

Abstract
Cell membranes have recently gained attention as a promising drug delivery system. Here, dendritic cell membrane vesicles (DC-MVs) are examined as a platform to promote T cell responses. Nano-sized DC-MVs are derived from dendritic cells (DCs) pre-treated with monophosphoryl lipid A (MPLA), an FDA-approved immunostimulatory adjuvant. These “mature” DC-MVs activate DCs in vitro and increase their expression of co-stimulatory markers. DC-MVs also promote cross-priming of antigen-specific T cells in vitro, increasing their survival and CD25 expression. In addition, these mature DC-MVs potently augment the expansion of adoptively transferred CD8+ T cells in vivo, generating 2-4 fold higher frequency of antigen-specific T cells, compared with other control formulations, including “immature” DC-MVs obtained without the MPLA pre-treatment. Taken together, these results suggest that DC-MVs are an effective delivery platform for T cell activation and may serve as a potential delivery system for improving adoptive T cell therapy.
Keywords: membrane vesicle, vaccine, peptide antigen, T cell activation
Graphical Abstract

Nano-sized dendritic cell membrane vesicles (DC-MVs) are examined as a platform to promote T cell responses. DC-MVs activate DCs and promote cross-priming of CD8+ T cells in vitro. DC-MVs potently amplify the expansion of adoptively transferred CD8+ T cells in vivo. DC-MVs may serve as a potential peptide delivery system for augmenting adoptive T cell therapy.
Cancer immunotherapy is transforming the current landscape of oncology.[1] Adoptive T cell therapy has recently shown great promise and produced effective clinical responses, leading to clinical approval for blood cancers.[2] Immune checkpoint blockade designed to release the brakes from the immunosuppressive tumor environment allows for anti-tumor therapeutic effects.[3] At the same time, advances in neo-antigen characterization in tumors has pushed for readily-synthesized and potent peptide-based vaccination.[4,5] These discoveries and a tremendous amount of work over the past decade had led to the development of effective cancer vaccines evaluated in clinical trials.[6] However, induction of effective immunity is often dependent on appropriate antigen delivery methods for optimal responses, with peptide-based vaccines being particulary susceptible to forming depots at the site of administration leading to diminished potency.[7]
In this study, we examined cell-derived vesicles for antigen delivery to promote antigen-specific T cell responses. Utilizing cell membrane preparation technologies that we and others have reported,[8–10] we generated dendritic cell membrane vesicles (DC-MVs) from pre-activated antigen-presenting cells (APCs) (Figure 1A). Here, we show that DC-MVs can be effectively loaded with antigen peptides and promote activation of antigen-specific T cells in vitro. We also demonstrate their potency to expand adoptively transferred T cells in vivo. Our results suggest that DC-MVs is a promising platform for augmenting adoptive T cell therapy and improving peptide-based cancer vaccination.
Figure 1. DC-MVs directly promote T cell activation and proliferation in vitro.

A. Schematic demonstrating preparation of DC-MVs. B. Western Blot analysis of MPLA-activated DCs and activation marker-enriched membrane fraction. C-D. Proliferation and activation of OT-I T cells are shown after seeding T cells at 50,000 or 10,000 cells per well in the presence of SIINFEKL alone or SIINFEKL mixed in with DC-MVs. Mean ± SD are shown. Statistical analysis was performed using two-way ANOVA comparison with Tukey's multiple comparison test comparing (MPLA)DC-MV-SIINFEKL to either DC-MV-SIINFKEL (#p < 0.05, ####p < 0.0001) or SIINFEKL alone (**p < 0.01, ****p < 0.0001).
