

SUMMARY
CRISPR-Cas immunity requires integration of short, foreign DNA fragments into the host genome at the CRISPR locus, a site consisting of alternating repeat sequences and foreignderived spacers. In most CRISPR systems, the proteins Cas1 and Cas2 form the integration complex and are both essential for DNA acquisition. Most type V-C and V-D systems lack the cas2 gene and have unusually short CRISPR repeats and spacers. Here, we show that a miniintegrase comprising the type V-C Cas1 protein alone catalyzes DNA integration with a preference for short (17–19 base pair) DNA fragments. The mini-integrase has weak specificity for the CRISPR array. We present evidence that the Cas1 proteins form a tetramer for integration. Our findings support a model of a minimal integrase with an internal ruler mechanism that favors shorter repeats and spacers. This minimal integrase may represent the function of the ancestral Cas1, prior to Cas2 adoption.
Graphical Abstract
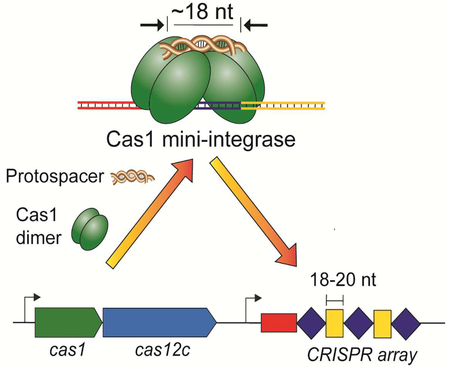
Wright et al. demonstrate the function of an integrase comprised of a single protein, Cas1, that inserts unusually short fragments of viral DNA into the bacterial genome as part of the CRISPRCas adaptive immunity pathway. This minimal integrase may represent the function of the ancestral Cas1, prior to Cas2 adoption.
INTRODUCTION
CRISPR-Cas (clustered regularly interspaced short palindromic repeats, CRISPR associated) systems function as a form of adaptive immunity for bacteria and archaea (Barrangou et al., 2007). In these systems, a dedicated integrase incorporates segments of foreign DNA, termed protospacers, into the host CRISPR locus in a process known as acquisition (Yosef et al., 2012, Wright et al., 2016b; Sternberg et al., 2016). The CRISPR locus, which consists of direct repeats separated by viral-derived spacers of similar length, is transcribed and processed into mature CRISPR RNAs (crRNAs) that are used by an effector complex to identify and destroy any phage or plasmid bearing sequence complementarity to the virally derived segment of the crRNA (Pourcel et al., 2005; Mojica et al., 2005; Bolotin et al., 2005; Brouns et al., 2008; Garneau et al., 2010; Hale et al., 2009).
While the Cas proteins and mechanisms involved in CRISPR immunity vary widely, the acquisition module is largely conserved (Koonin et al., 2017). In all studies conducted so far, Cas1 and Cas2 have been found to be necessary for acquisition (Sternberg et al., 2016). These two proteins form a heterohexameric complex that catalyzes the integration of new protospacers at one end of the CRISPR array (Barrangou et al., 2007; Nuñez et al., 2014; Nuñez et al., 2015b). Cas1 is the catalytic subunit, while Cas2 plays an important structural role by bridging the two Cas1 dimers and stabilizing both the bound protospacer DNA and the CRISPR repeat during integration (Arslan et al, 2014; Nuñez et al., 2014; Yosef et al., 2012; Datsenko et al., 2012; Wang et al., 2015; Wright et al., 2017; Xiao et al., 2017).
This architecture provides an internal ruler mechanism that dictates the 25–50 base pair length of the protospacer for integration (Nuñez et al., 2015a; Wang et al., 2015). Integration occurs when the 3’ ends of the protospacer make a nucleophilic attack at each end of the CRISPR repeat sequence, resulting in a half-site intermediate after the first attack that proceeds to a full-site product (Arslan et al., 2014; Nuñez et al., 2015b). Specific recognition of the CRISPR repeat is important to avoid damage to the genome resulting from off-target integration. Integration is specific to the CRISPR locus with the preferred site being the first repeat adjacent to the leader, an upstream sequence that also contains the promoter for the CRISPR array (Yosef et al., 2012; Nuñez et al., 2014; Nuñez et al., 2015b; Wright et al., 2016b; Wei et al., 2015). Cas1-Cas2 integrases have intrinsic target specificity, although host factors have been shown to improve integration specificity in some hosts (Nuñez et al., 2016; Yoganand et al., 2017; Nivala et al., 2018; Fagerlund et al., 2017; Rollie et al., 2018).
Since this Cas1-Cas2 module is conserved across most systems, the recent discovery of a potential non-canonical acquisition module provides an opportunity to investigate the activity of a more minimal integrase (Shmakov et al., 2015; Burstein et al., 2017). Two type V CRISPR-Cas subtypes, V-C and V-D, have been shown to lack cas2 genes entirely and have notably shorter CRISPR spacers and repeats. These observations are especially exciting given recent studies suggesting that Cas1 originated from a superfamily of DNA transposons called casposons (Koonin et al., 2017; Krupovic and Koonin, 2016; Krupovic et al., 2014; Hickman and Dyda, 2015). Casposons encode a Cas1 homolog, called a casposase, which has transposase activity and some sequence preference for its integration sites; notably, like the type V systems, casposons do not include Cas2 (Beguin et al., 2016; Krupovic et al., 2014). Determining whether and how the type V systems carry out acquisition without Cas2 and where these systems fit in the evolutionary history of CRISPR-Cas can shed light on the origins and diversity of CRISPR-based anti-viral immunity.
