

Abstract
Gastrointestinal diseases, specifically Crohn’s disease, ulcerative colitis, diverticular disease, and primary biliary cirrhosis are all characterized by complicated inflammation of the digestive tract. Their pathology is multifactorial, and risk factors encompass both genetic and environmental factors. Recent advances in the genetic component of inflammatory bowel diseases (IBDs) have revealed that the tumor necrosis factor superfamily member 15 (TNFSF15) contains a number of risk alleles associated not only with IBD but also with other diseases such as diverticular disease and primary biliary cirrhosis. These risk alleles in TNFSF15 and the altered expression of its gene product can serve as the common ground between these disorders by explaining at least some of the underlying processes that lead to a dysregulated immune response and subsequent chronic inflammation. Here, we aim to outline how the TNFSF15 gene is involved in the proliferation and cell fate of different populations of T cells and subsequently in the control of both pro- and anti-inflammatory cytokines. Furthermore, we summarize what is currently known of TNFSF15 control region variants, how they are associated with each mentioned disease, and how these variants can explain the autoimmune pathology of said diseases through altered TNFSF15 expression.
Keywords: Tumor necrosis factor superfamily member 15, Diverticular disease, Death receptor 3, Ulcerative colitis, Crohn’s disease, Primary biliary cirrhosis
Core tip: Tumor necrosis factor superfamily member 15 and the protein it encodes, tumor necrosis factor ligand-related molecule 1 play a vital role in the mucosal immunity. Expression of tumor necrosis factor ligand-related molecule 1 and death receptor 3-mediated signaling both exert their effects in Crohn’s disease, ulcerative colitis, diverticular disease, and primary biliary cirrhosis, which can serve to bridge the gap of knowledge regarding the genetic components of this group of inflammatory diseases as well as provide common ground for a putative targeted treatment.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic disorder that constitutes an important worldwide health problem. This group of diseases is multifactorial and characterized by chronic relapsing intestinal inflammation[1]. The two major subtypes of IBD are ulcerative colitis (UC) and Crohn’s disease (CD). IBD is a global disease with the highest prevalence in Western countries (North America, Europe, and Oceania) although recently there has been an accelerated incidence rate in the newly industrialized countries of Asia, South America, and Africa, where societies have become more westernized[1]. Although the exact etiology of IBDs is still unknown, numerous studies have revealed the multifactorial nature of IBDs that encompasses genetic susceptibility, environmental factors, intestinal microbiota, and the immune response system[2].
Another common gastrointestinal disorder, similar in its prevalence amongst western populations is colonic diverticulosis. The term “diverticulosis” refers to the occurrence of diverticula due to the formation of pouches by the mucosal wall of the intestine[3]. Colonic diverticulosis, or diverticular disease (DD), is a broad-spectrum term, as the condition involves a number of clinical manifestations that can range from the presence of constant abdominal systems without inflammation (symptomatic uncomplicated diverticular disease) to a significant and symptomatic inflammatory process (segmental colitis associated with diverticulosis and diverticulitis)[4].
Both diverticulitis and IBDs share overlapping characteristics and symptoms including, but not limited to: clinical presentation involving diarrhea, mucus in the stool, abdominal pain, weight loss, fistulae, bowel structuring, and inflammation[5,6]. This overlap can make diagnosis difficult for the attending clinician, although distinction can be achieved by endoscopical examination[5]. Despite this difficulty and in order to improve our understanding of the relation between inflammation and gastrointestinal disorders, we have to ask the question, what is the driving factor behind these shared attributes of CD, UC, and DD?
The common ground for the pathological signs of IBDs and DD appears to be a dysregulated mucosal immune response[7,8]. This dysregulation often results in impaired epithelial barrier function and damage to the surrounding epithelial tissue. Both pro- and anti-inflammatory cell lines and their respective secreted cytokines are involved in this response. In CD, T helper 1 (Th1)/Th17 cells and interleukin (IL)-12 as well as IL-23 are characteristic, whereas in UC the major factor is natural killer T (NKT) cells secreting IL-13 and IL-5[9].