To generate DC-MVs, we first obtained DCs from bone marrow of C57BL/6 mice with the use of granulocyte-macrophage colony-stimulating factor.[11] Immature bone marrow-derived dendritic cells (BMDCs) were lysed by freeze-thaw cycles and mild probe-tip sonication. After removing large debris and organelles via centrifugation, the resulting lysate samples were incubated with 20 mM CaCl2 for 1 hour to promote fusion and aggregation of cellular membranes, which allowed for isolation of DC-MVs with table-top centrifugation. We sought to pre-activate DCs before generating DC-MVs and studied their impact on the subsequent T cell cross-priming. Specifically, we pre-activated BMDCs with monophosphoryl lipid A (MPLA), an FDA-approved immunostimulatory Toll-like receptor 4 agonist.[12,13] We first confirmed upregulation of co-stimulatory markers, including CD80 and CD86, in whole cell lysate of MPLA-treated DCs by Western blotting (Figure 1B). Immature DCs without any MPLA treatment (NO TX) exhibited minimal expression of CD80 or CD86. In contrast, pre-treatment of DCs with MPLA increased the expression levels of co-stimulatory markers, in particular, CD86, on DC-MVs regardless whether BMDCs were harvested from culture dishes either by Accutase treatment and a cell scraper (MPLA) or by pipetting (MPLA-S) (Figure 1B). Based on these results and high yield of total protein (80%) from the Accutase-based harvest, we proceeded with this method for the source for MPLA-activated DC-MVs, which is henceforth termed (MPLA)DC-MVs. Dynamic light scattering analysis indicated that (MPLA)DC-MVs had an average hydrodynamic size of 130 ± 4 nm and a polydispersity index of 0.17 ± 0.01.
Next, we examined the ability of DC-MVs to present antigen peptides directly to T cells and promote their activation and proliferation through engagement with T cell receptor (TCR, signal 1) and CD28 (signal 2). We employed a model antigen peptide SIINFEKL, an immunodominant MHC-I epitope from ovalbumin (OVA). To examine the interactions between DC-MVs and antigen-specific T cells, we performed the commonly used carboxyfluorescein succinimidyl ester (CFSE) dilution assay[14] with SIINFEKL-specific CD8α+ T cells obtained from OT-I transgenic mice with the exception that CD8α+ T cells were directly induced with DC-MVs without the presence of any intact APCs. Using the standard 50,000 cells/well seeding density of CD8α+ OT-I T cells, we observed that SIINFEKL peptide alone promoted CFSE dilution of T cells; however, the number of OT-I T cells surviving at the end of the three day culture was low, with minimal expression of CD25, which is a subunit of the IL-2 receptor and a late marker of TCR-dependent T cell activation (Figure 1C). This may be due to direct peptide binding and epitope presentation in the context of MHC-I on the T cell surface itself, resulting in swift T cell cross-priming and cross-killing, a phenomenon termed fratricide.[15] In contrast, we observed strong proliferation of T cells with the increased expression of CD25 when we cultured CD8α+ OT-I T cells in the presence of 10 ng/ml SIINFEKL and immature DC-MVs (Figure 1C). For these in vitro studies, we added DC-MV formulations directly to SIINFEKL-containing media without column purification in order to ensure equivalent antigen dose without any variation across all groups. Notably, DC-MVs pre-activated with MPLA further amplified T cell responses; (MPLA)DC-MV-SIINFEKL significantly enhanced T cell survival, compared with the DC-MV-SIINFEKL group and the SIINFEKL control group (p < 0.0001, at 10 ng/mL SIINFEKL concentration, Figure 1C). In addition, (MPLA)DC-MV-SIINFEKL significantly improved the CD25 expression on CD8α+ OT-I T cells, compared with either control groups (p < 0.0001, at 10 ng/mL SIINFEKL concentration, Figure 1C). We also confirmed these results using a lower seeding density of OT-I T cells (10,000 cells/well) aimed to limit direct cell-cell interactions (Figure 1D). Taken together, these results suggested that MPLA pre-treatment increased the expression of co-stimulatory markers on DC-MVs and improved their efficacy to cross-prime antigen-specific T cell responses in vitro.
