

Abstract
Sensitivity and resolution are key considerations for NMR applications in general, and for metabolomics in particular, where complex mixtures containing hundreds of metabolites over a large range of concentrations are commonly encountered. There is a strong demand for advanced methods that can provide maximal information in the shortest possible time frame. Here we present the optimization and application of the recently introduced 2D real-time BIRD 1H-13C HSQC experiment for NMR-based metabolomics at 13C natural abundance of aqueous samples. For mouse urine samples, it is demonstrated how this real-time pure shift sensitivity improved Heteronuclear Single Quantum Correlation (HSQC-SI) method provides broadband homonuclear decoupling along the proton detection dimension and thereby significantly improves spectral resolution in regions that are affected by spectral overlap. Moreover, the collapse of the scalar multiplet structure of cross-peaks leads to a sensitivity gain of about 40% - 50% over a traditional 2D HSQC-SI experiment. The experiment works well over a range of magnetic field strengths and is particularly useful when resonance overlap in crowded regions of the HSQC spectra hampers accurate metabolite identification and quantitation.
Keywords: Pure shift NMR spectroscopy, BIRD decoupling, HSQC, NMR-based metabolomics in H2O, mouse urine, broadband homonuclear decoupling
Graphical Abstract
INTRODUCTION
Over the last decade metabolomics has rapidly gained increasing popularity for accurately describing the molecular phenotype of an organism, organ, cell line, or biofluid.1–6 Besides mass spectrometry (MS),7–10 NMR spectroscopy is a key analytical tool for this purpose.11–14 Although the use of two-dimensional (2D) NMR methods has recently been growing,15–18 one-dimensional (1D) 1H NMR is still the most frequently used experiment in metabolomics due to its relative simplicity and sensitivity.19–22 However, 1D 1H spectra of metabolite mixtures are often severely crowded, which makes it impossible to reliably identify many of the metabolites present, causing a loss of potentially important information. Although multidimensional NMR methods can overcome many of these issues for the more comprehensive and more accurate characterization of metabolomics samples, spectral overlap can still pose a formidable challenge for certain spectral regions. The development of experiments with optimal resolution and sensitivity therefore continues to be an important objective.
The 1H-13C Heteronuclear Single Quantum Correlation (HSQC)23,24 experiment has become a standard tool for untargeted metabolomics owing to its good resolving power due to the large chemical shift dispersion in the 13C dimension. However, 2D HSQC can still suffer from serious peak overlaps when applied to complex mixtures encountered in real-world metabolomics applications. On the one hand, non-uniform sampling approaches25–29 are increasingly utilized to optimize spectral resolution along the indirect 13C dimension for a given amount of total NMR measurement time. On the other hand, broadband homonuclear decoupled, so-called ‘pure shift’, methods30–33 are particularly promising to boost the resolution in the direct proton dimension, which is typically crowded due to the large number of resonances confined to a small 1H chemical shift range and the presence of J-splittings caused by homonuclear proton-proton scalar couplings. Pure shift methods suppress homonuclear J-couplings by collapsing multiplets into singlets, which leads to simpler spectra with increased resolution along the proton dimension. Over the last decade, significant efforts have been made to develop more efficient homonuclear decoupled pulse sequences34–41 and to pave the way for their everyday application in the structural elucidation of synthetic organic molecules and natural products.42–49 However, pure shift experiments have not found their way into routine metabolomics so far, although pure shift HSQC-based approaches have been proposed recently for targeted metabolomics.50,51 It should be also noted that for many pure shift experiments resolution enhancement is accompanied by a considerable sensitivity loss as a consequence of the application of selective J-refocusing elements, which select only a subset of spins to achieve homonuclear decoupling. These elements can be realized by frequency selection,52 the combination of spatial and frequency discrimination (Zangger-Sterk approach53), statistical selection (anti-z-COSY54 and PSYCHE55 methods), or isotope-based discrimination (BIRD pulse sequence56).
Reduced sensitivity often limits the application of pure shift methods in metabolomics where studies with a large size of samples containing metabolites at low concentration are typical. Until now, real-time BIRD HSQC57 is the only fully broadband homonuclear decoupled experiment that is free from any sensitivity penalty, which makes it the most promising candidate for everyday 2D HSQC-based metabolomics applications.
Here we report an untargeted metabolomics application of the real-time BIRD HSQC-SI experiment to mouse urine sample dissolved in H2O, which is one of the most complex and most challenging metabolite mixtures. The method was optimized for aqueous solution, which is the sample condition most frequently encountered in metabolomics. Simultaneous enhancement of resolution and sensitivity in the HSQC spectrum of mouse urine sample is demonstrated without necessitating longer NMR measurement times.