We hypothesized that the Cas1 protein of CRISPR type V-C and type V-D systems functions on its own as a DNA integrase whose architecture might confer a preference for the characteristic short spacer sequences in the CRISPR arrays of these systems. Here, we reconstitute the type V-C integration reaction and show that type V-C Cas1 is sufficient to catalyze full-site integration of 18-base pair DNA substrates. Furthermore, our experiments reveal that V-C Cas1 has an intrinsic mechanism to orient protospacers in a biased manner during integration. We observed considerably more off-target integration with this mini-integrase relative to other CRISPR integrases, suggesting the need for additional host factors to provide target site specificity. We present evidence for Cas1 multimerization triggered by association with half-site and full-site integration product mimics. Our findings support a model for a minimal CRISPR integrase in which Cas1 provides both DNA substrate length selectivity and catalytic activity necessary for foreign DNA acquisition and which may represent the ancestral CRISPR adaptation module.
RESULTS
Type V-C and V-D systems have CRISPR arrays with short repeats and spacers and lack cas2 genes
To date, type V-C and type V-D CRISPR-Cas systems have been found exclusively in uncultured microbes. Type V-C systems have been identified in marine and gut metagenomes, encoded on contigs which were too short to determine their host, and type V-D were identified in the genomes of small, putatively symbiotic bacteria from groundwater samples (Shmakov et al., 2015; Burstein et al., 2017). In both systems, adaptation modules lack cas2 and are characterized by short spacers (17–20 bp) (Fig. 1A, B). To better characterize type V-C and V-D systems, we searched for previously unidentified systems within assembled metagenomes available in the IMG/M database, which incorporates a wide variety of environments (Chen et al., 2016). We report here the identification of a new V-C system from a mouse cecum sample and a V-D system from beetle gut (Table S1). In these systems, cas2 genes were also absent and the spacers were considerably shorter compared to spacers from other CRISPR-Cas systems (Fig. 1C).
Figure 1. Type V-C and V-D CRISPR Systems Have Short Repeats and Spacers and Lack cas2 Genes.

(A) CRISPR-Cas loci of the effector proteins of the type V-C system from mouse cecum and V-D system from beetle gut with lengths of genes and repeats and spacers. See Table S1 for protein sequences.
(B) Distribution of mean spacer lengths of type V-C/D systems.
(C) Comparison of mean spacer length for V-C/D vs. other CRISPR-Cas systems. The blue density plot depicts the distribution of mean spacer length in CRISPR-Cas systems, excluding V-C/D systems. The mean spacer lengths of published V-C and V-D systems are noted in red and orange dots, respectively, with arbitrary y-values to set them apart.
Type V-C Cas1 catalyzes DNA integration without Cas2
We set out to determine whether type V-C CRISPR-Cas1 can catalyze DNA substrate integration into a target DNA molecule. We expressed and purified the Cas1 protein derived from both the new type V-C and type V-D CRISPR systems and conducted integration assays with the well-characterized Escherichia coli type I-E Cas1-Cas2 integrase as a positive control. These acquisition assays were performed with a supercoiled target plasmid containing the cognate CRISPR locus for each protein and a 33-bp or 18-bp DNA substrate for the E. coli Cas1-Cas2 complex or type V Cas1, respectively (Table S2). We evaluated integration by analyzing changes in plasmid topology, using gel electrophoresis to separate the open-circle integration products from the non-integrated supercoiled plasmid (Fig. 2A) (Nuñez et al., 2015b).
Figure 2. Type V-C Cas1 Integrates DNA Fragments of Expected Length In Vitro.

(A) Schematic of in vitro integration of protospacer into a target supercoiled pCRISPR plasmid. Nucleophilic attack by the protospacer yields two different open-circle products: the half-site and full-site integration products.
(B) Integration assay comparing type I-E, V-C, and V-D systems. A 33-nt protospacer is used for the type I-E system and an 18-nt protospacer for the type V-C and V-D systems. The open-circle integration products (OC), supercoiled target plasmid (SC), and topoisomers (Topo) are indicated.
(C) Integration assay with type V-D Cas1, radiolabeled protospacer, and plasmid target. Integration product and free protospacer are indicated and schematized.
(D) Integration assay with variable length fluorescent protospacers from 15-bp to 25-bp long. Star indicates 6-carboxyfluorescein label.
(E) Quantification of (D) demonstrating the effect of protospacer length on type V-C Cas1 integration. The fraction integrated is calculated as the fraction of the fluorescent protospacer that has been integrated into the target plasmid. Experiments were carried out in triplicate; the bars represent mean values, with error bars depicting standard deviations. See also Figure S1.
See Table S2 for nucleotide sequences.