While direct stimulation of T cells may be possible in the controlled in vitro conditions, it may be difficult to achieve this within the complex in vivo environment. To address this, we examined if DC-MVs taken up by APCs can indirectly enhance T cell activation. Also, we sought to compare DC-MVs pre-activated with MPLA or CpG (a TLR-9 agonist based on DNA oligonucleotide with unmethylated CpG motifs[16]). For this particular experiment, we prepared DC-MV formulations pre-incubated with SIINFEKL overnight, followed by column chromatography. This ensured that unbound antigen was separated out, thus eliminating the possibility of free SIINFEKL directly binding BMDCs and activating T cells. BMDCs were pulsed with various SIINFEKL-loaded DC-MV formulations overnight, washed extensively, and incubated with CFSE-labeled OT-I T cells for three days, followed by flow cytometry-based analysis. BMDCs incubated with (MPLA)DC-MV-SIINFEKL increased proliferation (Figure 2A-C) and CD25 expression of OT-I T cells in a dose-dependent manner (Figure 2D). (MPLA)DC-MV-SIINFEKL significantly enhanced T cell proliferation, compared with unactivated DC-MVs at 10 µg/ml protein dose of DC-MVs (p < 0.01), and there was a trend (although no statistical difference) for increased T cell proliferation with (MPLA)DC-MV-SIINFEKL, compared with (CpG)DC-MV-SIINFEKL (Figure 2B).
Figure 2. DC-MVs indirectly promote T cell activation and proliferation in vitro.
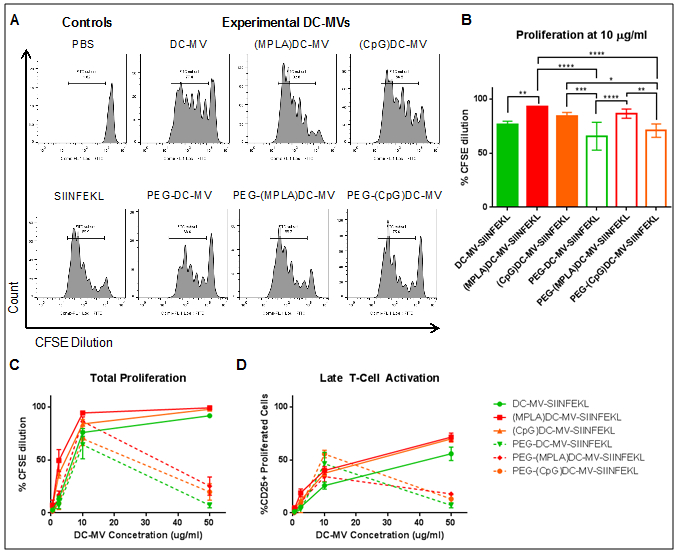
A-B. Representative histograms and summary of OT-I T cell proliferation in the presence of BMDCs and SIINFEKL-loaded DC-MVs at 10 µg/ml protein concentration of DC-MVs. C. Proliferation of OT-I T cells seeded with SIINFEKL-loaded DC-MVs at various concentrations. D. Activation was measured as the fraction of CD25-positive T cells following stimulation. Mean ± SD are shown. Statistical analysis was performed using two-way ANOVA comparison with Tukey's multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
We also examined whether introducing poly(ethylene glycol) (PEG) on the surfaces of DC-MVs affected their efficacy to activate T cells. To produce PEGylated DC-MVs, we incubated DC-MVs with 10 mg/ml DSPE-PEG in 100 mM EDTA solution and used a mild water bath sonication,[10] which promoted surface coating with PEG while removing excess calcium added during the purification of DC-MVs as detailed above.
PEGylated (MPLA)DC-MVs significantly improved T cell proliferation, compared with PEGylated DC-MVs from either unactivated or CpG-treated DCs at the 10 µg/ml dose (p < 0.0001 and p < 0.01, respectively, Figure 2B), and this trend was observed at DC-MV concentrations ranging from 2 to 10 µg/ml (Figure 2C-D). However, at higher concentration of 50 µg/ml, the PEGylated DC-MV formulations exhibited loss of bioactivity (Figure 2C-D), supposedly by interfering with antigen uptake and presentation. Comparing PEGylated versus non-PEGylated DC-MVs, we did not observe any statistical differences within each adjuvant-induced stimulation condition, except for CpG-treated DC-MVs that exhibited decreased T cell proliferation after PEGylation (p < 0.05, Figure 2A-B), potentially due to PEG-mediated interference of vesicle-cell interactions. Overall, these results demonstrated that DC-MVs could activate T cells via an indirect pathway of APC-mediated uptake and antigen presentation to T cells. Based on these results, we chose to focus on (MPLA)DC-MVs without PEGylation for the subsequent studies.
