

Abstract
Inositol 1, 4, 5-trisphosphate (IP3) binding at the N-terminus (NT) of IP3 receptor (IP3R) allosterically triggers the opening of a Ca2+-conducting pore located ~100 Å away from the IP3-binding core (IBC). However, the precise mechanism of IP3 binding and correlated domain dynamics in the NT that are central to the IP3R activation, remains unknown. Our all-atom molecular dynamics (MD) simulations recapitulate the characteristic twist motion of the suppressor domain (SD) and reveal correlated ‘clam closure’ dynamics of IBC with IP3-binding, complementing existing suggestions on IP3R activation mechanism. Our study further reveals the existence of inter-domain dynamic correlation in the NT and establishes the SD to be critical for the conformational dynamics of IBC. Also, a tripartite interaction involving Glu283-Arg54-Asp444 at the SD – IBC interface seemed critical for IP3R activation. Intriguingly, during the sub-microsecond long simulation, we observed Arg269 undergoing an SD-dependent flipping of hydrogen bonding between the first and fifth phosphate groups of IP3. This seems to play a major role in determining the IP3 binding affinity of IBC in the presence/absence of the SD. Our study thus provides atomistic details of early molecular events occurring within the NT during and following IP3 binding that lead to channel gating.
Introduction
Calcium is a universal and versatile intracellular messenger that regulates a diverse array of biological processes starting from fertilization to cell death1. In most cells, generation of calcium signals is initiated by agonist stimulation of cell surface receptors, which activates phospholipase C isoforms, thereby producing inositol 1, 4, 5-trisphosphate (IP3) in the plasma membrane. IP3 then rapidly diffuses into the cytosol and acts on IP3 receptors (IP3Rs). The latter represents a major class of intracellular Ca2+ channels, present primarily within the membrane of endoplasmic reticulum (ER). IP3 binding to the N terminus of IP3Rs leads to the opening of the distal pore and the resultant Ca2+ flux from the ER lumen elevates cytosolic free Ca2+ concentration. IP3-evoked Ca2+ signals are often spatio-temporally complex and used for regulating various cellular processes. Aberrant activity of these ion channels are known to underlie some pathological conditions2.
In mammals, there are three isoforms of IP3R (IP3R1, IP3R2 and IP3R3) which share ≥60% similarity in amino acid sequence and similar overall domain architecture. A functional IP3R is a homo- or hetero-tetramer of subunits, each comprising of ~2750 residues. Each subunit consists of a large cytosolic N-terminal region (NT), an intermediary regulatory domain and six transmembrane helices including the pore-forming region towards the C-terminal3. Crystal structure of NT of rat IP3R1 has been resolved in both apo and IP3-bound state4,5. The NT contains an IP3-binding core (IBC, residues 224–604, rat IP3R1) and the ‘suppressor domain’ (SD, residues 1–223, rat IP3R1). The IBC can further be envisaged to be made of IBC-β (residues 224–436) and IBC-α (residues 437–604) domains and the cleft formed by these domains accommodate the IP3 molecule (Fig. 1). The three NT domains – SD, IBC-β and IBC-α together form a triangular structure where the SD interacts with IBC through two interfaces, β-interface with IBC-β and α-interface with IBC-α. A 310-like turn (α5) extending to the β13 strand of the IBC-β position the IBC relative to the SD, where the SD is located behind the IP3-binding site in the IBC. A recent 4.7 Å full length structure of rat IP3R1 solved through cryoelectron microscopy (cryo-EM)6 shows that the IBC is located ~100 Å away from the pore region of the receptor. Quite intriguingly, the cryo-EM structure of rat IP3R1 also shows that, unlike the closely related ryanodine receptors (RyRs), the C-terminal tail of the IP3R directly interacts with the SD of NT and forms a connecting link between the pore-forming helices and the IP3-binding NT region. Very recently, crystallographic analysis also shed light on the long-range communication between IBC and the channel domain. The IP3-dependent conformational changes are transmitted to the channel through the three large α-helical domains located between the NT and the transmembrane channel domain7. These findings together reaffirm the existence of allosteric modulation of IP3-mediated channel gating in IP3R.
Figure 1.

Different sub-domains of IP3-bound NT of IP3R. The suppressor domain (SD, 1–224); IP3 binding core-β (IBC-β, 225–436) and IBC-α (437–604) are shown as cyan, orange and green, respectively. The HS loop (residue 165–180) of SD that interacts with the IBC-β of a neighboring monomeric subunit in the functional tetrameric form of IP3R is colored in red. IP3, which binds at the cleft formed between IBC-β and IBC-α is shown in stick representation.
Comparison of the apo and IP3-bound crystal structures4,5,7 provides static snapshots of IP3-evoked major domain movements in NT, where the IP3 binding brings IBC-β and IBC-α domains close together in a ‘clam closure’ type manner. In agreement with experimental findings8, large domain motions in the apo IBC as compared to the IP3-bound form, was observed from the molecular dynamics (MD) simulations of the IBC9, supporting this ‘clam closure’ during IP3 binding. Crystallographic studies further showed IP3-induced translational displacement of SD that causes the latter to rotate towards the IBC, measuring an angular displacement of ~9° between the arm helices (α2/α3 helix, Fig. 1) of the apo and IP3-bound NT5. Such a twist in SD causes translational movement of the conserved HS loop (residues 165–180, rat IP3R1) of the SD and the IP3 has been proposed to disrupt the inter-subunit interaction between adjacent NTs in the tetrameric IP3R that are mediated by IBC-β and HS loop of the SD, allowing the channel to open. Attenuation of IP3-evoked Ca2+ release in the Tyr167Ala mutant also provided insight into the prime role of the SD in mediating functional coupling between ligand binding and channel gating10. However, a dynamic view of the structural events during and immediately following IP3 binding at the NT that lead to the opening of a distal (located ~100 Å away) pore still remains to be elucidated.
As mentioned above, the SD plays a crucial role in traversing IP3-binding events at the NT to the channel pore. However, as the name (‘suppressor domain’) implies, the SD was found to suppress the IP3 binding affinity of IBC11,12. Purified protein constructs comprising only the IBC-β and IBC-α binds IP3 with higher affinity than the NT or even the whole protein. A cluster of 7 conserved amino acid residues located on one side of the SD were found to be critical for the suppression of IP3 binding affinity13. IP3R isoforms are well known to differ in their IP3-binding affinity3 and this has been attributed to the structural difference within their SD11. The Gibb’s free energy (∆G) of IP3 binding calculated from the thermodynamics analysis also reflected on the suppressing nature of SD, where ∆G of IP3 binding at 296 K was −37.1 ± 0.1 kJ/mol and −43.2 ± 0.1 kJ/mol in presence (i.e., NT) and absence (i.e., IBC) of the SD respectively14. Suppression of IP3-binding affinity of the IBC by the SD is thus well established by now, though the exact molecular basis underlying such phenomenon remains elusive.