For the E. coli proteins, both Cas2 and protospacer DNA were required for integration, consistent with previous experiments (Fig. 2B) (Nuñez et al., 2015b). For the type V-C Cas1, we observed efficient production of open-circle plasmid only in the presence of the 18-bp DNA substrate. These results show that the type V-C Cas1 catalyzes plasmid nicking and/or ligation of short DNA fragments to plasmid DNA in the absence of Cas2 (Fig. 2B). Notably, type V-C Cas1 does not generate any observable plasmid topoisomers, which are visible as a band that migrates ahead of the supercoiled target plasmid for the E. coli Cas1-Cas2 integration reactions (Nuñez et al., 2015b). These products result from abortive integration (integration followed by disintegration), and their absence suggests that the type V-C Cas1 is less prone to reversible integration under the tested conditions (Nuñez et al., 2015b). Together, these results show that the type V-C Cas1 protein alone possesses catalytic activity in the presence of short DNA fragments, consistent with its potential function as a mini-integrase.
We did not observe generation of open-circle plasmid products by the type V-D Cas1 in these experiments, which could indicate a complete lack of activity or simply a lower reaction efficiency. To increase the sensitivity of the assay, we repeated the experiment using radiolabeled 18-bp DNA substrates (Nuñez et al., 2015b). When the type V-D Cas1 was incubated with the labeled DNA and a target plasmid, the radiolabel was incorporated into a product the size of an open-circle plasmid, suggesting the V-D Cas1 is indeed capable of protospacer integration, albeit at reduced levels under the studied reaction conditions (Fig. 2C).
We next wondered whether the type V-C Cas1, like other previously studied CRISPR integrases, also has an internal ruler mechanism that dictates the spacer length and whether its preferred DNA substrates in vitro would match the in vivo distribution of spacers for these systems. We explored the DNA substrate length requirements for the type V-C Cas1 plasmidaddition activity by performing integration assays with fluorescent protospacers with lengths ranging from 15–25 bp (Table S2). The results showed that V-C Cas1 is most active with substrates that are 18 bp long, consistent with the predominant spacer length for native V-C CRISPR arrays (Fig. 1B, 2D, 2E, S1A). Although the extent of integration decreased as the DNA fragment length diverged from 18 bp, a significant amount of integrated protospacer was detected for substrates 16–21 bp. We observed a generally similar distribution when identifying integration through open-circle formation (Fig. S1B, C). In both assays, integration was also observed with substrates outside the range observed for spacers in vivo. While V-C may be able to integrate a range of protospacer lengths, an alternative possibility is that V-C Cas1 processes longer oligos to preferred length and then inserts them. To examine whether integration of long protospacers is processing-dependent, we carried out integration reactions with a 23-bp protospacer with phosphorylated 3’ ends and compared the results to regular 23bp protospacer integration. The 3’ phosphate is expected to prevent integration unless one or more terminal nucleotides are removed to expose a 3’ OH (Fagerlund et al., 2017). The results show very low levels of conversion to open-circle products (less than 5% after 60 minutes) with the phosphorylated substrate compared to much greater conversion rates (over 15% after 60 minutes) with the regular 23-bp substrate (Fig. S1D, E). This suggests that most integration of long protospacers is processing-independent, though this Cas1 is capable of protospacer processing. A more likely explanation for the integration of long protospacers is promiscuity of the half-site reaction, as previous work in a type II-A has shown that protospacer length preference is more pronounced for the full-site reaction (Wright and Doudna, 2016).
In addition to varying DNA fragment length, we also tested substrates with different single-stranded 3’ overhangs from 0–4 nt long, since other CRISPR integrases prefer substrates with single-stranded 3’ overhangs (Table S2) (Nuñez et al., 2015a; Wang et al., 2015; Wright and Doudna, 2016.). We found that substrates bearing 1–2 nt 3’ overhangs on either end were integrated into the target plasmid at faster rates than those with blunt ends, with 2-nt-overhang substrates yielding a 5-fold increase in the amount of open-circle plasmid relative to blunt substrates after a 10 minute reaction (Fig. S1F, G). No integration was observed for any protospacers with overhangs longer than 2 nt (Fig. S1F, G). From these results, we determined that the preferred substrate of type V-C Cas1 is an 18-bp protospacer with 2-nt overhangs. Together, our results suggest that V-C Cas1 has an internal ruler mechanism similar to Cas1-Cas2 complexes, but it favors shorter length substrates.
Promiscuous DNA integration by V-C Cas1
The Cas1-Cas2 integrase from other CRISPR systems catalyzes DNA insertion selectively at the leader-proximal repeat in the CRISPR array (Fagerlund et al., 2017; Rollie et al., 2018; Nuñez et al., 2015b; Wright et al., 2016b). In the type V-C CRISPR system under investigation here, two CRISPR arrays flank the two cas genes in the host genome (Fig. 1A). Based on sequence comparisons, we were unable to determine whether one array is preferred and which side of each array contains the leader sequence that directs new acquisition events. To examine the preferred integration sites for type V-C Cas1, we carried out high-throughput sequencing of the plasmid integration products (Nuñez et al., 2015b). To prepare the sequencing library, integration reactions were conducted with the type V-C Cas1, an 18-bp protospacer, and either pUC19 (a control lacking a CRISPR array) or a plasmid bearing shortened versions of both natural V-C CRISPR arrays (Table S2). After fragmentation of the integration products, an extension with a barcoded protospacer-specific primer was carried out to enrich for fragments containing integration sites (Fig. 3A). Following removal of PCR duplicates, we mapped ~2.8 million reads to pUC19 and the CRISPR array-bearing plasmid (Fig. 3B, C; Fig. S2A, B).