Tumor necrosis factor superfamily member 15 (TNFSF15), also known as tumor necrosis factor ligand-related molecule 1 (TL1A) and vascular endothelial growth inhibitor (VEGI) is a tumor necrosis factor (TNF) family member encoding a ligand produced by a variety of cell lines, including endothelial cells, macrophages, dendritic cells (DCs), and T cells[10]. First described in 2002 as a T-cell stimulatory cytokine[11], studies have discovered that it affects cell lines related to both the innate and adaptive immune responses by its receptor, death receptor 3 (DR3)[12]. Since then, the role of this cytokine-receptor pair has been linked to the immunomodulation and vascular endothelial function observed in IBDs[6].
TNFSF15 FUNCTION AND EXPRESSION
The gene product of TNFSF15, TL1A, is a TNF-like factor, which is expressed in endothelial cells (human umbilical vein endothelial cells, adult dermal microvascular endothelial cells, and uterus myometrial endothelial cells), gut lamina propria lymphocytes, and macrophages.
TL1A is a longer splicing variant of the coding gene TNFSF15 compared to the initially described protein TL1/VEGI. The difference between the two variants is that TL1A is encoded by all four coding exons, whereas a continuous DNA containing the fourth exon and its 5’ adjacent intron encodes TL1. As a result, the two variants have identical C-terminal regions while the N-terminal regions are different for the two proteins. TL1A is a type II transmembrane protein, containing 251 amino acids and has a molecular weight of 28 kDa. The transmembrane form of TL1A can be cleaved by enzymes and exists as a functional soluble protein[11,13]. This cleavage can vary depending on the cell of origin[14]. The soluble form is more abundantly synthesized by DCs, and the membrane-bound protein is expressed by both T cells and DCs. The different forms, like other members of the TNF superfamily, have different functions. Soluble TL1A can be detected after DC and monocyte stimulation in vitro, and increased levels have been detected in serum samples from patients with rheumatoid arthritis, an autoreactive disease[15].
The receptor for TL1A, DR3, was identified in the 1990s[16], and was later discovered to be highly homologous to TNF receptor 1 (TNFR1)[12]. Signaling by DR3 is facilitated primarily through the use of TNFR-associated death domain protein, which contains a TNF receptor associated factors-binding domain as well as a death domain. This combination allows DR3 to activate nuclear factor kappa B and mitogen-activated protein kinase[17], which allows it to play a role in both apoptosis and anti-apoptosis, cell survival, and proliferation[18].
The expression of TL1A is closely linked to the levels of inflammation over the course of IBD and is also correlated to areas affected by the disease[10]. While TL1A baseline expression can be low[19], proinflammatory stimulation seems to be the switch that increases TL1A expression[20]. Both TL1A and DR3 are expressed across all members of the T cell family[21,22], despite the original discovery of TL1A in endothelial cells. The action of TL1A-DR3 signaling is most profound in the differentiation and stimulation of T cell subtypes. Co-stimulation with TL1A increases IL-2 signaling[11,20,23], whereas TL1A itself stimulates the proliferation of T cells. Specific CD4+ T cells can up-regulate DR3 and produce interferon gamma in response to TL1A combined with IL-12 and IL-18 in the intestinal mucosa[23], suggesting a putative mechanism for TL1A gut signaling and expression[19]. TL1A also affects Th17 cells as DR3 expression is highly upregulated on this cell subset[24], although TL1A-mediated Th17 proliferation is achieved in a DR-3 independent manner[20]. Furthermore, TL1A plays a role in the development of regulatory T cells (Treg), as stimulation by the soluble form of TL1A increases Treg proliferation[25]. However, in vitro assays have shown that the increased numbers of Tregs also show reduced suppressive capability[26].