In the present study, we have used a computational approach, combining ab initio and homology modelling, all-atom MD simulations and principal component analyses to capture the landscape of IP3-mediated conformational dynamics in IP3R NT and to unearth the molecular details of SD-evoked differential IP3 binding affinity of the NT. Analysis of essential dynamics revealed that the binding of IP3 triggers a characteristic twist motion of SD, followed by ‘clam closure’ of IBC-β and IBC-α. Inspection of the atomic-level interaction profile led to the identification of a tripartite interaction involving Arg54,Glu283 and Asp444 at the SD-IBC interface, which potentially involved in triggering IP3-mediated receptor activation. Very intriguingly, hundreds of nanoseconds long MD simulations of IP3-bound IBC in presence and absence of the SD revealed that the ‘Arg269-flipping’ and its positional interaction either with 1st (P1) or 5th (P5) phosphate groups of IP3 largely govern the affinity of IBC for IP3. To the best of our knowledge, this is the first report that has unveiled the dynamic picture of IP3-invoked conformational changes in IP3R NT and more importantly, the molecular mechanism underlying the SD-dependent differential IP3 binding affinity of the receptor.
Results and Discussion
To elucidate the conformational dynamics associated with the activation of IP3R by the natural agonist IP3, we performed all-atom unbiased MD simulations of IP3R NT in its apo and IP3-bound states. Different systems simulated in this study are presented in Table 1 and detailed description of system preparation is provided in the methodology section. Each system was initially simulated for 300 ns of production run following 10 ns of equilibration at 300 K. The results from the simulation studies are discussed below.
Table 1.
Details of the simulated IP3R N-terminal systems.
| System | protein componenta,b | bound-ligand | duration of simulation |
|---|---|---|---|
| 1 |
 (NT, 1–604) (NT, 1–604) |
— | 300 ns |
| 2 |
 (NT, 1–604) (NT, 1–604) |
IP3 | 300 ns |
| 3 |
 (IBC, 225–604) (IBC, 225–604) |
— | 300 ns |
| 4 |
 IBC, 225–604) IBC, 225–604) |
IP3 | 300 ns |
| 5 |
 SD-knockout, 225–604) SD-knockout, 225–604) |
IP3 | 400 ns |
aB chain of the crystal structures - PDB ID 3UJ4 and 3UJ0 - were used for the simulation.
bSystem 5 was prepared by removing SD from the final conformation of the IP3-bound NT obtained from 300 ns of simulation of system 2.
IP3 driven twist motion of the suppressor domain
The root mean square deviation (RMSD) of NT with respect to the minimized starting structure as a function of simulation time shows that the IP3-bound NT exhibits significant structural deviation, marked by higher RMSDs (Fig. S1a). It is noteworthy that, compared to the apo form, IP3-bound NT fluctuates considerably throughout the 300 ns of simulation. This observation is further supported by the computed residue-wise root-mean-squared fluctuations (RMSFs), indicating increased dynamics of the NT due to the binding of IP3 (Fig. S1b). Although RMSDs and RMSFs reflect the conformational changes at NT while IP3 binds, it is important to understand the prominent and collective domain motions associated with IP3 binding, which can provide insight into the IP3-mediated activation of IP3R.
We performed principal component analysis (PCA) on simulation trajectories to capture the significant and concerted motions of different NT domains, in presence and absence of IP3. PCA identifies the top essential modes (eigenvectors) that represent the major part of collective motions. The top two eigenvectors (principal components, PCs) have captured the significant motions of IP3R NT (Fig. S2). To compare the conformational space sampled by apo (system 1) and liganded NT (system 2), the two-dimensional projection of the simulation ensembles onto the plane defined by the first two PCs were plotted (Fig. S3a). Although the regions explored by the two systems overlap, notable difference in conformational sampling can be seen between the apo and IP3-bound states along the PC1 and PC2. A similar trend was also noted from the PCA on the second set of simulations data. This observation is consistent with the results from RMSD and RMSF analyses (Fig. S1a,b), where the protein dynamics was found to be intensified in the presence of the IP3.
The principal motions of protein residues can be better visualized and interpreted by representing the eigenvectors as porcupine plots15. The porcupine plots showing the motion of SD and IBC of apo and IP3-bound NT, along the direction of PC1 and PC2, are presented in Fig. 2a–d respectively. In the apo state (Fig. 2a,b), loop regions in the NT experienced more flexibility compared to rest of the protein. Particularly, only the arm loop of the SD exhibits significant fluctuations whereas the remaining secondary structures show minimal motions. Similarly, the loop regions of the IBC-β and IBC-α showed comparatively more flexibility than the other part of the domain. Notably, in the apo state, the IBC-α exhibits directional movement towards the IBC-β, where the latter experiences negligible mobility (Movie S1).
Figure 2.

Ligand-induced domain dynamics in IP3R NT. The porcupine plots representing the principal motions along the direction of PC1 and PC2 respectively are shown for [(a,b)] apo NT and [(c,d)] IP3-bound NT. Different domains and the HS loop are colored as shown in Fig. 1. (e) The extent of twist motion of the SD measured in terms of twist angle (defined in the methodology) in the apo (black) and IP3-bound (red) states of IP3R NT, as a function of simulation time. (f) A plot of varying SD – IBC buried interfacial surface area in apo (black) and IP3-bound (red) NT, along the simulation time.
Interestingly, IP3 binding at the IBC substantially altered the dynamics of different structural units of NT (Fig. 2c,d). The binding of IP3 has directly influenced the dynamics of IBC. As in apo NT, IBC-α moved towards the IBC-β in the IP3-bound NT. Moreover, in contrast to the apo NT where IBC-β exhibits minimal displacement, the IBC-β of the IP3-bound NT underwent larger dynamics and moved towards the IBC-α. As shown by the vector direction in Fig. 2c,d, IP3 induced motions in the NT brings the IBC -β and -α close to each other. This observation is consistent with the findings from earlier studies. The crystallographic experiments showed that IP3 binding evokes ‘clam closure’, where the β and α domains of IBC get close to each other4,5,8. Such ligand-induced domain closure is also reported in other ligand-gated ion channels that notably include the ionotropic glutamate receptors16. Markedly, as seen from the porcupine plots, the binding of IP3 at IBC has allosterically influenced the dynamics of SD. The SD attains a typical twist motion, where the entire SD sweeps over the inner surface of the IBC (Fig. 2c,d, Movie S2). The SD anchored on the inner surface of the IBC through the IBC-β and the IP3 binding causes the SD to twist along the inner surface of IBC-α. The β2/β3, β5/β6, and β10/β11 loops of SD interact with α8 and α9 helices of IBC-α at the α-interface (see Fig. 1). Such a twist motion triggers the SD to move towards the IBC-α, which in turn causes an increased translational displacement of the conserved HS loop of SD. The IP3-evoked change in NT dynamics is consistent with the results from RMSD and RMSF analyses, where the increase in RMSD and RMSF in system 2 can be attributed to the IBC clam closure and twist motion of SD (Fig. S1a,b).