Figure 3. Integration of Protospacers Occurs at Many Off-Target Sites.

(A) Schematic of library preparation protocol for high-throughput sequencing. Integration products are fragmented, end-repaired, A-tailed, and ligated with Y-adapters. A protospacer-specific extension is carried out before amplification with Illumina primers.
(B and C) Integration sites along pCRISPR. Results are separated based on the orientation of the protospacer that is integrated: the plots show integration by the (+) strand of the protospacer (B) and the (-) strand of the protospacer (C). See also Figures S2 and S3.
(D and E) Magnified view of integration in the CRISPR arrays by the (+) strand of the protospacer (D) and the (−) strand of the protospacer (E).
See Table S2 for nucleotide sequences.
Surprisingly, most integration events did not occur at the CRISPR locus. Instead, we observed frequent off-target integration in the lacZ gene and the plasmid origin for both pUC19 and pCRISPR (Fig. 3B, C; Fig. S2A, B). The repeat borders did support levels of accurate integration above background, but several off-target sites within the repeats were even more frequently used (Fig. 3D, E). The repeat-specific integration was largely restricted to the upstream (relative to the cas1 gene) array and, in particular, the first repeat of the array, suggesting that this might be the preferred array and that the upstream sequence may be the leader (Fig. 1A). The overall lack of specificity suggests that a host factor or possibly sequence elements outside of the available assembled sequence are required for specificity. A sequence logo generated from all pCRISPR integration sites revealed a strong preference for G in both the −1 and +1 position relative to the integration site, while the sequence at the −1 – +1 site of the first repeat is “TC,” further suggesting that V-C Cas1 relies on factors other than its intrinsic sequence preference to recognize the repeat in vivo (Fig. S2C). We tested the ability of V-C Cas1 to insert fragments into short DNA targets containing either a single repeat or the sequence surrounding the dominant off-target peaks in lacZ and in both cases observed many products, consistent with a lack of strict selection for a target site (Fig. S3).
We observed trends in the sequencing data indicating that V-C Cas1 integrates DNA substrates with biased orientation. For repeat-specific integration events, the (+) strand of the protospacer (CTCTCCGAGGCCAGCGTG) was integrated primarily in the (+) strand of the plasmid, likely corresponding to leader-side integration, while integration of the (–) protospacer strand was biased toward spacer-side integration (Fig. 3D, E). We also noted paired integration peaks at the dominant off-target site in lacZ, with a spacing and strand orientation consistent with full-site integration. Data at this site also suggested biased orientation of the inserted sequence, with most protospacer (+) strand integration events occurring in the (–) strand of the plasmid (Fig. 3B, C).
Type V-C Cas1 catalyzes full-site integration
Given that low levels of half-site integration have been observed for Cas1 alone in at least one canonical Cas1-Cas2 system, we investigated whether type V-C Cas1 could complete the full integration reaction alone (Rollie et al., 2018). Since bulk integration assays and sequencing data cannot differentiate between half-site and full-site integration events and previous full-site assays require precise integration to be interpretable, we devised a different method to test whether V-C Cas1 can perform full-site integration (Wright and Doudna, 2016). We used a selection based on chloramphenicol resistance to recover plasmids with full-site integration events in the target region (Díez-Villaseñor et al., 2013). A reporter plasmid was designed with the 5’ CRISPR repeat and putative leader sequence immediately upstream of an out-of-frame chloramphenicol resistance gene (CmR + 2) such that the open reading frame is restored by the 43 bp insertion resulting from full-site integration (18-bp spacer + duplication of a 25-bp repeat) (Fig. 4A). In vitro integration reactions were carried out with this reporter plasmid and an 18-bp protospacer, and the products were electroporated into E. coli (Table S2). Transformants were plated on media containing chloramphenicol, and surviving colonies were sequenced to confirm full-site integration and determine the location of integration sites.
Figure 4. Detection of Full-Site Integration by Type V-C Cas1.

(A) Schematic representation of the chloramphenicol resistance turn-on screen to detect full-site integration near the leader-repeat junction. The construct contains a CRISPR repeat and leader upstream of an out-of-frame chloramphenicol resistance gene (CmR + 2). Translation of the transcript generated by Pcat begins upstream of the repeat (arrow) and ends in the leader (stop sign). Full-site integration of an 18-nt protospacer restores the open reading frame for the CmR coding sequence. Transforming and plating on chloramphenicol plates allows for positive selection of clones that have the inserted spacer. Sanger sequencing is used to confirm full-site integration.