EXPERIMENTAL SECTION
Sample Preparation
After their collection, the urine samples of 1-month healthy mice were immediately frozen in liquid N2 and stored at −80 °C. Before NMR measurements, the mouse urine samples were thawed on ice. For each NMR sample, 178 μl mouse urine was mixed with 20 μl phosphate buffer (from 500 mM pH 7.4 stock solution prepared in D2O) and 2 μl DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid from 10 mM stock solution prepared in D2O). It was then homogenized to have a final concentration of 50 mM of phosphate buffer and 0.10 mM of DSS. 200 μl of the final samples were transferred to 3 mm NMR tubes for NMR data collection.
NMR Experiments and Processing
NMR spectra were collected at 298 K on Bruker AVANCE III HD 600 and 850 MHz spectrometers, both equipped with a cryogenically cooled TCI probe. Each 1H-13C HSQC-SI spectrum of mouse urine was recorded with 4096 total data points in the 1H t2 dimension and 1024 total points in the 13C t1 dimension. 8 scans per t1 total points were used and the relaxation delay between consecutive scans was 1.5 s. The spectral widths along 1H and 13C dimensions were 12 ppm (17 ppm for experiments measured on the 600 MHz instrument) and 165 ppm, respectively and the transmitter frequency offsets were ~4.7 ppm and 77.5 ppm, respectively. Data acquisition in the real-time BIRD 1H-13C HSQC-SI experiments was divided into 8 chunks and the length of each chunk was 25.088 ms. INEPT and BIRD delays were adjusted according to 1JCH of 145 Hz. All NMR data was zero-filled, Fourier-transformed, phase-corrected and plotted using Bruker Topspin 3.5 software. Chemical shift database query and identification of metabolites from the 2D NMR spectra were done using the COLMARm58 NMR webserver (http://spin.ccic.ohio-state.edu/index.php/colmar).
RESULTS AND DISCUSSION
In a first step, we optimized the real-time BIRD 1H-13C HSQC-SI method for aqueous metabolite samples and for high-field NMR instruments (600 MHz field and higher), which are typical for metabolomics studies, but less so for NMR studies of synthetic organic molecules and natural products. The optimized pulse sequence is displayed in Figure 1. In this gradient-enhanced sensitivity-improved (SI) HSQC sequence,24 signal acquisition is performed by a real-time BIRD acquisition scheme37 instead of the conventional one. This means that the FID detection is periodically interrupted in order to refocus proton-proton J-evolution between FID portions, so-called ‘chunks’, by a double spin echo pulse sequence element containing a BIRD (BIlinear Rotational Decoupling)56,59 pulse cluster and a non-selective 180° proton pulse. The BIRD isotope-selective module can distinguish among protons attached to 13C and 12C in natural isotopic abundant samples due to the large mismatch between one- and multiplebond 1H-13C coupling constants. It also means that BIRD fails to discriminate among diastereotopic methylene protons because they are attached to the same 13C atom. Consequently, the real-time BIRD acquisition scheme can remove all proton-proton couplings except for the geminal ones. In the HSQC experiment, BIRD works without sensitivity loss, since the basic HSQC pulse sequence preselects only those protons that are directly attached to 13C nuclei.
Figure 1.

The NMR pulse sequence scheme of the real-time pure shift 1H-13C HSQC-SI optimized for aqueous metabolomics samples. Narrow and wide, filled, black bars correspond to 90° and 180° hard pulses, respectively, with phase x unless indicated otherwise. A trim pulse during the initial INEPT step is shown as striped wide bar. Smoothed CHIRP inversion and refocusing 13C pulses are shown as blue trapezoids. Broadband inversion 13C pulses (BIP) are illustrated as green half-ellipses. Phases other than x are ϕ1 = y; ϕ2 = x, -x; ϕ3 = x, x, -x, -x; ϕ4 = y, y, -y, -y; ϕ5 = -x; and ϕrec = x, -x, -x, x. Delays are set as follows: τ = 1/(4 1JCH), τ’ = 1/(4 1JCH) for CH or 1/(8 1JCH) for all multiplicities, τa = p16 + d16 + 8 μs. Coherence order selection and echo-antiecho phase sensitive detection in the 13C-dimension are achieved with gradient pulses G1 and G4 in the ratio of 80 to 20.1. Purging gradient pulses G2 and G3 are set to 11% and −5% of the maximum gradient strength (53 G/cm), typically with 600 μs (p19) and 1 ms (p16) duration followed by a recovery delay of 200 μs (d16). Coherence transfer pathway selection gradient pulses (G5 = 31% and G6 = 49%) are used around the BIRD clusters and the 180° proton hard pulse in the J-refocusing blocks with 500 μs duration followed by a 200 μs recovery delay. Composite pulse decoupling (CPD) according to the p5m4sp180.2 supercycle is applied during data acquisition.