TNFSF15 AND INFLAMMATION
Genetic studies attempting to evaluate the role of TNFSF15 have only begun recently following previously suggested hints of genetic factors involved in IBD[27,28]. The first genome-wide association studies (GWAS) conducted in 2005 discovered an association between TL1A and CD in a Japanese cohort of patients[29]. Subsequent studies have replicated and confirmed the association of TNFSF15 in European populations, for patients with both CD or UC[30]. Further investigation on specific patient subsets confirmed the protective haplotype[31] and revealed that TL1A expression is increased in carriers of the risk haplotype in a Jewish cohort of patients with CD and Escherichia coli exposure[32].
The findings of the previously mentioned studies and the data obtained allowed for the further investigation of TNFSF15 single nucleotide polymorphisms (SNPs) and their role not only in CD and UC, but also in DD. The first case study aiming to investigate how these SNPs can exert an effect revealed that the SNP rs7848647 and specifically, the risk allele G conferred an additive higher risk towards DD requiring surgical intervention[33]. As a follow-up, another study aimed to increase the number of participants and to include six other SNPs, four of which had been previously associated with CD[29] as a risk haplotype and to reveal if such an association could be found for DD as well. Results demonstrated not only that the CD risk haplotype was associated with DD, but two protective haplotypes emerged as well[6]. Although both studies provided hopeful results, they also suggest that there might be further undiscovered SNPs in DD pathology.
TNFSF15 variants have also been associated with primary biliary cirrhosis (PBC), a chronic and progressive liver disease, leading to hepatic failure and liver cirrhosis. One of the hallmarks of PBC is an autoimmune reaction towards biliary epithelial cells. Combined with data from twin studies, this has driven research into a possible genetic component of PBC. The first GWAS studies demonstrated the association of TNFSF15 rs4979462 in Asian populations with PBC[34] as the second strongest susceptibility gene. Specifically, rs4979462 has been found to be one of the main causal variants in the gene, due to a creation of a novel nuclear factor 1 binding site, a finding further strengthened by the increased TNFSF15 mRNA expression[35]. Other large-scale GWAS studies have also demonstrated that TNFSF15 is a part of a multitude of PBC risk loci involved in T cell, B cell, and natural killer (NK) cell stimulation and proliferation[36].
TL1A expression has also proven to be one of the key hallmarks of IBD. It was first observed in CD and UC[10,21,37] with both increased protein and mRNA levels compared to healthy controls[19]. This expression is regulated in a two-fold manner in gastrointestinal disorders. First, single-nucleotide polymorphisms have been found to correlate with TL1A expression[32,38,39] in a variety of immune cells. Second, TL1A can be induced by a number of gut-specific bacteria[22] with expression levels adjusting accordingly to the presence or absence of bacteria. Localized increased inflammation correlating with increased TL1A expression and exposure to bacteria in patients with CD, UC, and DD appears to be the common ground between these gastrointestinal disorders (Figure 1).
Figure 1.
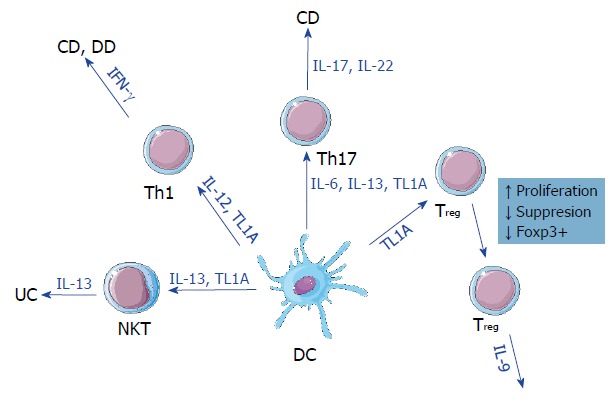
Effect of tumor necrosis factor ligand-related molecule 1 (TL1A) signaling across different subtypes of leukocytes. TL1A acts by promoting cytokine signaling, cell differentiation, and proliferation. These effects vary between different subtypes. While TL1A promotes natural killer T cells and T helper Th1/Th17 proliferation, it can also suppress regulatory T cell functions and forkhead box P3 positive expression. Regardless, TL1A is not associated exclusively with any form. CD: Crohn’s disease; DC: Dendritic cell; DD: Diverticular disease; IFN-γ: Interferon gamma; IL: Interleukin; NKT: Natural killer T cells; TL1A: Tumor necrosis factor Ligand-related molecule 1; Th: T helper; Treg: Regulatory T cell; Foxp3: Forkhead box P3; UC: Ulcerative colitis.