The extent of translational displacement of SD due to its twist motion in IP3-bound NT is further amplified by calculating the twist angle along the simulation trajectories (described in the methodology). Figure 2e shows the evolution of twist angle along the simulation time in apo and IP3-bound systems. It can be noted that the SD twists minimally in the apo system with the twist angle fluctuating over a range of 0°–10°, with an average of ~5°. In accordance with the observation from essential dynamics (Fig. 2c,d), the angle calculation also captured the increased twist motion of SD caused by the IP3 binding at IBC (Fig. 2e). In contrast to the apo system, the angle underwent much larger fluctuation (2°–23°, with an average of ~13°) during simulation. Such a wide range of twist angle shows the intensive translational displacement experienced by the SD during IP3 binding to the IBC. Recent structural studies also support the observed IP3 driven twist motion of the SD, where superimposition of apo and IP3-bound NT revealed ~9° angular displacement between the SD arm helices5. However, in contrast to the finding from static crystal structures, MD simulations here provided a dynamic view of IP3 mediated binding events at the IP3R NT. As depicted in Fig. 2e, the SD twist extensively during IP3 binding, where the domain sampled through the conformations with lowest twist angle value of 2°, as seen in apo system, as well as explored the conformations exclusive to the IP3-bound state, where the SD shows highest degree of twist (~23°). The change in buried interfacial surface area at SD – IBC interface, illustrated in Fig. 2f, also support the observed twist motion of SD. The computed average buried interfacial surface area is 1999 ± 232 Å2 in the apo system whereas the buried area reduced to 1239 ± 134 Å2 in the IP3-bound system. Such a reduction in SD – IBC interfacial buried area in IP3-bound system implies that the interface has weakened during the IP3 binding and many of the residues at the interface are exposed to the solvent. This can be attributed to the enhanced twist motion of SD that affected the SD – IBC interfacial packing. Taking together, the analyses of apo and IP3-bound systems clearly indicate that the IP3 binding at IBC allosterically influences the SD dynamics and the latter experiences a characteristic twist motion.
It is intriguing to note that kinetic analyses of single IP3Rs activity recorded using the nuclear patch-clamp technique have shown rapid intra-burst flickers that, unlike the much longer inter-burst intervals, remain largely insensitive to the variation of concentrations of IP3 as well as other cytosolic modulators (e.g. free Ca2+ or ATP)17,18. Such stereotypical, ligand-insensitive flickers were also observed for a mutant rat IP3R1 reconstituted in planar lipid bilayers19. The flickers were also noticeable, albeit with much lower frequency, in spontaneously active IP3Rs recorded from the Xenopus oocyte at ultra-low (<5 nM) level of cytosolic free Ca2+ concentration20. Based on the PCA and twist angle analysis (Fig. 2), we hypothesise that the basal twist motions observed in the SD in our simulation with the apo NT relate to conformations that probably underlie the ligand-insensitive intra-burst flickers of IP3Rs, which, however, requires further experimental validation.
IP3 binding breaks the triad of interaction at SD – IBC interface
In the earlier section, we have shown that the SD sweeps over the inner surface of IBC at the SD – IBC interface when IP3 binds at the IBC pocket. As observed in earlier structural studies4,5,13,21, our simulations also identified crucial residue contacts at the SD – IBC interface in apo and IP3-bound NT. Several residues from the SD and IBC involved in interface communication are shown in Fig. S4. A salt bridge between Lys225 and Asp228 positions the SD relative to the IBC and stabilizes the β-interface by helping in arranging the residues at the interface. Notably, a network of hydrophobic and electrostatic interactions via Val33, Leu32, Arg54, and Lys127 from SD and Asp444, Phe445, Asp448, Ala449, Val452, and Leu476 from IBC-α are involved in maintaining the α-interface.
Besides identifying residue contacts seen in static crystallographic structures, MD simulations of the NT provided further insight into the allosteric rearrangements in interfacial residue network during IP3 binding. The detailed analysis of the apo system revealed the formation of a bidentate hydrogen bond (H-bond), involving residues from all the three NT domains – SD, IBC-β and IBC-α. In the apo NT, Arg54 of SD forms a bidentate H-bond with Glu283 and Asp444 of IBC-β and IBC-α, respectively (Fig. 3a). It can be noted from the figure that Arg54 is situated at the center of the triangulated structure formed by the three NT domains. The guanidinium side chain of Arg54 H-bonds with the side chain carboxylate group of Glu283 and Asp444, with an average distance of 2.8 Å and 3.2 Å respectively. The −NH group of guanidinium interacts with the −COO group of Glu283 while the guanidinium −NH2 group H-bonds with the −COO group of Asp444. Interestingly, IP3 binding at the IBC breaks this triad of interaction at the SD – IBC interface (Fig. 3b). The binding of IP3 at the IBC allosterically disrupts the interaction of Arg54 with Asp444 and the former then rearranges its side chain in such a way that the guanidinium −NH2 group H-bonds with the −COO of Glu283. To further evaluate the stability of this bidentate interaction, we calculated the probability distribution of H-bond distance in Arg54 – Glu283 and Arg54 – Asp444 interaction during the entire span of simulation. In apo NT (Fig. 3c), Arg54 – Asp444 distance distribution was nearly Gaussian, peaking at the average distance of 2.8 Å, whereas the Arg54 – Glu283 interaction distance was distributed with two peaks, first at 3.2 Å and the second one at 5.1 Å. IP3 binding shifts this equilibrium interaction of Arg54 towards Glu283, with the measured Arg54 – Glu283 average distance of 2.8 Å (Fig. 3d). On the other hand, Arg54 was found to be H-bonding with Asp444 (peak at a distance of 2.8 Å) during only about 20% of the simulation time while the Arg54-Glu283 interaction persist in about 80% of the simulation trajectories. Moreover, the distribution clearly indicates that, in the apo NT, though Arg54 shows comparatively equal affinity towards Glu283 and Asp444, the interaction with Asp444 is found to be stronger than that with Glu283. The energy of Arg54 – Glu283 and Arg54 – Asp444 interaction was calculated as −5.8 ± 2.8 kcal/mol and −15.3 ± 0.6 kcal/mol respectively. Notably, Arg54 – Asp444 interaction energy was decreased to −4.7 ± 0.9 kcal/mol in IP3-bound NT (system 2) while the Arg54 – Glu283 interaction energy was −6.6 ± 1.3 kcal/mol, further supporting the IP3-driven residue rearrangements at the SD – IBC interface. The IP3 mediated shift in Arg54 – Glu283/Asp444 bidentate interaction can also support the IP3-evoked twist motion of SD. In the apo state, Arg54 of SD H-bonds with Glu283 and Asp444 and such a bidentate interaction at SD – IBC interface can lock and restrict the conformational flexibility of SD. The IP3 binding breaks this triad of interaction by disrupting the Arg54 – Asp444 interaction at the α-interface and stabilises the Arg54 – Glu283 H-bond at the β-interface. Such a residue rearrangement weakens the α-interface allowing the SD to slide over the inner surface of IBC-α and at the same time, anchor the SD immensely on the inner surface of β-interface. The absence of Arg54 – Glu283 H-bonding in the IP3-bound crystal structure (PDBID: 3UJ0)5 suggests that the crystal architecture may be representing one of the IP3-bound NT conformation explored during the simulation where Arg54 failed to make H-bond with Glu283. Biochemical studies in the past revealed that the mutation in Arg5413 or Asp44422 affected IP3-binding affinity of the receptor. However, the functional consequences of all these three residues are yet to be known to the best of our knowledge and future studies may look into this aspect.