As for other NMR pulse sequences, in real-time pure shift experiments satisfactory water suppression is a challenge. Since in the original real-time BIRD HSQC study57 deuterated solvent was used, water suppression was not an issue. Later, gradient pulse pairs were proposed flanking the 180° rotations to reduce residual water signal in real-time pure shift 15N HSQC experiments of proteins.60 However, the gradient pulses extend the length of the J-refocusing blocks, and undesired heteronuclear coupling evolution during this extra time causes additional line-broadening effects and spectral artifacts. To minimize such unwanted behavior, the BIRD sequence was modified by inserting an extra 13C broadband inversion pulse (BIP).61 The modified pulse sequence permits the application of even longer gradient pulses and recovery delays during the J-refocusing blocks for appropriate water suppression without decreasing the signal-to-noise or the spectral quality. Accordingly, in the present mouse urine study gradient pulses around the BIRD cluster and the 180° proton hard pulse in the J-refocusing blocks were applied with extended duration (500 μs) and larger strength (31% and 49%) than typically applied in organic solvent experiments.
The proper choice of two experimental parameters, namely the number (n) and the duration (aq/n) of the FID chunks, are also crucial to attain optimal signal-to-noise and spectral quality. The two parameters are dependent on each other as their product defines the total acquisition time, aq. If the chunks are chosen to be too long, undesired proton J-coupling evolution causes imperfect decoupling and the appearance of sidebands at multiples of n/aq in the direct dimension. On the other hand, if the FID chunks are too short, the frequent application of J-refocusing blocks causes additional signal loss and line broadening due to T2 relaxation and the cumulative effect of pulse imperfections. Moreover, if the BIRD delay does not match all one-bond heteronuclear coupling constants, which in metabolomics applications is the case, for example for aromatic vs. aliphatic resonances, the NMR spectral quality will further deteriorate. Based on these considerations and tests with different types of metabolomics samples, the use of 8 chunks with a duration of 25 ms each, which yields a total acquisition time of 200 ms, was found to represent a suitable choice for the collection of real-time pure shift 1H-13C HSQC-SIs of metabolomics samples.
The proper selection of a suitable heteronuclear decoupling sequence during acquisition is another aspect that needed further consideration. On the one hand, the bilevel adiabatic decoupling sequence, which is routinely used for standard experiments on modern high-field NMR spectrometers, could not be applied (at least on our current Bruker Avance III HD systems) in combination with real-time windowed acquisition used for a pure shift HSQC. On the other hand, GARP sequences do not work perfectly for the entire 13C spectral window on high-field instruments under safe power handling conditions, but the normal adiabatic sequence (p5m4sp180.2) worked effectively for the full 13C frequency range.
After fine-tuning of the pulse sequence and experimental parameters described above for typical metabolomics samples, we used the real-time pure shift 1H-13C HSQCSI method for an untargeted metabolomics study of mouse urine. Urine in general is one of the chemically most complex biofluids, because kidneys secrete all the soluble waste material from the bloodstream including metabolites from a large variety of endogeneous biochemical pathways, foods, drinks, drugs, environmental chemicals, and bacterial byproducts. For example, the recent human urine metabolome database contains 2651 confirmed metabolite species, which was curated by combining the results of different analytical platforms used by multiple studies.62 Owing to the thousands of cross-peaks of NMR spectra of urine, even standard 1H-13C HSQC spectra are often severely crowded, hindering the unambiguous identification and assignment of all resonances and the precise quantitation of the underlying metabolites. This makes urine samples an excellent test bed for pure shift experiments to assess their effectiveness for resolution enhancement and sensitivity.
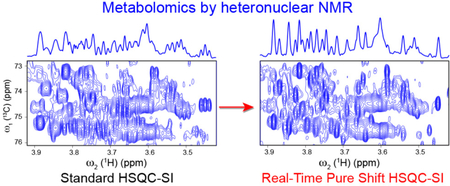
Some of the most crowded regions of a standard 1H-13C HSQC-SI spectrum acquired on a mouse urine sample is depicted in Figure 2B,D,F. It clearly shows that even at high magnetic field (20 T or 850 MHz 1H frequency) the 2D heteronuclear correlation map is severely overlapped, hindering both metabolite identification and quantitation. By contrast, the optimized real-time pure shift 1H-13C HSQC-SI experiment of the same spectral regions (Figure 2A, C and E) has cross-peaks with their proton-proton splittings removed and, as a result, multiplet signals collapsed to singlets thereby significantly increasing spectral resolution. The only exceptions are the diastereotopic methylene protons because they are attached to the same 13C atom and BIRD does not remove the geminal couplings between them. If the analysis of these methylene protons is crucial, we recommend using the PerfectBIRD HSQC experiment,41 which tackles this limitation of BIRD, however at the expense of a prolonged measurement time as it requires the measurement of a pseudo-3D experiment. Figure 2 also demonstrates that in the pure shift HSQC-SI spectrum (Panels A,C,E) numerous resonances are well-separated from each other compared to the standard HSQC-SI spectrum (Panels B,D,F), significantly facilitating the unambiguous identification of individual cross-peaks along with the accurate determination of their chemical shifts, intensities and volumes. The full HSQC spectra are depicted in Figure S1 and S2 in the Supporting Information.