As naturally-produced membrane vesicles are known to transduce cell-to-cell signals,[17,18] we sought to determine if the artificially produced DC-MVs can activate live DCs in vitro. BMDCs were pulsed with various DC-MV formulations and analyzed for activation markers, including CD40 and CD80, via flow cytometry. (MPLA)DC-MVs significantly upregulated both CD40 and CD80 on BMDCs, compared with unactivated DC-MVs (p < 0.0001 and p < 0.001, respectively, at 200 µg/ml dose, Figure 3A), demonstrating their potency to activate DCs.
Figure 3. DC-MVs promote expansion of adoptively transferred T cells in vivo.

A. BMDCs were cultured in vitro with DC-MV formulations, and the expression of maturation markers was determined by flow cytometry. B. Loading of FITC-labeled SIINFEK(FITC)L peptide on various DC-MV formulations was determined using a microplate-based quantification. C. Congenic Thy1.2+ C57BL/6 mice were adoptively transferred with OT-I CD8α+Thy1.1+ T cells on day 0, immunized with various DC-MV formulations on day 1, and analyzed for the frequency of Thy1.1+ T cells on day 6. Mean ± SD are shown. Statistical analysis was performed using two-way ANOVA comparison with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Next, we examined the ability of DC-MVs to load SIINFEKL peptide in an MHC-I haplotype-specific manner. As SIINFEKL has a high affinity for H-2Kb from C57BL/6 mouse strain,[19] we examined the amount of SIINFEKL peptide bound on DC-MVs derived from C57BL/6 (H-2Kb, H-2Db) and compared to that from BALB/c (H-2Kd, H-2Dd, H-2Ld) as the control group. To quantify peptide loading, we utilized SIINFEKL peptide with a covalently bound fluorophore (FITC) to the lysine amino acid of the peptide (SIINFEK(FITC)L), which can effectively bind to H-2Kb MHC-I.[19,20] We generated (MPLA)DC-MVs or unstimulated DC-MVs and incubated them with SIINFEK(FITC)L at various membrane protein concentrations, followed by desalting column chromatography to remove any unbound peptide. We observed a similar amount of SIINFEKL peptide loaded onto DC-MVs regardless of the MHC-I haplotypes (C57BL/6 versus BALB/c) or pre-activation status with MPLA (Figure 3B). This outcome suggests that the peptide is likely loaded onto DC-MVs via non-specific interactions; however, we cannot rule out the possibility that a small fraction of peptide is specifically bound to and presented by H-2Kb MHC-I molecules on DC-MVs due to small quantity of pMHC-I complexes in DC-MV formulations.
Based on our in vitro results, we next evaluated if DC-MVs can promote T cell activation in vivo. We utilized adoptive cell transfer of OT-I T cells to provide a basal, equivalent frequency of antigen-specific T cells within each animal. On day 0, OT-I CD8α+Thy1.1+ T cells (5 × 105 cells per animal) were adoptively transferred via intravenous administration into recipient, congenic Thy1.2+ C57BL/6 mice, and on day 1, the animals were immunized with various DC-MV formulations. On day 6, peripheral blood samples were analyzed for the frequency of Thy1.1+ T cells. DC-MV formulations were incubated with 10 µg SIINFEKL and used directly without column purification in order to ensure equivalent antigen dose without any variation across all groups. Subcutaneous administration of soluble SIINFEKL with or without MPLA resulted in a weak expansion of adoptively transferred OT-I T cells, with 15–20% Thy1.1+ T cells among CD8α+ T cells (Figure 3C). Unactivated DC-MV-SIINFEKL did not enhance the expansion of OT-I T cells, compared with the soluble controls. In contrast, (MPLA)DC-MV-SIINFEKL derived from C57BL/6 mice markedly improved the expansion of OT-I T cells, achieving 4.1-fold, 2.4-fold, and 2.1-fold increases, compared with the SIINFEKL, SIINFEKL+MPLA, and DC-MV-SIINFEKL groups (p < 0.05, p < 0.01, and p < 0.05, respectively, Figure 3C), thus demonstrating that MPLA-induced pre-activation of BMDCs played a critical role in T cell activation by (MPLA)DC-MVs. Notably, (MPLA)DC-MV-SIINFEKL derived from BALB/c mice also induced robust expansion of OT-I T cells, compared with other control groups (Figure 3C); however, there was no statistical difference between (MPLA)DC-MV-SIINFEKL formulations derived from C57BL/6 or BALB/c mice.