Figure 3.

A triad of interactions involving Arg54, Glu238 and Asp444 at the SD – IBC interface found to be crucial for IP3-mediated conformational dynamics. [(a,b)] Ensemble averaged structure of IP3R NT highlighting the Arg54 interaction with Glu283 and Asp444. (a) Arg54 forms a bidentate interaction with Glu283 and Asp444. (b) IP3 binding breaks the Arg54 – Asp444 interaction while maintaining the interaction Arg54 – Glu283. The average distance between the residues is shown. [(c,d)] Probability distribution of Arg54 – Glu283 and Arg54 – Asp444 distance in (c) apo and (d) IP3-bound NT. Distribution is computed over the entire span of simulation for each system. Color scheme: Arg54 – Glu283, black; and Arg54–Asp444, red.
To summaries, the present all-atom MD simulation of IP3R NT provides a dynamic view of conformational landscape of NT during and immediately following IP3 binding and sheds light on the mechanism of IP3-mediated IP3R activation leading to the pore opening. In a biologically relevant, functional tetrameric form of IP3R, the monomeric units are arranged in such a way that the SD of one monomer interacts with the IBC-β of the neighboring monomer through the HS loop of SD (Fig. S5)6. It is proposed that the HS loop – IBC-β interaction holds the tetrameric IP3R in a closed state. The IP3 binding evokes the IBC clam closure and causes the SD to twist towards IBC, disrupting the HS loop – IBC-β interaction that allosterically opens the ion conducting pore, which is located ~100 Å away from the IP3 binding site6. In the present study, analyses of essential dynamics of the apo and IP3-bound NT domains clearly indicate that the IP3 binding at IBC allosterically affect the SD dynamics and the latter experiences a characteristic twist motion. The twist motion causes the SD to swing towards IBC-α, which eventually leads to the translational displacement of the conserved HS loop of SD. The residue Tyr167 of the HS loop was found to play a prime role in coupling the IP3 binding to gating and mutation of Tyr167Ala had shown to attenuate the IP3-evoked Ca2+ release10. It is noteworthy that, during simulation, Tyr167 experienced a maximum displacement of ~13 Å in the IP3-bound system while the displacement was about ~8 Å in the apo system (Fig. S6a,b) and such dynamics of the HS loop can break the SD – IBC interaction at the inter-monomeric interface (Fig. S5).
IP3 stabilizes IBC domain in the absence of the SD
So far, with the aid of extensive MD simulations, we have explored the IP3 driven conformational dynamics in IP3R NT. To further investigate the effect of hitherto identified SD twist motion on the IBC dynamics, we simulated two additional systems of apo and IP3-bound IBC in the absence of the SD (system 3 and 4, Table 1). As the RMSD in Fig. S7 shows, the apo IBC (system 3), in the absence of the SD, underwent significant conformational fluctuation from its starting configuration during the course of simulation (average RMSD of 8.4 ± 1.9 Å). However, in the presence of the SD (system 1), IBC gets stabilized after an initial equilibration phase of 40 ns with minimal structural deviation during rest of the simulation (average RMSD of 4.8 ± 1.2 Å). This suggests that, in the apo state, IBC experiences conformational constraints in presence of the SD (i.e., as a part of NT, system 1) whereas the IBC becomes increasingly dynamic in the absence of the SD. Interestingly, IP3 binding reverses this dynamic behavior of IBC. As shown in Fig. S7, the IP3-bound IBC of NT (i.e., in presence of SD, system 2) fluctuates significantly along the 300 ns of simulation, whereas the binding of IP3 stabilized the IBC in the absence of the SD (system 4 in Table 1). The RMSD values from the figure clearly indicate that, the liganded IBC is highly dynamic in presence of the SD (average RMSD of 7.1 ± 1.6 Å). On the other hand, in the absence of the SD, RMSD of IP3-bound IBC reached a stable plateau after an initial 40 ns of equilibration phase suggesting that the domain is stabilized during rest of the simulation (average RMSD of 5.2 ± 1.4). To further examine the local structural transformations of IBC in the presence and absence of the SD, we compared the residue-wise Cα RMSF of systems 1–4. The calculated RMSF values also corroborate the observation from RMSD analyses. As illustrated in Fig. 4a, in the absence of the SD, the apo and IP3-bound IBC exhibit distinct dynamic properties. The IBC residues were comparatively more flexible in the apo state (system 3) than the IP3-bound state (system 4). However, as seen from RMSD analyses, residue-wise RMSF also shows that the presence of the SD has influenced the dynamic behavior of IBC residues (Fig. 4a, black and red lines). In the presence of the SD (ie., NT), IBC residues experienced increased thermal fluctuations in the IP3-bound state as compared to the apo state.
Figure 4.

Conformational dynamics of the IBC domains depend on the presence of the SD. (a) Residue-level RMSF of IBC residues in different simulated systems. Color scheme: apo NT, black; IP3-bound NT, red; apo IBC, green; and IP3-bound IBC, blue. The porcupine plots representing the principal motions along the direction of PC1 and PC2 respectively are shown for [(b,c)] apo IBC and [(d,e)] IP3-bound IBC. Different domains are colored as shown in Fig. 1.
Furthermore, to understand the domain movements associated with differential dynamics of IBC in the presence and absence of the SD, we resorted to PCA to identify the essential motions in the protein. As shown in Fig. S2, the top two eigenvectors (PCs) have captured significant motions of apo and IP3-bound IBC from each simulation ensemble. The projection of MD ensemble onto the 2D plane defined by the top two PCs plotted in Fig. S3b provides a comparison of conformations sampled by IBC of system 3 and 4. As the figure shows, although conformational overlap is observed, apo IBC spans a larger space in contrast to that of the IP3-bound IBC. This suggests that the dynamics of IBC is restricted in the liganded state as compared to the apo state. This is consistent with the results from the RMSF analysis, indicates that residues with high RMS fluctuations contribute significantly to the dominant motions of the IBC.
The porcupine plots showing the directional motion of apo and IP3-bound IBC along the direction of PC1 and PC2 in presence and absence of the SD are presented in Fig. 4b–e. Noticeably, in the apo state (Fig. 4b,c), all the secondary structure elements of IBC were found to be profoundly dynamic in the absence of the SD (system 3), whereas the IBC exhibits comparatively more restricted motions in presence of the SD (system 1, Fig. 2a,b). In the absence of the SD, apo IBC underwent a bending motion where the β- and α- domains of IBC moved towards each other (Movie S3). However, presence of the SD (i.e., in apo NT, system 1) has hampered the bending motion of IBC by severely attenuating the dynamics of IBC-β (Fig. 2a,b, Movie S1). Interestingly, IP3 binding reverses this characteristic motion observed in the IBC in presence and absence of the SD. In the absence of the SD, binding of IP3 has restricted the overall dynamics of IBC (system 4, Fig. 4d,e, Movie S4). The IP3 in the binding site prevented the bending motion of IBC domains, which was observed in the apo IBC (system 3, Fig. 4b,c). This is expected as the binding site of IP3 located at the IBC-β – IBC-α interface and the bound IP3 imparts local stability by preventing the collapse of the interface that was observed in apo IBC. A similar observation was also made from an earlier simulation study with IBC, where the dissociation of IP3 conferred larger domain fluctuations to the protein9. However, in contrast to the reduced dynamics of IBC in system 4, IP3-bound IBC in presence of the SD (i.e., IBC of IP3-bound NT in system 2, Fig. 2c,d) shows increased domain movements, potentially due to the clam closure dynamics and twist motion of SD in the IP3-bound NT. The binding of IP3 at the IBC allosterically weakens the SD – IBC interface that frees the IBC whereas the stronger SD – IBC interface in the apo NT limits the IBC motions. Together, differential dynamics of IBC observed here suggests that a strong inter-domain communication prevails between SD and IBC where both of them influence each other’s dynamic behavior and the binding of ligand have long-range influence on domain dynamics.