Figure 2.

Comparison of crowded aliphatic regions of 2D 1H-13C real-time pure shift HSQC-SI (A, C, E) and standard HSQC-SI (B, D, F) of mouse urine at 850 MHz proton frequency. Panels G and H show the 1D 1H traces extracted from pure shift HSQC-SI (panel C) and standard HSQC-SI (panel D). The 13C chemical shift position (75.3 ppm) of the 1D 1H traces are indicated by blue arrows in Panels C and D. For an objective comparison, 2D contour plots and 1D cross-sections of the pure shift and standard HSQC-SI pairs were processed and displayed with the same parameters.
Because 14.1 Tesla (600 MHz 1H frequency) is a commonly used magnetic field strength for NMR-based metabolomics studies, we investigated the performance of the real-time pure shift HSQC-SI pulse sequence (Figure 1) on a 600 MHz instrument for comparison. 2D contour plots from the aromatic region of the mouse urine sample are displayed in Figure 3. Using the pure shift HSQC-SI method (Figure 3A), singlet signals appear for each distinct resonance, which are in contrast to the much broader multiplets observed in the standard HSQC-SI spectrum (Figure 3B). Our experiments demonstrate that the real-time pure shift HSQC-SI method performs exceedingly well also at 600 MHz. Moreover, the experiment is easily transferable between instruments requiring minimal pulse sequence parameter optimization.
Figure 3.

Aromatic region comparison of 2D 1H-13C real-time pure shift HSQC-SI (A) and standard HSQC-SI (B) of mouse urine at 600 MHz proton frequency. Panels C and D show the 1D 1H traces extracted from the pure shift HSQC-SI (panel A) and standard HSQC-SI (panel B). The 13C chemical shift position (131.7 ppm) of the 1D 1H traces are indicated by blue arrows in Panels C and D. For an objective comparison, 2D contour plots and 1D cross-sections of the pure shift and standard HSQC-SI pairs were processed and displayed with the same parameters.
As mentioned above, since the basic HSQC pulse sequence preselects those protons that are directly attached to a 13C nucleus, for samples at 13C natural abundance BIRD can be applied without sensitivity loss. This is in contrast to other fully broadband homonuclear decoupled experiments where a loss in sensitivity is unavoidable and can be quite significant. Moreover, signal-to-noise (S/N) improvement can be achieved by realtime BIRD in HSQC as a result of collapsing multiplets into singlets. Theoretically, the sensitivity enhancement could be two-fold or greater depending on the multiplet structure of a given resonance. However, in practice some coherence loss occurs because of T2 relaxation and pulse imperfections during the J-refocusing blocks. Extracted rows from pure shift and standard HSQC-SI are shown in Figures 2G, 3C and Figures 2H, 3D, respectively. Spectra were recorded with identical parameters and with the same total measurement time and are displayed with the same scaling factor for an objective comparison. For all cross-sections (Figures 2G, H and 3C, D) the S/N is on average 4050% larger for the homonuclear decoupled spectrum than for the conventional spectrum, though the actual signal-to-noise improvement varies from resonance to resonance because of differences in multiplet patterns, J-coupling strengths, and natural linewidths. To get additional insight into the maximal achievable sensitivity gain in real-time pure shift HSQC-SI experiments, we also compared the intensities of some signals that are naturally singlets in both HSQC spectra. The average signal intensity for such singlet resonances is in the real-time pure shift spectrum about 80% of the standard spectrum.
In summary, application of real-time pure shift HSQC-SI method to mouse urine samples in H2O offers a significant boost in both resolution and sensitivity compared to standard HSQC-SI. Therefore, the 2D real-time pure shift 1H-13C HSQC-SI experiment has the potential to become a standard for 2D HSQC experiments for metabolomics studies when resolution or sensitivity is a limiting factor.
Identification of metabolites with COLMARm webserver
Next, the suitability of 2D real-time pure shift 1H-13C HSQC-SI spectra was tested for the identification of metabolites by database query. For this purpose 2D HSQC-SI spectra of mouse urine were subjected to chemical shift database query using the COLMARm webserver.58 During this process, the first noticeable advantage of the pure shift HSQC-SI compared to the standard HSQC-SI is the better performance in automatic peak-picking, especially for crowded regions such as the carbohydrate region. In the standard HSQC-SI spectrum, all resolved multiplet components are picked, whereas only one peak per resonance (except for diastereotopic CH2 resonances) is picked in the pure shift spectrum due to proton-proton decoupling. It also means that fewer false negative peaks are selected in the pure shift HSQC-SI resulting in improved efficiency of automatic peak-picking. Identification of the compound fucose in Figure 4 clearly illustrates the improved peak-picking and more accurate metabolite identification by pure shift HSQC-SI. Figure 4B shows that all components of proton-proton coupled crosspeak multiplets are picked in the standard HSQC-SI, which could cause ambiguities for chemical shift matching against NMR metabolomics databases. In contrast, Figure 4A illustrates that due to spectral simplification (most multiplet signals collapsed to singlets) and improved spectral resolution, automatic peak-picking and chemical shift matching are more accurate and less ambiguous for the pure shift spectrum.
