngeal, or trunk dystonia. These body parts were involved only in patients with moderate to severe dystonia. Most of the patients had dystonia in the mild and mild‐to‐moderate severity range. When comparing both genetic subgroups, patients who fell towards the severe spectrum were patients with POLR3A mutations.
Moreover, the only three patients who had severe dystonia requiring treatment were carrying POLR3A mutations, correlating with the expected more severe and rapidly progressive course of previous reports.16
Current literature in the treatment of dystonia is sparse and relies mainly on symptomatic treatment. Treatment decisions are based on the distribution and severity of dystonia, together with the functional impact on the patient's daily life, and possible complications such as pain or hip dislocation in severe cases. First‐line treatment for generalized and multifocal dystonia is the oral medication trihexyphenidyl, an anticholinergic drug. The second‐line is tetrabenazine, a reversible dopamine‐depleting agent. Other agents used include baclofen, benzodiazepines, or L‐dopa in select cases. If a specific muscle group is affected, botulinum toxin injections can be used with high success rates. Finally, deep brain stimulation remains an option for refractory patients even though there is no systematic evidence for this patient population to date. In our small number of patients who needed treatment (three patients), two patients with moderate dystonia were well controlled on a single medication (L‐dopa and trihexyphenidyl). In the other patient who had severe dystonia with important paroxysmal exacerbations, three oral medications were tried, including trihexyphenidyl, tetrabenazine, and oxcarbazepine; despite these medications and the treatment of his concurrent spasticity, only a sub‐optimal response was obtained.
Finally, the authors acknowledge some limitations of this study including the retrospective nature of the project, which limits the quality of information gathered regarding the timing of onset and evolution of the dystonia. Another limiting factor is that the research team saw the patients at different stages of their disease. Lastly, the incomplete agreement between raters, which was adjusted statistically, is another limitation that could be addressed in future studies by having a greater number of reviewers. A prospective natural history study is required to understand the progression of dystonia, its impacts, and whether early detection and management could alter the course of the disease, and improve the patients’ quality of life.
Conclusions
Dystonia is a common, yet underdiagnosed clinical manifestation in POLR3‐related leukodystrophy; it tends to be evident as the disease progresses and in most individuals has no impact on their functional level. When treatment is needed, a good response to typically used medications is noted in some cases. Further studies are needed to assess the evolution of the dystonia over time, its functional impact on patients, and patients’ response to treatment.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
G.A.Y.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
L.T.: 1A, 1B, 1C, 3B
K.G.: 3B, Sanger sequencing
A.V.: 1A, 1B, 1C, 3B
R.S.: 1A, 1B, 1C, 3B
N.I.W.: 1A, 1B, 1C, 3B
S.C.: 1A, 1B, 1C, 3B
G.B.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
Disclosures
Ethical Compliance Statement: This study was approved by research ethics boards at the Montreal Children's Hospital of the McGill University Health Center, Canada; and the Myelin Disorders Bioregistry Project at The Children's National Medical Center, Washington DC. All patients' guardians/patients signed a consent form/assent form detailing the nature of the detailed phenotyping study, including the authorization to videotape and publish the examinations. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This study was supported by grants from the Canadian Institutes of Health Research (201610PJT‐377869 and MOP‐G2‐341146‐159133‐BRIDG), the Fondation les Amis d'Elliot, the Fondation Lueur d'Espoir pour Ayden, and the Réseau de Médecine Génétique Appliquée of the Fonds de Recherche en Santé du Québec. G.B. has received a Research Scholar Junior 1 award from the Fonds de Recherche du Québec en Santé (FRQS) 2012‐2016 and a Canadian Institute of Health Research New Investigator salary award (2017–2022; 201512MSH‐360766‐171036).
The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosure for previous 12 months: G.A.Y. is funded by the Ministry of Saudi Arabian National Guard Forces and the Ministry of Higher Education of Saudi Arabia.
A.V. receives in kind and research funding support from Illumina, Shire, Gilead, Eli Lilly. G.B. received research grants from Bluebird Bio; and has received funds to organize conferences from Foruna Fix, Allergan, Retrophin, Actelion Pharmaceuticals, NeuroVia and Genzyme.
Supporting information
Supporting Table 1. Details of the confirmed genetic mutations in the study cohort.