SD suppresses the IP3 affinity by shuffling the mode of interaction between the IBC and IP3
Though the SD is critical for coupling the IP3-induced events to channel-opening, it is well known to suppress the affinity of the IBC or the full IP3R protein for IP3 binding11,12. The IP3 binding affinity of the NT or the whole receptor is reduced by more than one order of magnitude compared to that of IBC alone. Experimentally measured dissociation constants (Kd) of IP3 from different studies showed the higher affinity of IBC for IP3 in the absence of the SD11,12,14,23. The binding free energy (∆G) of IP3 at 296 K is calculated to be 37.1 ± 0.1 kJ/mol for the NT whereas the IBC, in the absence of the SD, shows higher binding affinity with ∆G of −43.2 ± 0.1 kJ/mol14. Furthermore, study with different IP3R isoforms revealed that IP3 binding affinity of the IBC is almost identical in the absence of the SD suggesting the critical role of SD in allosterically modulating the IP3 binding to the receptor24. Though previous studies revealed the inhibitory action of the SD, the precise molecular mechanism underlying the reduced binding affinity of IP3 in presence of the SD is not yet understood clearly. As discussed in the following sections, the present study with the aid of sub-microsecond MD simulations unveiled the molecular details of ‘suppressing action’ of the SD.
Firstly, to get a qualitative comparison of binding affinity and to complement the experimental observation, we computed the free energy of binding of IP3 to NT (i.e., presence of the SD, system 2) and IBC alone (i.e., absence of the SD, system 4) using MM/PBSA approach (see methodology section). The calculated binding ∆G was found to be −70.4 ± 1.2 kcal/mol and −79.5 ± 1.1 kcal/mol for NT and IBC respectively. In agreement with the experimental findings, it is evident from the binding free energy that the IBC, in the absence of the SD, shows comparatively higher affinity for IP3 than the whole NT. In order to unravel the molecular mechanism underlying such differential binding affinity, we further analyzed the interaction of individual protein residues with IP3 in terms of pairwise interaction energy. The pairwise interaction energy of residues lining the IP3-binding pocket is presented in Fig. 5a. Surprisingly, most of the pocket residues interact with IP3 with relatively similar strength except Arg269. The interaction energy of Arg269 with IP3 was about 15 units more in the absence of the SD as compared to that in the presence of the SD. In presence of SD (system 2), Arg269 – IP3 interaction energy was calculated to be −17.3 ± 0.6 kcal/mol. Markedly, in the absence of the SD (system 4), Arg269 of IBC interacts strongly with IP3 with energy of −33.8 ± 0.8 kcal/mol. In brief, the energy calculations suggest that the Arg269 – IP3 interaction plays a crucial role in determining IP3R’s affinity for IP3 in presence and absence of the SD.
Figure 5.

The SD influences the interaction of IP3 with the IBC. (a) Residue-level decomposition of interaction energy between IBC pocket residues and IP3. Color scheme: IP3-bound NT (system 2), red; IP3-bound IBC (system 4), blue; and SD-knockout (system 5), green. [(b,c)] Time evolution of Arg269 guanidinium – PO3 center of mass distance in (b) IP3-bound NT and (c) IP3-bound IBC. The inset shows the distance in IP3-bound NT during the initial 9 ns of simulation. Color scheme: Arg269 – P1, green and Arg269 – P5, red.
To obtain a better understanding of the receptor – IP3 interaction, we compared the ensemble-averaged structures of IP3-bound NT (system 2) and IBC (system 4) complexes. This is shown in Fig. S8a,b. For clarity, only the ligand and the adjacent protein residues that are involved in direct and consistent interactions are shown. It can be noted from the figure that the sugar moiety and the phosphate groups of the IP3 efficiently interact with the residues spanning the IBC -β and -α domains25. As expected, the binding pocket is lined by a number of basic and polar residues to accommodate a highly negatively charged molecule like IP3. Three phosphate groups - P1, P4, and P5 - of the IP3 molecule are involved in an extensive network of H-bonds with five arginine residues - Arg265, Arg269, Arg504, Arg511 and Arg568. The Lys508 that lies at the interior of binding pocket also forms H-bond with the P5 of IP3. During the entire span of simulation, Thr267 and backbone amino group of Gly268 was found to interact with P4 phosphate group. The C6′-OH of the IP3 forms H-bond with either Arg504 or Tyr567 in both system 2 and system 4, whereas other hydroxyl groups fail to make any direct interaction with protein residues. Such a less significant role of IP3 hydroxyl groups in its binding with IP3R is reported from the biophysical studies with IP3 derivatives26.
As discussed above, most of the pocket residues show similar pattern of interaction with IP3 in the presence and absence of the SD. However, very interestingly, the SD seemed to influence the mode of interaction of Agr269 with IP3. In the presence of the SD (system 2), the guanidinium group of Arg269 was found to interact effectively with the P1 (phosphate group attached to the C1′ of the inositol sugar ring) of the bound IP3 molecule (Fig. S8a). On the other hand, in the absence of the SD (system 4), Arg269 of the IBC makes multiple interactions with the bound IP3, where the guanidinium group of the residue H-bonds with P5 (phosphate group attached to the C5′ of the inositol sugar ring) and C6′-OH of the IP3 molecule (Fig. S8b). In both systems, the guanidinium −NH/−NH2 groups were involved in H-bond with the PO32−/C6′-OH groups of the IP3 molecule. As evident from the figure, Arg269, which interacts with the P1 of the IP3 in the presence of the SD (ie., in NT), is repositioned in the absence of the SD and forms H-bond with the P5 and C6′-OH groups of IP3 that lies deep inside the pocket. We further examined the stability of Arg269 – IP3 interaction by measuring the Arg269 – PO32− distance along the 300 ns simulation trajectories (Fig. 5b,c). In the presence of the SD, the guanidinium group of Arg269 interacts with the P1 having a H-bonding distance of ~3.8 Å throughout the simulation whereas the P5 was found to be located ~8 Å away from the Arg269 (Fig. 5b). Strikingly, Arg269 flipped its mode of interaction in the absence of the SD (system 4). As shown in Fig. 5c, rather than interacting with the P1 (distance of ~7 Å) as seen in the presence of the SD, the guanidinium group of the residue H-bonds with P5 in the absence of the SD, maintains a distance of ~3.8 Å. A similar analysis of time evolution of Arg269 – C6′-OH distance during simulation is depicted in Fig. S9. The guanidinium −NH2 – C6′−OH average distance was measured to be 5.8 ± 1.0 Å in the IP3-bound NT (system 2) as compared to the H-bond distance of 3.9 ± 0.5 Å in the IP3-bound IBC (system 4).