(B) Visual representation of identified full-site integration events near the leader-repeat junction. The colored arrowheads designate the position of the integration sites, with the number written above the arrowhead representing the number of base pairs from the leader-repeat junction. The corresponding colored lines designate the sequence that is duplicated upon spacer insertion. The arrowhead height is scaled to the total integration events at that site.
(C) Number of integration events with specified spacer orientation at each integration site. The number of non-integration events (as a result of deletions) is also indicated. The mean and standard deviation of three independent replicates are shown. See also Figure S4.
See Table S2 for nucleotide sequences.
The results from the selection show that type V-C Cas1 carries out full-site integration in vitro. The sequencing alignments show insertion of the expected 18-bp spacer and duplication of the 25 bp directly adjacent to the spacer, which is consistent with full-site integration in other CRISPR systems. The locations of the integration events are scattered near the leader-repeat junction, with peaks at the expected integration site as well as 15, 27, and 36 nucleotides upstream (Fig. 4B, C). The full-site integration results appear to show a strong bias for protospacer orientation, with all integration events at the leader-repeat junction having the protospacer oriented with the TT end toward the leader (Fig. 4C). To further explore the potential sequence dependence of the protospacer orientation, we varied the 3’ terminal nucleotides of the protospacer and also tested another protospacer with a completely randomized sequence (Table S2). Of the new protospacers designed, only the protospacer with the TA 3’ end has the possibility to generate a stop codon upon integration and was therefore excluded from our analysis. For all of the other protospacers, both orientations result in an open reading frame. For all of the tested 3’ ends, integration at the repeat occurred almost exclusively with the same relative orientation as observed for the initial protospacer (Fig. S4). Different 3’ end nucleotides led to different apparent orientation preferences at off-target sites, but the orientation bias at the repeat appears to depend on the internal sequence rather than terminal nucleotides. The randomized sequence, however, resulted in no integration events at the repeat. The orientation of integrated spacers is critical in vivo for ensuring that the resulting crRNA targets a protospacer with a PAM on the correct side (Mojica et al., 2009; Sashital et al.; 2012; Deveau et al., 2008). This data suggest that V-C Cas1 has an intrinsic mechanism to correctly orient protospacers during integration, similar to what has been observed for the E. coli integrase, though it is unclear how this mechanism functions in the native context to ensure fidelity (Wang et al., 2015). Surprisingly, the 3’ terminal nucleotides do appear to influence specific target recognition of the leader-repeat junction. The protospacers with the AA + TT 3’ ends and TC + AG 3’ ends were integrated at the leader-repeat junction in more than 40% of integration events on average, while some other protospacers were properly integrated in less than 5% of integration events (Fig. S4). The relevance of this variable specificity for the in vivo reaction is unclear, but it further supports the model that the protospacer sequence affects the integration reaction.
The Cas1 mini-integrase forms a tetramer for integration
Given that Cas1-Cas2 integrases function as heterohexameric complexes containing four copies of Cas1 and two copies of Cas2, we wondered whether the V-C Cas1 integrase also functions as a multimeric complex (Nuñez et al., 2014; Wang et al., 2015). While V-C Cas1 presumably associates with the protospacer in the conditions used for the integration reaction, the salt concentrations necessary to maintain solubility in the presence of protospacer prevent stable association for analytical measurement of the complex. When run on a size-exclusion column, the protospacer and protein are observed to elute as separate species (Fig. 5A). In an effort to capture the active form of the type V-C Cas1, we designed a full-site product mimic (pseudo-full-site substrate) and a half-site product mimic that might result in a more stable complex (Wright et al., 2017). The pseudo-full-site substrate includes a section of the leader sequence, the repeat, and a protospacer with a break in the middle to give the integrase access to the repeat. The half-site substrate has only one end of the protospacer covalently connected at the leader-repeat junction (Table S2).
Figure 5. Type V-C Cas1 Forms a Multimeric Complex for Integration.

(A) Chromatograms of size-exclusion runs of type V-C Cas1, Cas1 with protospacer, Cas1 bound to half-site substrate, and Cas1 bound to pseudo-full-site substrate. Dashed black line indicates elution volume of free Cas1 dimer. Dashed green line indicates elution volume of Cas1 complex bound to full-site substrate.
(B) Molecular weight characterization of the apo Cas1 and Cas1 bound to pseudo-full-site substrate by size exclusion chromatography coupled with dual angle light scattering. The experimental Mp for each peak is indicated.
See Table S2 for nucleotide sequences.
Size-exclusion chromatography suggests that V-C Cas1 elutes as a dimer when in isolation but elutes as a larger multimer when bound to either the pseudo-full-site or the half-site substrate (Fig. 5A). The shift in size after association with DNA is further supported by dynamic light scattering results. The average radius calculated across three runs was 3.8 ± 0.8 nm for the apo V-C Cas1 sample and 8.2 ± 2.0 nm for the V-C Cas1-pseudo-full-site substrate complex, with a polydispersity index between 0.39 and 0.45 (Fig. S5). These results show that in the presence of pseudo-full-site substrate, V-C Cas1 forms a complex that is a little more than twice the size of its apo state. Dual angle light scattering analyses of the apo Cas1 and the Cas1 bound to pseudo-full-site substrate reveal peak molecular weights (91.2 and 191.1 kDa) that correspond closely to free dimer and tetramer bound to DNA (86 and 207 kDa), respectively (Fig. 5B). The intense light scattering peak at 7 mL is due to small amounts of aggregate in the void. Together, these results are consistent with the conclusion that the apo V-C Cas1 primarily exists as a dimer but, in the presence of the pseudo-full-site DNA substrate, and to a lesser extent the half-site substrate, forms a tetramer.