Figure 4.

Comparison of metabolite identification results of mouse urine sample based on 2D 1H-13C real-time pure shift HSQC-SI (A, C) and standard HSQC-SI (B, D) using the COLMARm webserver (http://spin.ccic.ohio-state.edu/index.php/colmarm/index). Panels A and B show the concatenated HSQC-SI regions of fucose and Panels C and D show the corresponding spectral regions of compound N-acetyl-galactosamine. Orange dots denote the cross-peaks that were picked by the automatic peak-picker embedded in the COLMARm webserver. Red dots denote the peaks that match the database peaks of the identified compounds, i.e., fucose and N-acetyl-galactosamine. Red circles in panel D denote missing peaks of N-acetyl-galactosamine in the standard HSQC-SI due to low sensitivity.
As a consequence, the combination of enhanced sensitivity and resolution in realtime pure shift HSQC-SI spectra substantially improves automatic metabolite identification. When querying HSQC spectra measured on multiple mouse urine samples against the COLMAR chemical shift database, more metabolites could be identified in the pure shift spectra and metabolites were identified with higher confidence compared to standard spectra due to a matching ratio58 between the number of observed and expected cross-peaks for a given compound close to 1. Figures 4C and 4D illustrate the sensitivity improvement in real-time pure shift HSQC-SI. All seven peaks of N-acetylgalactosamine are picked and identified in the pure shift HSQC-SI, while four peaks are missing in the standard HSQC-SI. In the real-time pure shift HSQC-SI, we could unambiguously (with a perfect matching ratio of 1) identify seven metabolites, namely 4-hydroxy-benzoic acid, alanine, N-acetyl-galactosamine, phosphoethanolamine, salicyluric acid, serine, sulfanilic acid, which could not be observed or identified in the standard HSQC-SI spectrum of the same sample under identical experimental conditions and measurement time.
CONCLUSIONS
The correct and complete identification of metabolites is a pivotal step in most metabolomics applications. Multidimensional NMR is a powerful tool for this purpose.15 However, sensitivity and resolution limitations can be challenging in the case of complex samples, such as urine, containing several hundred detectable metabolites. Over the last few years, important developments have emerged for improving sensitivity, resolution and spectral simplicity in NMR spectra. Among these, pure shift methods are particularly promising to further boost the power of NMR-based metabolomics.
In the present work, we optimized the pure shift (real-time BIRD) 1H-13C HSQCSI experiment for typical conditions in metabolomics, such as aqueous solutions and high-field instruments. The improved real-time pure shift HSQC-SI method achieves simultaneous enhancement of resolution and sensitivity in HSQC spectra without increasing NMR measurement time for mouse urine. It was also shown that pure shift HSQC-SI significantly facilitates automatic peak-picking and chemical shift matching against NMR metabolomics databases. Thus, it allows the more effective and more accurate identification of metabolites. We hope that this work paves the way for the routine use of real-time pure shift HSQC-SI spectra for everyday metabolomics.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by a postdoctoral fellowship from former President of Hungary Dr. László Sólyom (to I. T.), a graduate fellowship from Foods for Health, a focus area of the Discovery Themes Initiative at OSU (to C.W.), and the National Institutes of Health (grants R01 GM 066041 (to R.B.), and R01AG055059 (to S.O.Y.). All NMR experiments were performed at the Campus Chemical Instrument Center NMR facility at the Ohio State University.