A closer look at the initial configurations from the simulation trajectories has also provided further insight into the SD-invoked Arg269 flipping. In the crystal structures, Arg269 was found to interact with the P5 of the IP3 in both IP3-bound IBC (PDB ID: 1N4K) as well as NT (PDB ID: 3UJ0) with a distance of about 2.7 Å. It is to be noted that the region spanning residues 268–270 that includes Arg269 is not resolved in any of the apo-NT crystal structures (PDB IDs: 3UJ4, 3T8S apo, 5 × 9Z, 5XA0) as well as an IP3-bound NT structure (PDB ID: 3T8S). It is, however, apparently resolved in the IP3-bound NT structures of the Cys-less rat IP3R1 (PDB ID: 3UJ0)5 as well as the mouse IP3R1 (PDB ID: 5GUG, 5XA1)7. But, for all these structures, the crystal structure of mouse IP3R1 IBC (PDB: 1N4K) that is clearly devoid of the SD, effectively served as the reference for the initial molecular replacement5,7. We suspect that this region containing Arg269 is very flexible and this is supported by our simulation as well as close inspection of the available crystal structures of the NT and full length IP3R14,5,7. Intriguingly, within the initial 3 ns of the simulation in our study, Arg269 within the IP3 bound NT breaks its interaction with P5 and flips towards P1, measuring a reduction in Arg269 – P1 distance from 8 Å to ~3.8 Å (Fig. 5b inset) and retained the distance of ~3.8 Å during rest of the simulation. It can be postulated that, in the starting NT configuration, the SD influenced the overall IBC architecture where the loop bearing the Arg269 was unable to occupy in its initial conformation, forcing the Arg269 to reposition itself to interact with the P1 of the IP3.
To strengthen our finding further that the SD modulates Arg269 flipping and interaction with IP3, we created a new simulation system (system 5) by removing the SD from the final conformation of IP3-bound NT obtained after the 300 ns simulation of system 2. Hence, the newly generated system consists of IP3-bound IBC of the previously simulated NT where the Arg269 interact with the P1 of the IP3. Here, our aim was to examine whether the IBC can sense the absence of the SD and reorient the Arg269 in such a way that it interacts with P5 as seen in IP3-bound IBC (i.e., in the absence of the SD, system 4). As in the above section, we analysed the Arg269 – IP3 interaction in system 5 by monitoring the guanidinium – PO32− distance along the simulation time. In Fig. 6, we combined the calculated distance from system 2 and 5. To our surprise, IBC in system 5 senses the absence of the SD during the course of simulation that eventually leads to the necessary structural rearrangements in the IBC with flipping of the Arg269 towards P5. As shown in Fig. 6, after ~280 ns of simulating the SD-removed NT (i.e., IBC), Arg269 – P5 distance decreased from about 10 Å to the H-bonding distance of ~3.8 Å and, at the same time, Arg269 – P1 distance increased to about 8 Å. This suggests that Arg269 breaks its interaction with P1 and flips towards P5 after ~280 ns of simulating the SD-removed IP3-bound IBC. Remarkably, the flipping of Arg269 was also reflected in the IP3 binding affinity. As expected, the IP3 binding affinity of IBC for IP3 was increased in the absence of the SD, with a rise in ∆G of binding from −71.9 ± 2.1 kcal/mol, in the IP3-bound NT, to −80.9 ± 0.9 kcal/mol, after the removal of the SD. The flipping has also altered the Arg269 – IP3 interaction energy (Fig. 5a). By shifting the site of interaction from P1, in presence of the SD, to P5, after the removal of the SD, Arg269 has strengthened its interaction with IP3 with an increase in energy from −17.3 ± 0.6 kcal/mol to −32.6 ± 0.1 kcal/mol. It is noteworthy that the calculated energies are in good agreement with that calculated from IP3-bound IBC in system 4 (Fig. 5a, blue bar). A movie showing the flipping mechanism of Arg269, in the absence of the SD, in system 5 is presented in Movie S5. Together, these analyses suggest that the SD allosterically modulates the IBC interaction with IP3 and Arg269 plays a crucial role in determining the binding affinity of IBC for IP3 in presence and absence of the SD. Higher IP3 binding affinity in the absence of the SD can be ascribed to the multiple interactions of Arg269 with IP3, involving Arg269 – P5 and Arg269 – C6′-OH, in contrast to the Arg269 – P1 interaction in the presence of the SD (Figs 5b,c, S8 and S9).
Figure 6.
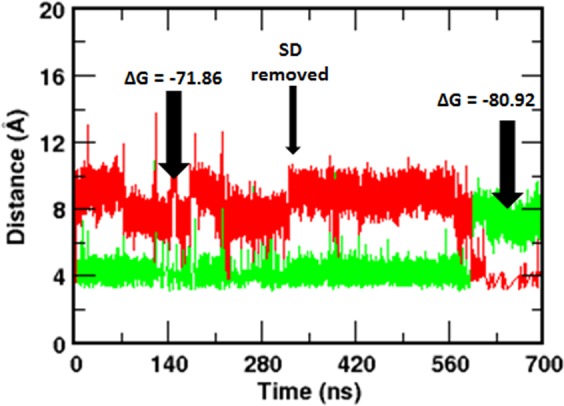
Removal of the SD from the IP3-bound NT flips the Arg269 interaction with the IP3. Time evolution of Arg269 guanidinium – PO3 center of mass distance combined from system 2 and system 5 (see Table 1 for system definition). The SD was removed from IP3-bound NT (system 2) after the initial 300 ns of simulation and thus generated IP3-bound IBC (system 5), which was simulated for another 400 ns. Corresponding free energy of IP3 binding before and after the removal of SD are shown. Color scheme: Arg269 – P1, green and Arg269 – P5, red.
Conclusions
As with most other ligand-gated ion channels, a detailed understanding of the activation gating of IP3Rs remains a holy grail in the field. Although, the currently available structural and biochemical information has significantly advanced our understanding of the structure-to-function relationship in IP3R, details of the dynamics of IP3-mediated domain movements within IP3R NT remained largely unexplored. In the present study, extensive MD simulations of apo and IP3-bound NT and analysis of essential dynamics have provided a detailed picture of correlated domain movements during and immediately following IP3 binding. Compared to the apo form, SD of IP3-bound NT showed characteristic twist motion, where the SD move towards the IBC-α. During the binding process, IP3 has brought together the β and α domains of IBC, leading to the ‘clam closure’. Our results demonstrated a dynamic view of unique structural plasticity acquired by the IP3R NT as a consequence of IP3 binding and support the observations inferred from crystallographic studies5. The H-bond and energetic analyses revealed the IP3-induced molecular rearrangements at the SD – IBC interface, where Glu283-Arg54-Asp444 triad of interaction breaks during the IP3 binding. The Arg54-Asp444 interaction breaks while the Arg54 maintains its interaction with Glu283. MD simulations further show the existence of inter-domain dynamic correlation in the IP3R NT and reveal that the SD is critical to the conformational dynamics of IBC. Very interestingly, the study unraveled the SD-dependent flipping of interaction of Arg269 between P1 and P5 phosphate groups of IP3, which seems to play a major role in governing the IP3 binding affinity of IBC in the presence/absence of the SD. It is to be noted that the residues identified in this study (Arg54, Arg269, Glu283 and Asp444) are conserved across different IP3R isoforms and in different species (Fig. S10). To conclude, the present study has brought out the importance of alternate conformations of IP3R NT needed for its function and the comprehensive understanding of the IP3-binding mechanism of IP3R derived here could help to understand the physiological activation and pharmacological regulation of IP3Rs.