Further proof for the involvement of TNFSF15 comes in the form of studies investigating the association of risk variants within the gene and the requirement of surgical intervention as part of the treatment plan[33,40]. Cases that do not respond to medical treatment and present themselves with severe colonic inflammation ultimately require surgical resection, which represents a higher risk for the patient.
Because of its mode of action, and by having a very specific niche of activity and expression, TNFSF15 can be considered a putative therapeutic target. Studies have investigated the effect of anti-TL1A antibodies on sodium-sulfate induced colitis[41] and T-cell transfer models[42]. Some have even successfully managed to reverse fibrosis in these models[43]. As TL1A expression can vary, depending on genetic variations in its control region, it remains to be seen whether a targeted anti-TL1A therapy can be applied in a clinical setting.
CONCLUSION
TNFSF15 and the protein it encodes share a unique position amongst other genetic factors for gastrointestinal disorders. It enhances the T cell responses in CD, UC, and DD acting as a bridge between these disorders. It also plays a role in the immune response of mucosal tissue against bacterial infections, another common factor in gastrointestinal disorders. Furthermore, genetic variations in this gene have been associated as risk factors for all of the diseases examined here. All of these characteristics point towards the further study of TNFSF15 and the TL1A-DR3 interaction among patients with IBD and DD, both as a putative therapeutic target and a risk prediction factor.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to declare.
Manuscript source: Invited manuscript
Peer-review started: August 3, 2018
First decision: August 30, 2018
Article in press: October 18, 2018
Specialty type: Gastroenterology and hepatology
Country of origin: Bulgaria
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
P- Reviewer: Hitomi Y, Ramakrishna BS S- Editor: Ma RY L- Editor: Filipodia E- Editor: Wu YXJ
Contributor Information
Tanya Kadiyska, Department of Medical Chemistry and Biochemistry, Sofia Medical University, Sofia 1431, Bulgaria; Genetic Medico-Diagnostic Laboratory Genica, Sofia 1612, Bulgaria. kadiyska_t@yahoo.com.
Ivan Tourtourikov, Genetic Medico-Diagnostic Laboratory Genica, Sofia 1612, Bulgaria.
Ana-Maria Popmihaylova, University of Montpellier, Montpellier 34090, France.
Hilda Kadian, Bulgarian Association for Inflammatory Bowel Diseases, Sofia 1527, Bulgaria.
Ani Chavoushian, Department of Gastroenterology, Acibadem City Clinic Oncology Center, Sofia 1784, Bulgaria.