To confirm whether the multimer was indeed a tetramer bound to a single DNA substrate, we used native nanoelectrospray ionization mass spectrometry (nanoESI-MS) measurements. Apo Cas1 and the Cas1-DNA complex were initially cross-linked before measuring by nanoESI-MS. While the predominant detected species was cross-linked dimer for both samples, we observed tetramer in both samples as well, with much more cross-linked tetramer in the Cas1-DNA complex sample (Fig. 6A, B). This suggests V-C Cas1 may form a tetramer in the absence of DNA, but the presence of DNA favors additional tetramer formation. A possible explanation for the significant amount of cross-linked dimer and free DNA detected in the Cas1-DNA sample is that the two Cas1 dimers in the tetramer may not support efficient cross-linking at the interface and are only weakly associated, resulting in disassembly during buffer exchange to the nanoESI-MS buffer or during the nanoESI-MS measurements.
Figure 6. Detection of Type V-C Cas1 Tetramer by Native Mass Spectrometry.

(A) NanoESI mass spectra of cross-linked samples of apo Cas1. Measured molecular masses and corresponding cartoons of each species are listed on the top right corner. Ions of different species are labeled with different colors.
(B) NanoESI mass spectra of cross-linked samples of Cas1 after complexing with pseudo-full-site substrate. See also Figure S6.
(C) Model for type V-C mini-integrase compared to canonical Cas1-Cas2 integrase.
See Table S2 for nucleotide sequences.
In an effort to capture the tetramer bound to DNA in its native state without cross-linking, we also performed native MS on complexes purified by size-exclusion chromatography. For the Cas1-DNA complex, we detected masses that correspond exactly to tetramer bound to a single copy of pseudo-full-site substrate (Fig. S6). Observed in greater abundance were free dimer, dimer bound to a single copy of pseudo-full-site substrate, free monomer, and free DNA, which again suggests that the Cas1 tetramer-DNA complex disassembles either during the buffer exchange to the nanoESI-MS buffer or during the nanoESI-MS measurements. A small amount of tetramer was also observed for the apo Cas1 sample, but the amount detected was lower than that of tetramer bound to pseudo-full-site substrate in the DNA-bound sample. Notably, no free tetramer was detected from the DNA-containing sample. Together with the size-exclusion and light scattering results, the data suggest that there exists an equilibrium between the dimer and tetramer in solution that is biased toward the dimer in the apo state but, in the presence of the pseudo-full-site DNA substrate, shifts to a tetramer.
Together, our results support a model in which the type V-C Cas1 mini-integrase tetramerizes for integration, providing the architecture for a flexible internal ruler mechanism that favors shorter DNA integration substrates (Fig. 6C). For the Cas1-Cas2 integrases, the Cas2 protein dimer at the center of the integrase complex acts as a bridge between the two catalytic Cas1 dimers. A condensed tetramer structure without a Cas2 bridge explains why the type V-C integrase prefers ~18-bp protospacers rather than the longer protospacers that are preferred by the other Cas1-Cas2 integrases (Nuñez et al., 2015a; Wang et al., 2015).
DISCUSSION
Type V-C and V-D CRISPR systems are unique among microbial adaptive immune systems in having unusually short CRISPR array spacers and repeats and lacking a cas2 gene previously thought to be essential for CRISPR sequence acquisition (Shmakov et al., 2015; Burstein et al., 2017). Although both Cas1 and Cas2 are essential for new DNA integration in most CRISPR systems, we demonstrate that type V-C and V-D Cas1 alone can carry out integration of DNA substrates, including full-site integration in the case of V-C Cas1. Furthermore, type V-C Cas1 appears to have an intrinsic mechanism to orient protospacers during integration, though further work is required to understand how this activity cooperates with protospacer processing to result in PAM-specific orientation in vivo. Our experiments revealed that type V-C Cas1 has many off-target integration sites, even for full-site integration, which typically has greater substrate specificity than half-site integration in other CRISPR systems (Wright et al., 2016b). However, we have reason to suspect that varying the 3’ end nucleotides of the protospacer can yield more specific target recognition. Our results strongly suggest that additional host factors or mechanisms are involved either in the protospacer selection or during integration to achieve specific target recognition in vivo and avoid deleterious off-target integration.
Our findings are consistent with a model in which the V-C Cas1 minimal integrase forms a tetramer for the integration of new protospacers. Without a Cas2 dimer acting as a bridge, the tetramer model for the type V-C Cas1 would result in a shortened internal ruler, thus explaining the preference for short protospacers of ~18 bp. While V-C Cas1 exists predominantly as a dimer in solution, binding to the protospacer might stabilize transient tetramerization events to produce an active complex, resulting in the tetramer observed in the intermediate- and productbound complexes.