Materials and Methods
A series of unbiased all-atom MD simulations of IP3R NT (residues 1–604) in its apo and IP3-bound states (Table 1) were performed. The crystal structure of the IP3R NT was used to model the full-length NT with residues 1–604. Thus, the atomic coordinates of the apo (PDB ID: 3UJ4) and IP3-bound (PDB ID: 3UJ0) IP3R NT obtained from the protein data bank were used in this study. The missing residues; 1–6, 76–84, 204, 269–270, 294–298, 373–381, 407, 426–427, 486–500, 525–550 and 578–604 were incorporated with the help of MODELLER9v1627 using crystal structures of IBC (PDB ID:1N4K) and SD (PDB ID: 1XZZ) as templates. Additionally, the crystal configurations were also missing a sequence of residues 319–351. Since we noted that a very low sequence similarity with available structures in databases, the 3D model of this sequence was constructed using the ab initio protein structure predictor, QUARK28. Quark builds the models based on structures of small fragments of 1–20 residues, which was later assembled by replica exchange Monte Carlo simulations using an atomic-level knowledge-based force field. The modeled structure of the amino acid sequence 319–351 was merged with the crystal conformation of the apo (system 1) and liganded NT (system 2) and optimized by the conjugate gradient energy minimization method in Modeler program27. Further, the apo (system 3) and IP3-bound (system 4) IBC-only structures of IP3R NT were generated from the full-length NT by deleting the N-terminal SD of residues 1–224.
In each of the above protein structures, hydrogen atoms were added using H++ server29, maintaining the ionization state of the residues at the pH of 7.4. A set of partial atomic charges for IP3 was obtained via quantum calculations. A B3LYP geometry optimization procedure was performed using Gaussian 09 with the 6–311 + G* basis set30. The atom-centered RESP charges were calculated via fits to the electrostatic potentials obtained from the calculated wave functions31. The interaction parameters for the IP3 were adopted from a previous work, where the forcefield parameters for phosphorylated inositols were developed based on OPLS-AA/AMBER framework32. Subsequently, the structures were energy minimized for 1000 steps using the steepest descent algorithm and another 1000 steps by the conjugate gradient method, using AMBER 14.0 simulation package33. After relaxing the added atoms in gas phase, each structure was solvated in cubic periodic box of explicit water with water molecules extending 14 Å outside the protein complex on all sides. The 3-site TIP3P water model was chosen to define the water molecules. In system 1 and 2, the simulation box contained ~45,000 water molecules, while ~22000 water molecules were present in the systems 3 and 4. Charge neutrality was maintained by adding one Cl− ion in system 1 and five, three and four Na+ ions in system 2–4 respectively. The solvated systems were subjected to extensive energy minimization and thermalization with raising the temperature gradually to 300 K in canonical ensembles, maintaining harmonic restraints on crystallized heavy atoms of protein and ligand. Afterwards, solvent density was adjusted under isobaric and isothermal conditions at 1 atm and 300 K and the harmonic restraints were slowly reduced to zero. To enable the volume variation, simulations were performed in an NPT ensemble using the Berendsen thermostat and barostat. The systems were equilibrated for 10 ns, with a 2 fs simulation time step. The long-range Coulombic interactions were treated using Particle Mesh Ewald sum with a cut off of 12 Å applied to Lennard-Jones interactions and all bonds involving hydrogen atoms were constrained using the SHAKE algorithm. Each system was later subjected to 300 ns of production run, saving the trajectories at an interval of 2 ps for further analysis. All simulations were performed using the AMBER 14.0 molecular dynamics simulation package with the AMBER ff14SB force field33. Additionally, a new simulation system (system 5, Table 1) was generated from the final conformation of IP3-bound NT obtained from 300 ns of simulation of system 2. This ‘SD- knockout’ system was prepared by removing the N-terminal SD of 224 residues from the IP3-bound NT. Like other systems, simulation of system 5 was performed for 400 ns following a similar protocol as stated above. Together, a total of 1.6 µs of atomistic simulations were carried out in this work.
Principal component analysis (PCA) is employed to explore the dominant motions of apo and IP3-bound IP3R NT region. PCA tool has been used in the past to locate the essential motions in complex systems with high dimensionality, like in proteins, and the detailed methodology can be found elsewhere34. PCA was performed in this study using pyPcazip program package35. The eigenvectors from PCA were visualized as porcupine plots using VMD36, where the direction of the porcupine corresponds to the direction of the motion and the length of the porcupine denotes the magnitude of the motion. The twist of the SD was calculated by measuring the angle defined by the two structures, the crystal structure and a snapshot from the simulation. A plane was defined on each structure by three points that is located on the center of mass of each domain (SD, IBC-β and IBC-α). The ‘twist angle’ defined as the dihedral angle between the two planes was calculated. PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4, Schrodinger, LLC) and VMD36 was used to generate all structural figures.
Free Energy Calculations
Binding free energies of IP3 were calculated using Molecular Mechanics Poisson Boltzmann Surface Area (MMPBSA) method implemented in Amber tools package33. The block averaged ∆G values were calculated from five independent windows of 5 ns generated by taking the last 25 ns trajectory and each window has 100 snapshots from the trajectory. In general, the free energy of ligand binding to protein in solvent is estimated as follows,
where Gcomplex, Gprotein, and Gligand are the free energies of the protein-ligand complex, unbound protein, and free ligand, respectively. The ∆Gbinding can also be calculated from the changes in the molecular mechanical gas-phase energy (ΔEMM), entropic contribution, and solvation free energy:
where ΔEMM is the vacuum potential energy that considers the bonded and non-bonded interactions between the protein and ligand. The solvation free energy, ΔGsolv, is estimated by solving the linearised Poisson Boltzman equation for each of the three states (ΔGpolar) and adding an empirical term for hydrophobic contributions to it (ΔGnonpolar). The hydrophobic contribution is calculated from the solvent accessible surface area. As in practice, the entropic contribution (TΔS) was omitted since the calculations involve binding of similar ligand to the same protein. Therefore, the computed values will be termed as the relative binding free energies.
Supplementary information
Acknowledgements
This work was supported by a Royal Society research grant and Royal Society University Research Fellowship (to T.R.). A.C. was a Royal Society Newton Fellow. D.L.P. was supported by the Biotechnology and Biological Sciences Research Council UK. X.C. was a recipient of the Cambridge Trust-IDB Malaysia scholarship. AC thank Prof. Saraswathi Vishveshwara (Molecular Biophysics Unit, Indian Institute of Science Bangalore, India) for critically reading the manuscript.