Taken together, these results suggest that DC-MVs are an effective delivery platform for peptide-based vaccines and may serve as potential nanomaterials for cancer immunotherapy. In particular, these nano-sized DC vesicles are expected to effectively drain to target lymphatic tissues and may promote the maintenance of adoptive T cell therapies through the functional presentation of co-stimulatory markers or antigen presentation by APCs. Additionally, DC-MVs generated from a patient’s blood-sourced cells are expected to be fully biocompatible without triggering anti-vector immunity, thus providing a new material platform for cancer immunotherapy.
Experimental Section
DC-MV Preparation and Peptide Loading:
BMDCs were generated as previously described.[11] DC membrane vesicles (DC-MVs) were generated from sonicated cell lysate, following the removal of large debris and organelles via centrifugation (10,000 × g, 10 minutes). Lysate samples were adjusted to 6 mg/ml concentration and incubated with 20 mM CaCl2 for 1 hour to promote fusion and aggregation of membranes, which allowed for washing with table-top centrifugation (20,000 × g, 5 minutes). Where indicated, DC-MVs were then resuspended with mild water bath sonication in 10 mg/ml DSPE-PEG in 100 mM EDTA solution in PBS, which promoted surface coating with PEG and removal of excess calcium by chelation. Finally, DC-MVs were passed through PBS-equilibrated Zeba desalting column to remove excess EDTA, calcium, and DSPE-PEG to generate PEGylated DC-MVs. SIINFEKL or fluorescently labeled SIINFEK(FITC)L peptides (Genscript) were loaded at 100 µg/ml onto DC-MVs by incubation at 37°C at varying concentrations of DC-MVs (10.0, 2.5, and 1.0 mg/ml in PBS). DC-MV concentrations were measured by Pierce BCA assay (ThermoFisher). Peptide loading efficiency was determined after samples were passed two times through 40 kDa Zeba desalting column and quantified by a microplate-based fluorescence assay.
T Cell Proliferation In Vitro:
T cell proliferation was assessed using SIINFEKL-specific primary T cells obtained from OT-I transgenic mice. Briefly, spleens from 6- to 12-week old OT-I transgenic mice were harvested and processed into single cell suspension. Red blood cells were removed by three-minute incubation with ACK lysis buffer (Gibco) and CD8α+ T cells were separated using a CD8α+ T cell negative selection kit (StemCell Technologies). Cells were then labeled with 1 µM CFSE solution by incubating at 37°C for ten minutes and, after washing, resuspended in complete T cell media (RPMI supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/ml streptomycin, 50 μM β-mercaptoethanol, 1 mM HEPES buffer, and 1X non-essential amino acid solution (Gibco)). Cells were then plated in 96-wells at 50,000 or 10,000 cells/well and treated with 0.01 to 10 ng/ml SIINFEKL peptide with or without 50 µg/ml of DC-MVs. After three days of culture, T cells were collected, washed, blocked with FACS buffer containing anti-CD16/32 antibodies, and stained with anti-CD8α, anti-CD25, and DAPI, followed by flow cytometry. The results were analyzed by FlowJo software.
Animal Studies:
Animals were cared for following the federal, state, and local guidelines. All work performed on animals was in accordance with and approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan, Ann Arbor. OVA-specific, Thy1.1+ OT-I CD8α+ T cells were obtained as described above, and 5 × 105 cells were adoptively transferred into naïve congenic Thy1.2+ C57BL/6 mice (female, 6–8 weeks, Envigo, USA) via intravenous tail vein injection. One day after the transfer, the animals were immunized with SIINFEKL peptide (10 µg per mouse) with or without DC-MVs (250 µg protein per mouse). After 5 days, peripheral blood samples were obtained using submandibular bleeds and red blood cells removed via ACK lysis to yield peripheral blood mononuclear cells (PBMCs). Samples were processed for flow cytometry by washing, blocking CD16/32 Fc receptor, and staining with anti-CD8α and anti-Thy1.1. Cells were then resuspended in FACS DAPI solution and examined via flow cytometry.