Although it is possible that the type V-C and V-D systems are the unique outcome of a loss event of the cas2 gene, a recent study examining the Cas1 phylogeny indicated that type V-C and V-D Cas1 proteins may represent an important stage in the evolutionary history of CRISPR-Cas adaptation (Makarova et al., 2018). V-C and V-D Cas1 genes were found to form a branch along with solo Cas1 genes, found in the absence of apparent CRISPR systems or casposons, that was rooted near the casposon branch. This phylogeny is consistent with a model wherein the type V-C and V-D Cas1 proteins represent an extant version of an ancestral Cas1 that evolved from the casposase, which has integrase activity and similar target specificities as CRISPR-Cas spacer acquisition, but does not require Cas2 (Beguin et al., 2016; Krupovic et al., 2014). If this is correct, then the first CRISPR systems likely appeared much like extant V-C/D systems, with an effector protein or complex alongside an isolated Cas1 integrase, as opposed to the previous model wherein Cas2 was a part of the ancestral casposon prior to the evolution of a functional CRISPR system (Krupovic et al., 2017). This minimal integrase could have later adopted Cas2, potentially from a toxin-antitoxin system, for greater structural stability, resulting in the canonical Cas1-Cas2 integrase complex used by type I and II systems (Krupovic et al., 2017). The addition of Cas2 to the complex also allows for the acquisition of longer spacers, which could in turn allow for greater targeting specificity of crRNA-effector complexes, giving systems bearing a Cas1-Cas2 integrase an advantage. A greater knowledge of the structure and mechanisms of these different integrases gives us more insight into the evolutionary history of CRISPR adaptation modules and their unique functions.
STAR METHODS
Cloning/Protein purification
CRISPR loci from the Cas12c system from mouse cecum (ORF Ga0073908_100052011 from IMG genome 3300005460) and the Cas12d system from Passlidae beetle gut (ORF IMNBL3_1000107225 from IMG genome 3300000114) were ordered as G-blocks. Protein-coding genes were codon-optimized for E. coli expression and arrays were reduced. To generate target plasmids, arrays and flanking intergenic sequences were amplified by PCR and inserted into a pUC19 backbone by Gibson Assembly. Cas1 genes were PCR amplified and inserted into a pET-3a plasmid with an N-terminal 10xHis-MBP-TEV tag. Proteins were expressed in BL21(DE3) Star cells. Cells were grown to an OD600 of ~0.6 and induced overnight at 16°C with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG). Cells were harvested and resuspended in lysis buffer (20 mM HEPES, pH 7.5, 1 M NaCl, 10 mM imidazole, 0.1% Triton X-100, 2 mM Tris (2-carboxyethyl)phosphine (TCEP), Complete EDTA-free protease inhibitor (Roche) and 10% glycerol. Cells were lysed by sonication, and the lysate was cleared by centrifugation. The supernatant was incubated on Ni-NTA resin (Qiagen), and the resin was washed with buffer containing 20 mM HEPES, pH 7.5, 1 M NaCl, 10 mM imidazole, 1 mM TCEP, and 5% glycerol. The protein was eluted with wash buffer supplemented to 300 mM imidazole. The protein was digested with TEV protease overnight. The salt concentration was reduced by dilution to 400 mM NaCl, the cleaved MBP tag was removed with an MBPTrap column (GE Healthcare), and the protein was bound to a HiTrap heparin HP column (GE Healthcare). The protein was eluted with a gradient from 400 mM to 1 M NaCl. The proteins were further purified on a Superdex 200 (16/60) column with Storage Buffer (20 mM HEPES, pH 7.5, 500 mM KCl, 1 mM TCEP, and 5% glycerol).
Integration assays
All integration assays were performed in buffer containing 20 mM HEPES, pH 7.5, 25 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.01% Nonidet P-40, and 10% DMSO and carried out at room temperature unless otherwise noted. Pre-complexing of the E. coli Cas1-Cas2 was carried out as previously described (Nuñez et al., 2015b). For experiments with unlabeled protospacer, 50 nM protospacer was added to 250 nM Cas1 (or Cas1-Cas2) and incubated at room temperature for 15 minutes, followed by addition of target plasmid (pVC_CRISPR1 or pVD_CRISPR) to 20 ng/μL (~10 nM). For the initial experiment alongside E. coli Cas1-Cas2, the reaction was carried out at 37°C for one hour. The protospacer length variation experiment with unlabeled substrates was carried out at room temperature for 30 minutes. For all assays using unlabeled substrates, reactions were quenched by the addition of 0.4% SDS and 25 mM EDTA and analyzed on a 1% ethidium bromide-stained agarose gel. Quantifications were made using Image Lab (BioRad). Integration of radiolabeled protospacer into plasmid by V-D Cas1 was carried out with 400 nM Cas1, 1 nM protospacer that was previously 5’ labeled with [γ−32P]ATP (PerkinElmer) and T4 polynucleotide kinase (New England Biolabs), and 20 ng/μL plasmid at 37°C for one hour. Products were separated on a 1.5% agarose gel. The gel was dried and visualized through phosphorimaging. Integration of radiolabeled protospacer into oligonucleotide targets by V-C Cas1 was carried out at room temperature with 100 nM Cas1, 10 nM 5’radiolabeled protospacer, and 100 nM double-stranded target DNA. Reactions were quenched by the addition of an equal volume of 95% formamide and 50 mM EDTA, and products were separated on 12% urea-PAGE. The gel was dried and imaged with phosphorimaging. For the fluorescent protospacer experiments, one strand of each protospacer was ordered with a 5’ 6-carboxyfluorescein (FAM) attachment and hybridized with its complementary unlabeled strand. Integration of fluorescent protospacers was carried out at room temperature with 250 nM Cas1, 10 nM protospacer, and 20 ng/μL plasmid for 1 hour. Reactions were quenched by the addition of 0.4% SDS and 25 mM EDTA and treated with proteinase K for 30 minutes at room temperature before analysis on a 1.5% agarose gel. The gel was imaged with a Typhoon FLA gel imaging scanner.