Author Contributions
A.C. and T.R. conceived the project. A.C. performed the research and analysed data with input from X.C. and D.L.P., A.C. and T.R. wrote the paper.
Data Availability
Most of the data generated or analyzed during this study are included in this article (and its Supplementary information files).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Aneesh Chandran, Email: aneeshc@iisc.ac.in.
Taufiq Rahman, Email: mtur2@cam.ac.uk.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-39301-3.
References
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiological reviews. 2016;96:1261–1296. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 3.Seo MD, Enomoto M, Ishiyama N, Stathopulos PB, Ikura M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochimica et biophysica acta. 2015;1853:1980–1991. doi: 10.1016/j.bbamcr.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Lin CC, Baek K, Lu Z. Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nature structural & molecular biology. 2011;18:1172–1174. doi: 10.1038/nsmb.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seo MD, et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature. 2012;483:108–112. doi: 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan G, et al. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature. 2015;527:336–341. doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamada K, Miyatake H, Terauchi A, Mikoshiba K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:4661–4666. doi: 10.1073/pnas.1701420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan J, et al. Ligand-induced conformational changes via flexible linkers in the amino-terminal region of the inositol 1,4,5-trisphosphate receptor. Journal of molecular biology. 2007;373:1269–1280. doi: 10.1016/j.jmb.2007.08.057. [DOI] [PubMed] [Google Scholar]
- 9.Ida Y, Kidera A. The conserved Arg241-Glu439 salt bridge determines flexibility of the inositol 1,4,5-trisphosphate receptor binding core in the ligand-free state. Proteins. 2013;81:1699–1708. doi: 10.1002/prot.24304. [DOI] [PubMed] [Google Scholar]
- 10.Yamazaki H, Chan J, Ikura M, Michikawa T, Mikoshiba K. Tyr-167/Trp-168 in type 1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. The Journal of biological chemistry. 2010;285:36081–36091. doi: 10.1074/jbc.M110.140129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwai M, Michikawa T, Bosanac I, Ikura M, Mikoshiba K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. The Journal of biological chemistry. 2007;282:12755–12764. doi: 10.1074/jbc.M609833200. [DOI] [PubMed] [Google Scholar]
- 12.Yoshikawa F, et al. Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. The Journal of biological chemistry. 1996;271:18277–18284. doi: 10.1074/jbc.271.30.18277. [DOI] [PubMed] [Google Scholar]
- 13.Bosanac I, et al. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Molecular cell. 2005;17:193–203. doi: 10.1016/j.molcel.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 14.Ding Z, et al. Binding of inositol 1,4,5-trisphosphate (IP3) and adenophostin A to the N-terminal region of the IP3 receptor: thermodynamic analysis using fluorescence polarization with a novel IP3 receptor ligand. Molecular pharmacology. 2010;77:995–1004. doi: 10.1124/mol.109.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haider S, Parkinson GN, Neidle S. Molecular dynamics and principal components analysis of human telomeric quadruplex multimers. Biophysical journal. 2008;95:296–311. doi: 10.1529/biophysj.107.120501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Twomey EC, Sobolevsky AI. Structural Mechanisms of Gating in Ionotropic Glutamate Receptors. Biochemistry. 2018;57:267–276. doi: 10.1021/acs.biochem.7b00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taufiq UR, Skupin A, Falcke M, Taylor CW. Clustering of InsP3 receptors by InsP3 retunes their regulation by InsP3 and Ca2+ Nature. 2009;458:655–659. doi: 10.1038/nature07763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner LE, II, Yule DI. Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J Physiol. 2012;590:3245–3259. doi: 10.1113/jphysiol.2012.228320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos-Franco J, Galvan D, Mignery GA, Fill M. Location of the permeation pathway in the recombinant type 1 inositol 1,4,5-trisphosphate receptor. The Journal of general physiology. 1999;114:243–250. doi: 10.1085/jgp.114.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mak DO, McBride SM, Foskett JK. Spontaneous channel activity of the inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R). Application of allosteric modeling to calcium and InsP3 regulation of InsP3R single-channel gating. The Journal of general physiology. 2003;122:583–603. doi: 10.1085/jgp.200308809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi AM, et al. Synthetic partial agonists reveal key steps in IP3 receptor activation. Nature chemical biology. 2009;5:631–639. doi: 10.1038/nchembio.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sienaert I, et al. Localization and function of a calmodulin-apocalmodulin-binding domain in the N-terminal part of the type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 2002;365:269–277. doi: 10.1042/BJ20020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossi AM, Sureshan KM, Riley AM, Potter VL, Taylor CW. Selective determinants of inositol 1,4,5-trisphosphate and adenophostin A interactions with type 1 inositol 1,4,5-trisphosphate receptors. British journal of pharmacology. 2010;161:1070–1085. doi: 10.1111/j.1476-5381.2010.00947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan J, et al. Structural studies of inositol 1,4,5-trisphosphate receptor: coupling ligand binding to channel gating. The Journal of biological chemistry. 2010;285:36092–36099. doi: 10.1074/jbc.M110.140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosanac I, et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 26.Kozikowski AP, Ognyanov VI, Fauq AH, Nahorski SR, Wilcox RA. Synthesis of 1 D -3-deoxy-, 1 D -2,3-dideoxy-, and 1 D -2,3,6-trideoxy-myo-inositol 1,4,5-trisphosphate from quebrachitol, their binding affinities, and calcium release activity. J Am Chem Soc. 1993;115:4429–4434. doi: 10.1021/ja00064a002. [DOI] [Google Scholar]
- 27.Webb B, Sali A. Protein Structure Modeling with MODELLER. Methods in molecular biology. 2017;1654:39–54. doi: 10.1007/978-1-4939-7231-9_4. [DOI] [PubMed] [Google Scholar]
- 28.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic acids research. 2012;40:W537–541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaussian 09 (Gaussian Inc.: Wallingford CT, 2009).
- 31.Cornell WD, Ciepark P, Baylay CI, Kollman PA. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc. 1993;115:9620–9631. doi: 10.1021/ja00074a030. [DOI] [Google Scholar]
- 32.Rosen SA, Gaffney PR, Gould IR. A theoretical investigation of inositol 1,3,4,5-tetrakisphosphate. Physical chemistry chemical physics: PCCP. 2011;13:1070–1081. doi: 10.1039/c0cp00956c. [DOI] [PubMed] [Google Scholar]
- 33.Case, D. A. et al. AMBER 14. University of California: San Francisco (2014).
- 34.Chandran A, Vishveshwara S. Exploration of the conformational landscape in pregnane X receptor reveals a new binding pocket. Protein science: a publication of the Protein Society. 2016;25:1989–2005. doi: 10.1002/pro.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ardita S, et al. pyPcazip: A PCA-based toolkit for compression and analysis of molecular simulation data. SoftwareX. 2016;5:44–50. doi: 10.1016/j.softx.2016.04.002. [DOI] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14(33-38):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most of the data generated or analyzed during this study are included in this article (and its Supplementary information files).