Statistical analyses:
Sample sizes were chosen based on preliminary data from pilot experiments and previously published results in the literature. Statistical analysis was performed with Prism 6.0 software (GraphPad Software) by two-way ANOVA with Tukey’s comparisons post-test, as indicated. Data were normally distributed and variance between groups was similar. All values are reported as means ± SD. Statistical significance is indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Acknowledgements and financial conflict of interest
This work was supported in part by NIH (R01AI127070, R01EB022563, and R01CA210273) and Emerald Foundation. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16–1-0369), and NSF CAREER Award (1553831). L.J.O. is supported by pre-doctoral fellowships from UM Rackham and AFPE. Opinions interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. The authors declare no conflict of interest.
Contributor Information
Dr. Lukasz J. Ochyl, Department of Pharmaceutical Sciences, University of Michigan, Biointerfaces Institute, University of Michigan, 2800 Plymouth Drive, Ann Arbor, MI 48109, USA
Prof. James J. Moon, Department of Biomedical Engineering, University of Michigan, 2800 Plymouth Drive, Ann Arbor, MI 48109, USA; Department of Pharmaceutical Sciences, University of Michigan, Biointerfaces Institute, University of Michigan, 2800 Plymouth Drive, Ann Arbor, MI 48109, USA, moonjj@umich.edu
References
- [1].Chen DS, Mellman I, Immunity. 2013, 39, 1. [DOI] [PubMed] [Google Scholar]
- [2].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, N Engl J Med. 2014, 371, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen LP, Han X, Journal of Clinical Investigation. 2015, 125, 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schumacher TN, Schreiber RD, Science. 2015, 348, 69. [DOI] [PubMed] [Google Scholar]
- [5].Gubin MM, Artyomov MN, Mardis ER, Schreiber RD, J Clin Invest. 2015, 125, 3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ott PA, Hu ZT, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang WD, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li SQ, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, Nature. 2017, 547, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang X-F, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Schluns KS, Davis RE, Hwu P, Overwijk WW, Nature Medicine. 2013, 19, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fang RH, Jiang Y, Fang JC, Zhang L, Biomaterials. 2017, 128, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, Zhang L, Adv Mater. 2017, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ochyl LJ, Bazzill JD, Park C, Xu Y, Kuai R, Moon JJ, Biomaterials. 2018, 182, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lutz MB, Kukutsch N, Ogilvie ALJ, Rossner S, Koch F, Romani N, Schuler G, Journal of Immunological Methods. 1999, 223, 77. [DOI] [PubMed] [Google Scholar]
- [12].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC, Science. 2007, 316, 1628. [DOI] [PubMed] [Google Scholar]
- [13].Kuai R, Sun X, Yuan W, Ochyl LJ, Xu Y, Hassani Najafabadi A, Scheetz L, Yu MZ, Balwani I, Schwendeman A, Moon JJ, J Control Release. 2018, 282, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Quah BJC, Warren HS, Parish CR, Nature Protocols. 2007, 2, 2049. [DOI] [PubMed] [Google Scholar]
- [15].Su MW, Pyarajan S, Chang JH, Yu CL, Jin YJ, Stierhof YD, Walden P, Burakoff SJ, Eur J Immunol. 2004, 34, 2459. [DOI] [PubMed] [Google Scholar]
- [16].Krieg AM, Annu Rev Immunol. 2002, 20, 709. [DOI] [PubMed] [Google Scholar]
- [17].Pucci F, Pittet MJ, Clin Cancer Res. 2013, 19, 2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Raposo G, Stoorvogel W, J Cell Biol. 2013, 200, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Falk K, Rotzschke O, Faath S, Goth S, Graef I, Shastri N, Rammensee HG, Cellular Immunology. 1993, 150, 447. [DOI] [PubMed] [Google Scholar]
- [20].Saini SK, Ostermeir K, Ramnarayan VR, Schuster H, Zacharias M, Springer S, Proc Natl Acad Sci U S A. 2013, 110, 15383. [DOI] [PMC free article] [PubMed] [Google Scholar]