References
- 1.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 2.Kamm MA. Rapid changes in epidemiology of inflammatory bowel disease. Lancet. 2017;390:2741–2742. doi: 10.1016/S0140-6736(17)32669-7. [DOI] [PubMed] [Google Scholar]
- 3.Ceresoli M, Lo Bianco G, Gianotti L, Nespoli L. Inflammation management in acute diverticulitis: current perspectives. J Inflamm Res. 2018;11:239–246. doi: 10.2147/JIR.S142990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strate LL, Modi R, Cohen E, Spiegel BM. Diverticular disease as a chronic illness: evolving epidemiologic and clinical insights. Am J Gastroenterol. 2012;107:1486–1493. doi: 10.1038/ajg.2012.194. [DOI] [PubMed] [Google Scholar]
- 5.Peppercorn MA. The overlap of inflammatory bowel disease and diverticular disease. J Clin Gastroenterol. 2004;38:S8–10. doi: 10.1097/01.mcg.0000123993.13937.ec. [DOI] [PubMed] [Google Scholar]
- 6.Connelly TM, Choi CS, Berg AS, Harris L 3rd, Coble J, Koltun WA. Diverticulitis and Crohn’s disease have distinct but overlapping tumor necrosis superfamily 15 haplotypes. J Surg Res. 2017;214:262–269. doi: 10.1016/j.jss.2017.02.030. [DOI] [PubMed] [Google Scholar]
- 7.Tursi A. Diverticular disease: A therapeutic overview. World J Gastrointest Pharmacol Ther. 2010;1:27–35. doi: 10.4292/wjgpt.v1.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu XR, Liu CQ, Feng BS, Liu ZJ. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2014;20:3255–3264. doi: 10.3748/wjg.v20.i12.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamias G, Martin C 3rd, Marini M, Hoang S, Mishina M, Ross WG, Sachedina MA, Friel CM, Mize J, Bickston SJ, Pizarro TT, Wei P, Cominelli F. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–4874. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 11.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 12.Chinnaiyan AM, O’Rourke K, Yu GL, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 13.Zhan C, Yan Q, Patskovsky Y, Li Z, Toro R, Meyer A, Cheng H, Brenowitz M, Nathenson SG, Almo SC. Biochemical and structural characterization of the human TL1A ectodomain. Biochemistry. 2009;48:7636–7645. doi: 10.1021/bi900031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferdinand JR, Richard AC, Meylan F, Al-Shamkhani A, Siegel RM. Cleavage of TL1A Differentially Regulates Its Effects on Innate and Adaptive Immune Cells. J Immunol. 2018;200:1360–1369. doi: 10.4049/jimmunol.1700891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siakavellas SI, Sfikakis PP, Bamias G. The TL1A/DR3/DcR3 pathway in autoimmune rheumatic diseases. Semin Arthritis Rheum. 2015;45:1–8. doi: 10.1016/j.semarthrit.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Screaton GR, Xu XN, Olsen AL, Cowper AE, Tan R, McMichael AJ, Bell JI. LARD: a new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Natl Acad Sci USA. 1997;94:4615–4619. doi: 10.1073/pnas.94.9.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J Biol Chem. 2003;278:39251–39258. doi: 10.1074/jbc.M305833200. [DOI] [PubMed] [Google Scholar]
- 18.Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66:1403–1408. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 19.Richard AC, Ferdinand JR, Meylan F, Hayes ET, Gabay O, Siegel RM. The TNF-family cytokine TL1A: from lymphocyte costimulator to disease co-conspirator. J Leukoc Biol. 2015;98:333–345. doi: 10.1189/jlb.3RI0315-095R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meylan F, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D, Bundoc V, Hodges M, Shevach EM, Keane-Myers A, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prehn JL, Thomas LS, Landers CJ, Yu QT, Michelsen KS, Targan SR. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J Immunol. 2007;178:4033–4038. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]
- 22.Shih DQ, Kwan LY, Chavez V, Cohavy O, Gonsky R, Chang EY, Chang C, Elson CO, Targan SR. Microbial induction of inflammatory bowel disease associated gene TL1A (TNFSF15) in antigen presenting cells. Eur J Immunol. 2009;39:3239–3250. doi: 10.1002/eji.200839087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin S, Chin J, Seeber S, Niewoehner J, Weiser B, Beaucamp N, Woods J, Murphy C, Fanning A, Shanahan F, et al. TL1A/TNFSF15 directly induces proinflammatory cytokines, including TNFα, from CD3+CD161+ T cells to exacerbate gut inflammation. Mucosal Immunol. 2013;6:886–899. doi: 10.1038/mi.2012.124. [DOI] [PubMed] [Google Scholar]