Footnotes
Additional information showing standard and real-time pure shift 1H-13C HSQC-SI NMR spectra of mouse urine, and the real-time pure shift HSQC-SI pulse sequence code for Bruker spectrometers. This information is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
REFERENCES
- (1).Holmes E; Loo RL; Stamler J; Bictash M; Yap IKS; Chan Q; Ebbels T; De Iorio M; Brown IJ; Veselkov KA; Daviglus ML; Kesteloot H; Ueshima H; Zhao L; Nicholson JK; Elliott P Human metabolic phenotype diversity and its association with diet and blood pressure Nature 2008, 453, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhang B; Halouska S; Schiaffo CE; Sadykov MR; Somerville GA; Powers R NMR Analysis of a Stress Response Metabolic Signaling Network J. Proteome Res 2011, 10, 3743–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nicholson JK; Holmes E; Kinross JM; Darzi AW; Takats Z; Lindon JC Metabolic phenotyping in clinical and surgical environments Nature 2012, 491, 384. [DOI] [PubMed] [Google Scholar]
- (4).Patti GJ; Yanes O; Siuzdak G Metabolomics: the apogee of the omics trilogy Nat. Rev. Mol. Cell Biol 2012, 13, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Emwas AH; Luchinat C; Turano P; Tenori L; Roy R; Salek RM; Ryan D; Merzaban JS; Kaddurah-Daouk R; Zeri AC; Nagana Gowda GA; Raftery D; Wang Y; Brennan L; Wishart DS Standardizing the experimental conditions for using urine in NMR-based metabolomic studies with a particular focus on diagnostic studies: a review Metabolomics 2015, 11, 872–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gelman SJ; Naser F; Mahieu NG; McKenzie LD; Dunn GP; Chheda MG; Patti GJ Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress Cell Rep 2018, 22, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lenz EM; Wilson ID Analytical Strategies in Metabonomics J. Proteome Res 2007, 6, 443–458. [DOI] [PubMed] [Google Scholar]
- (8).Nikolskiy I; Mahieu NG; Chen Y Jr.; Tautenhahn R; Patti GJ An Untargeted Metabolomic Workflow to Improve Structural Characterization of Metabolites Anal. Chem 2013, 85, 7713–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bingol K; Bruschweiler-Li L; Yu C; Somogyi A; Zhang F; Brüschweiler R Metabolomics Beyond Spectroscopic Databases: A Combined MS/NMR Strategy for the Rapid Identification of New Metabolites in Complex Mixtures Anal. Chem 2015, 87, 3864–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Spalding JL; Cho K; Mahieu NG; Nikolskiy I; Llufrio EM; Johnson SL; Patti GJ Bar Coding MS2 Spectra for Metabolite Identification Anal. Chem 2016, 88, 2538–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nagana Gowda GA; Gowda YN; Raftery D Expanding the Limits of Human Blood Metabolite Quantitation Using NMR Spectroscopy Anal. Chem 2015, 87, 706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fan TWM; Lane AN Applications of NMR spectroscopy to systems biochemistry Prog. Nucl. Magn. Reson. Spectrosc 2016, 92-93, 18–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nagana Gowda GA; Raftery D Recent Advances in NMR-Based Metabolomics Anal. Chem 2017, 89, 490–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Markley JL; Brüschweiler R; Edison AS; Eghbalnia HR; Powers R; Raftery D; Wishart DS The future of NMR-based metabolomics Curr. Opin. Biotechnol 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bingol K; Brüschweiler R Multidimensional Approaches to NMR-Based Metabolomics Anal. Chem 2014, 86, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bingol K; Brüschweiler R Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods Curr. Opin. Biotechnol 2017, 43, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Marchand J; Martineau E; Guitton Y; Dervilly-Pinel G; Giraudeau P Multidimensional NMR approaches towards highly resolved, sensitive and highthroughput quantitative metabolomics Curr. Opin. Biotechnol 2017, 43, 49–55. [DOI] [PubMed] [Google Scholar]
- (18).Bhinderwala F; Lonergan S; Woods J; Zhou C; Fey PD; Powers R Expanding the Coverage of the Metabolome with Nitrogen-Based NMR Anal. Chem 2018, 90, 4521–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wishart DS Quantitative metabolomics using NMR TrAC, Trends Anal. Chem 2008, 27, 228–237. [Google Scholar]