Video 1. Moderate multifocal dystonia in POLR3‐related leukodystrophy. The first part of the video shows focal eyelid dystonia, demonstrated by forced closure of the eyelids. The second part shows head tremor, and while the arms are being held outstretched, moderate dystonic posturing and a combination of cerebellar and dystonic tremor of the hands. The third part shows dystonic postures of the feet at rest, which are worsening with repetitive foot tapping. The fourth part of the video demonstrates the inability to draw spirals secondary to hand dystonia. The last part shows marked loss of truncal posture secondary to cerebellar involvement, and moderate foot posturing while attempting to walk. Both of the cerebellar involvement and the dystonia are resulting in marked difficulties with standing and walking.
Acknowledgments
The authors would like to thank the patients and their families for their participation.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Wolf NI, Harting I, Boltshauser E, et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 2005; 64:1461–1464. [DOI] [PubMed] [Google Scholar]
- 2. Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 2006; 67:2066–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vázquez‐López M, Ruiz‐Martín Y, de Castro‐Castro P, Garzo‐Fernández C, Martín‐del Valle F. Central hypomyelination, hypogonadotrophic hypogonadism and hypodontia: a new leukodystrophy. Rev Neurol 2008; 47:204–208. [PubMed] [Google Scholar]
- 4. Wolf NI, Harting I, Innes AM, et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics 2007; 38:64–70. [DOI] [PubMed] [Google Scholar]
- 5. Wolff A, Koch MJ, Benzinger S, et al. Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr Dent 2010; 32:386–392. [PubMed] [Google Scholar]
- 6. Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 2011; 89:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernard G, Thiffault I, Tétreault M, et al. Tremor‐ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3‐10q23.31. Neurogenetics 2010; 11:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tétreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet 2011; 89:652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tétreault M, Putorti ML, Thiffault I, et al. TACH leukodystrophy: locus refinement to chromosome 10q22.3‐23.1. Can J Neurol Sci 2012; 39:122–123. [DOI] [PubMed] [Google Scholar]
- 10. Atrouni S, Darazé A, Tamraz J, Cassia A, Caillaud C, Mégarbané A. Leukodystrophy associated with oligodontia in a large inbred family: fortuitous association or new entity? Am J Med Genet A 2003; 118:76–81. [DOI] [PubMed] [Google Scholar]
- 11. Chouery E, Delague V, Jalkh N, et al. A whole‐genome scan in a large family with leukodystrophy and oligodontia reveals linkage to 10q22. Neurogenetics 2011; 12:73–78. [DOI] [PubMed] [Google Scholar]
- 12. Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal‐recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011; 89:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009; 31:582–587. [DOI] [PubMed] [Google Scholar]
- 14. Potic A, Brais B, Choquet K, Schiffmann R, Bernard G. 4H syndrome with late‐onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol 2012; 69:920–923. [DOI] [PubMed] [Google Scholar]
- 15. Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6:7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wolf N, Vanderver A, Van Spaendonk RM, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014; 83:1898–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Comella CL, Leurgans S, Wuu J, Stebbins GT, Chmura T. Dystonia Study Group. Rating scales for dystonia: a multicenter assessment. Mov Disord 2003; 18:303–312. [DOI] [PubMed] [Google Scholar]
- 18. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013; 28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geyer HL, Bressman SB. The diagnosis of dystonia. Lancet Neurol 2006; 5:780–790. [DOI] [PubMed] [Google Scholar]
- 20. Neychev VK, Gross RE, Lehericy S, et al. The functional neuroanatomy of dystonia. Neurobiol Dis 2011; 42(2):185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Table 1. Details of the confirmed genetic mutations in the study cohort.
Video 1. Moderate multifocal dystonia in POLR3‐related leukodystrophy. The first part of the video shows focal eyelid dystonia, demonstrated by forced closure of the eyelids. The second part shows head tremor, and while the arms are being held outstretched, moderate dystonic posturing and a combination of cerebellar and dystonic tremor of the hands. The third part shows dystonic postures of the feet at rest, which are worsening with repetitive foot tapping. The fourth part of the video demonstrates the inability to draw spirals secondary to hand dystonia. The last part shows marked loss of truncal posture secondary to cerebellar involvement, and moderate foot posturing while attempting to walk. Both of the cerebellar involvement and the dystonia are resulting in marked difficulties with standing and walking.