- 24.Pappu BP, Borodovsky A, Zheng TS, Yang X, Wu P, Dong X, Weng S, Browning B, Scott ML, Ma L, et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J Exp Med. 2008;205:1049–1062. doi: 10.1084/jem.20071364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, Malek TR, Levy RB, Podack ER. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest. 2010;120:3629–3640. doi: 10.1172/JCI42933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taraban VY, Slebioda TJ, Willoughby JE, Buchan SL, James S, Sheth B, Smyth NR, Thomas GJ, Wang EC, Al-Shamkhani A. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011;4:186–196. doi: 10.1038/mi.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonen DK, Cho JH. The genetics of inflammatory bowel disease. Gastroenterology. 2003;124:521–536. doi: 10.1053/gast.2003.50045. [DOI] [PubMed] [Google Scholar]
- 28.Brant SR, Shugart YY. Inflammatory bowel disease gene hunting by linkage analysis: rationale, methodology, and present status of the field. Inflamm Bowel Dis. 2004;10:300–311. doi: 10.1097/00054725-200405000-00019. [DOI] [PubMed] [Google Scholar]
- 29.Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, Cardon L, Takazoe M, Tanaka T, Ichimori T, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 30.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Büning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H; International IBD Genetics Consortium (IIBDGC), Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Picornell Y, Mei L, Taylor K, Yang H, Targan SR, Rotter JI. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–1338. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michelsen KS, Thomas LS, Taylor KD, Yu QT, Mei L, Landers CJ, Derkowski C, McGovern DP, Rotter JI, Targan SR. IBD-associated TL1A gene (TNFSF15) haplotypes determine increased expression of TL1A protein. PLoS One. 2009;4:e4719. doi: 10.1371/journal.pone.0004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Connelly TM, Berg AS, Hegarty JP, Deiling S, Brinton D, Poritz LS, Koltun WA. The TNFSF15 gene single nucleotide polymorphism rs7848647 is associated with surgical diverticulitis. Ann Surg. 2014;259:1132–1137. doi: 10.1097/SLA.0000000000000232. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, Nakamura H, Komori A, Nakamuta M, Zeniya M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. 2012;91:721–728. doi: 10.1016/j.ajhg.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hitomi Y, Kawashima M, Aiba Y, Nishida N, Matsuhashi M, Okazaki H, Nakamura M, Tokunaga K. Human primary biliary cirrhosis-susceptible allele of rs4979462 enhances TNFSF15 expression by binding NF-1. Hum Genet. 2015;134:737–747. doi: 10.1007/s00439-015-1556-3. [DOI] [PubMed] [Google Scholar]
- 36.Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, Dai Y, Gong Y, Wang L, Xu P, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun. 2017;8:14828. doi: 10.1038/ncomms14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Takayama T, Kitazume MT, Okamoto S, Koganei K, Sugita A, et al. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn’s disease. Inflamm Bowel Dis. 2010;16:568–575. doi: 10.1002/ibd.21124. [DOI] [PubMed] [Google Scholar]
- 38.Hedl M, Abraham C. A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8-induced IL-1. Proc Natl Acad Sci USA. 2014;111:13451–13456. doi: 10.1073/pnas.1404178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kakuta Y, Ueki N, Kinouchi Y, Negoro K, Endo K, Nomura E, Takagi S, Takahashi S, Shimosegawa T. TNFSF15 transcripts from risk haplotype for Crohn’s disease are overexpressed in stimulated T cells. Hum Mol Genet. 2009;18:1089–1098. doi: 10.1093/hmg/ddp005. [DOI] [PubMed] [Google Scholar]
- 40.Haritunians T, Taylor KD, Targan SR, Dubinsky M, Ippoliti A, Kwon S, Guo X, Melmed GY, Berel D, Mengesha E, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis. 2010;16:1830–1840. doi: 10.1002/ibd.21293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D, Braun J, Targan SR. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng L, Zhang X, Chen J, Ichikawa R, Wallace K, Pothoulakis C, Koon HW, Targan SR, Shih DQ. sustained tlla (tnfsf15) expression on both lymphoid and myeloid cells leads to mild spontaneous intestinal inflammation and fibrosis. Eur J Microbiol Immunol (Bp) 2013;3:11–20. doi: 10.1556/EuJMI.3.2013.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, Wallace KL, Chen J, Pothoulakis C, Koon HW, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014;7:1492–1503. doi: 10.1038/mi.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]