- (20).Nagana Gowda GA; Raftery D Whole Blood Metabolomics by 1H NMR Spectroscopy Provides a New Opportunity To Evaluate Coenzymes and Antioxidants Anal. Chem 2017, 89, 4620–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Takis PG; Schäfer H; Spraul M; Luchinat C Deconvoluting interrelationships between concentrations and chemical shifts in urine provides a powerful analysis tool Nat. Commun 2017, 8, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vignoli A; Ghini V; Meoni G; Licari C; Takis PG; Tenori L; Turano P; Luchinat C High-Throughput Metabolomics by 1D NMR Angew. Chem. Int. Ed 2018, 57, 2–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bodenhausen G; Ruben DJ Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy Chem. Phys. Lett 1980, 69, 185–189. [Google Scholar]
- (24).Kay L; Keifer P; Saarinen T Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity J. Am. Chem. Soc 1992, 114, 10663–10665. [Google Scholar]
- (25).Kazimierczuk K; Stanek J; Zawadzka-Kazimierczuk A; Koźmiński W Random sampling in multidimensional NMR spectroscopy Prog. Nucl. Magn. Reson. Spectrosc 2010, 57, 420–434. [DOI] [PubMed] [Google Scholar]
- (26).Kazimierczuk K; Orekhov VY Accelerated NMR Spectroscopy by Using Compressed Sensing Angew. Chem. Int. Ed 2011, 50, 5556–5559. [DOI] [PubMed] [Google Scholar]
- (27).Hansen AL; Li D; Wang C; Brüschweiler R Absolute Minimal Sampling of Homonuclear 2D NMR TOCSY Spectra for High‐Throughput Applications of Complex Mixtures Angew. Chem. Int. Ed 2017, 56, 8149–8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schlippenbach T.v., Oefner PJ; Gronwald W Systematic Evaluation of Non-Uniform Sampling Parameters in the Targeted Analysis of Urine Metabolites by 1H,1H 2D NMR Spectroscopy Sci. Rep 2018, 8, 4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li D; Hansen AL; Bruschweiler-Li L; Brüschweiler R Non-Uniform and Absolute Minimal Sampling for High-Throughput Multidimensional NMR Applications Chem. Eur. J 2018, 24, 11535–11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Adams RW In eMagRes; John Wiley & Sons, Ltd, 2014, pp 295–310. [Google Scholar]
- (31).Castañar L; Parella T Broadband 1H homodecoupled NMR experiments: recent developments, methods and applications Magn. Reson. Chem 2015, 53, 399–426. [DOI] [PubMed] [Google Scholar]
- (32).Zangger K Pure shift NMR Prog. Nucl. Magn. Reson. Spectrosc 2015, 86–87, 1–20. [DOI] [PubMed] [Google Scholar]
- (33).Castañar L Pure shift 1H NMR: what is next? Magn. Reson. Chem 2017, 55, 47–53. [DOI] [PubMed] [Google Scholar]
- (34).Nilsson M; Morris GA Pure shift proton DOSY: diffusion-ordered 1H spectra without multiplet structure Chem. Commun 2007, 43, 933–935. [DOI] [PubMed] [Google Scholar]
- (35).Sakhaii P; Haase B; Bermel W Experimental access to HSQC spectra decoupled in all frequency dimensions J. Magn. Reson 2009, 199, 192–198. [DOI] [PubMed] [Google Scholar]
- (36).Aguilar JA; Faulkner S; Nilsson M; Morris GA Pure shift 1H NMR: a resolution of the resolution problem? Angew. Chem. Int. Ed 2010, 49, 3901–3903. [DOI] [PubMed] [Google Scholar]
- (37).Lupulescu A; Olsen GL; Frydman L Toward single-shot pure-shift solution 1H NMR by trains of BIRD-based homonuclear decoupling J. Magn. Reson 2012, 218, 141–146. [DOI] [PubMed] [Google Scholar]
- (38).Castañar L; Nolis P; Virgili A; Parella T Full Sensitivity and Enhanced Resolution in Homodecoupled Band-Selective NMR Experiments Chem. Eur. J 2013, 19, 17283–17286. [DOI] [PubMed] [Google Scholar]
- (39).Meyer NH; Zangger K Simplifying proton NMR spectra by instant homonuclear broadband decoupling Angew. Chem. Int. Ed 2013, 52, 7143–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Foroozandeh M; Adams RW; Nilsson M; Morris GA Ultrahigh-Resolution Total Correlation NMR Spectroscopy J. Am. Chem. Soc 2014, 136, 11867–11869. [DOI] [PubMed] [Google Scholar]
- (41).Kaltschnee L; Kolmer A; Timári I; Schmidts V; Adams RW; Nilsson M; Kövér KE; Morris GA; Thiele CM “Perfecting” pure shift HSQC: full homodecoupling for accurate and precise determination of heteronuclear couplings Chem. Commun 2014, 50, 15702–15705. [DOI] [PubMed] [Google Scholar]
- (42).Timári I; Kaltschnee L; Kolmer A; Adams RW; Nilsson M; Thiele CM; Morris GA; Kövér KE Accurate determination of one-bond heteronuclear coupling constants with “pure shift” broadband proton-decoupled CLIP/CLAP-HSQC experiments J. Magn. Reson 2014, 239, 130–138. [DOI] [PubMed] [Google Scholar]
- (43).Reinsperger T; Luy B Homonuclear BIRD-decoupled spectra for measuring onebond couplings with highest resolution: CLIP/CLAP-RESET and constant-timeCLIP/CLAP-RESET J. Magn. Reson 2014, 239, 110–120. [DOI] [PubMed] [Google Scholar]
- (44).Aguilar JA; Cassani J; Delbianco M; Adams RW; Nilsson M; Morris GA Minimising Research Bottlenecks by Decluttering NMR Spectra Chem. Eur. J 2015, 21, 6623–6630. [DOI] [PubMed] [Google Scholar]
- (45).Castañar L; Roldán R; Clapés P; Virgili A; Parella T Disentangling Complex Mixtures of Compounds with Near-Identical 1H and 13C NMR Spectra using Pure Shift NMR Spectroscopy Chem. Eur. J 2015, 21, 7682–7685. [DOI] [PubMed] [Google Scholar]
- (46).Rachineni K; Kakita VMR; Dayaka S; Vemulapalli SPB; Bharatam J Precise Determination of Enantiomeric Excess by a Sensitivity Enhanced Two-Dimensional Band-Selective Pure-Shift NMR Anal. Chem 2015, 87, 7258–7266. [DOI] [PubMed] [Google Scholar]
- (47).Saurí J; Bermel W; Buevich AV; Sherer EC; Joyce LA; Sharaf MHM; Schiff PL; Parella T; Williamson RT; Martin GE Homodecoupled 1,1- and 1,n-ADEQUATE: Pivotal NMR Experiments for the Structure Revision of Cryptospirolepine Angew. Chem. Int. Ed 2015, 54, 10160–10164. [DOI] [PubMed] [Google Scholar]
- (48).Timári I; Kövér KE Broadband homonuclear decoupled HSQMBC methods Magn. Reson. Chem 2018, 56, 910–917. [DOI] [PubMed] [Google Scholar]
- (49).Mishra SK; Suryaprakash N Orchestrated approaches using pure shift NMR: Extraction of spectral parameters, ultra-high resolution, and sensitivity enhancement Magn. Reson. Chem 2018, 56, 893–909. [DOI] [PubMed] [Google Scholar]
- (50).Mauve C; Khlifi S; Gilard F; Mouille G; Farjon J Sensitive, highly resolved, and quantitative 1H–13C NMR data in one go for tracking metabolites in vegetal extracts Chem. Commun 2016, 52, 6142–6145. [DOI] [PubMed] [Google Scholar]
- (51).Farjon J; Milande C; Martineau E; Akoka S; Giraudeau P The FAQUIRE Approach: FAst, QUantitative, hIghly Resolved and sEnsitivity Enhanced 1H, 13C Data Anal. Chem 2018, 90, 1845–1851. [DOI] [PubMed] [Google Scholar]
- (52).Brüschweiler R; Griesinger C; Sørensen OW; Ernst RR Combined use of hard and soft pulses for ω1 decoupling in two-dimensional NMR spectroscopy J. Magn. Reson 1988, 78, 178–185. [Google Scholar]
- (53).Zangger K; Sterk H Homonuclear broadband-decoupled NMR spectra J. Magn. Reson 1997, 124, 486–489. [Google Scholar]
- (54).Pell AJ; Edden RAE; Keeler J Broadband proton-decoupled proton spectra Magn. Reson. Chem 2007, 45, 296–316. [DOI] [PubMed] [Google Scholar]
- (55).Foroozandeh M; Adams RW; Meharry NJ; Jeannerat D; Nilsson M; Morris GA Ultrahigh-resolution NMR spectroscopy Angew. Chem. Int. Ed 2014, 53, 6990–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Garbow JR; Weitekamp DP; Pines A Bilinear rotation decoupling of homonuclear scalar interactions Chem. Phys. Lett 1982, 93, 504–509. [Google Scholar]
- (57).Paudel L; Adams RW; Király P; Aguilar JA; Foroozandeh M; Cliff MJ; Nilsson M; Sándor P; Waltho JP; Morris GA Simultaneously enhancing spectral resolution and sensitivity in heteronuclear correlation NMR spectroscopy Angew. Chem. Int. Ed 2013, 52, 11616–11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bingol K; Li D-W; Zhang B; Brüschweiler R Comprehensive Metabolite Identification Strategy Using Multiple Two-Dimensional NMR Spectra of a Complex Mixture Implemented in the COLMARm Web Server Anal. Chem 2016, 88, 12411–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Uhrin D; Liptaj T; Kövér KE Modified BIRD Pulses and Design of Heteronuclear Pulse Sequences J. Magn. Reson., Ser A 1993, 101, 41–46. [Google Scholar]
- (60).Kiraly P; Adams RW; Paudel L; Foroozandeh M; Aguilar JA; Timári I; Cliff MJ; Nilsson M; Sándor P; Batta G; Waltho JP; Kövér KE; Morris GA Real-time pure shift 15N HSQC of proteins: a real improvement in resolution and sensitivity J. Biomol. NMR 2015, 62, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kiraly P; Nilsson M; Morris GA Practical aspects of real-time pure shift HSQC experiments Magn. Reson. Chem 2018, 56, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bouatra S; Aziat F; Mandal R; Guo AC; Wilson MR; Knox C; Bjorndahl TC; Krishnamurthy R; Saleem F; Liu P; Dame ZT; Poelzer J; Huynh J; Yallou FS; Psychogios N; Dong E; Bogumil R; Roehring C; Wishart DS The Human Urine Metabolome PLOS ONE 2013, 8, e73076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.