High-throughput sequencing
Integration reactions into pUC19 or pVC-CRISPR were carried out with 250 nM Cas1, 50 nM protospacer, and 20 ng/μL plasmid at room temperature for one hour. Reactions were quenched by the addition of phenol-chloroform-isoamyl alcohol, chloroform extracted, and ethanol precipitated. The products were purified with a DNA Clean & Concentrator column (Zymo) to remove free protospacer. Products were fragmented with dsDNA fragmentase (New England Biolabs) and end-repaired. A single round of extension with Taq polymerase was performed on the products with one of the two protospacer-specific primers. Free primer was removed with Exonuclease I (New England Biolabs, RRID:AB_2293772). Annealed adapters were ligated with T4 DNA ligase (New England Biolabs). Libraries were amplified with Q5 polymerase and NEBNext universal and index primers for Illumina (New England Biolabs), size-selected with AMPure XP beads (Beckman Coulter), and sequenced on an Illumina MiSeq with 150-nt single reads.
3’-adapter sequences were removed from reads with Cutadapt (Martin, 2011). Reads with identical barcodes and read sequences were removed. The barcode and protospacer sequence were removed with Cutadapt, and the resulting reads were mapped to the appropriate plasmid with Bowtie (Langmead et al., 2009). Consensus integration sequences were generated with WebLogo from the 5 nucleotides upstream and 30 nucleotides downstream of an integration site, weighted by how many reads were measured at the integration site (Crooks et al., 2004).
Full-site selection
The reporter plasmid construct pVC_CRISPR_Cat was designed by cloning a cat promoter and chloramphenicol resistance gene into a pUC19 plasmid followed by subsequent insertion of a sequence carrying 59 bp of the 5’ leader from pVC-CRISPR, the adjacent CRISPR repeat, 2 bp of the spacer adjacent to the repeat, and a start codon by Gibson Assembly. Twelve 50 uL integration reactions were carried out as described above with the reporter construct for 1 hr. The integration products were purified using the Qiagen MinElute PCR Purification Kit and electroporated into DH10B cells. Transformants were plated on LB agar containing chloramphenicol and 50 of the surviving colonies were sequenced.
Protein complex formation / light scattering / native nanoelectrospray ionization mass spectrometry
DNA substrates were made as previously described (Wright et al., 2017). Cas1 and DNA substrates were complexed by mixing 50 μM Cas1 and 12.5 μM DNA half-site or pseudo-fullsite substrates in Storage Buffer and dialyzing in Complex Buffer (10 mM HEPES, pH 7.5, 5 mM EDTA, 250 mM KCl, and 1 mM TCEP) for 2 hours using the Slide-A-Lyzer MINI Dialysis Devices and concentrated to 100 μM Cas1.
To characterize the molecular weight of the peaks off size-exclusion by light scattering, the samples were run on a Superdex 200 (16/60) column attached to an Agilent 1260 Infinity Multi-Detector system. Light scattering was performed using a 658nm laser. The UV signals at 280 nm and 260 nm and the light scattering signal at 90° were collected. Data analysis and Mp calculations were performed using the Bio-SEC software (Agilent).
Dynamic light scattering measurements were collected on a Malvern Zetasizer Nano instrument. Cas1 and DNA substrates were complexed as described above in 35 μM final Cas1 concentration and 8.75 μM final DNA concentration before filtering with a 0.22 μm Corning SpinX Centrifuge Tube Filter. Three runs with 70 measurements per run were collected for each sample. The following parameters were set up: viscosity, 0.891 cP; temperature, 25°C; refractive index, 1.375 (Mevel et al., 2008).
To cross-link the protein samples for native nanoelectrospray ionization mass spectrometry, Cas1 and DNA substrates were complexed as described above by dialyzing in Complex Buffer using 10 μM Cas1 and 2.5 μM DNA half-site or pseudo-full-site substrates. The samples were then cross-linked with 1 mM BS(PEG)5 (PEGylated bis(sulfosuccinimidyl)suberate) final concentration for 30 minutes before quenching with 30 mM final concentration Quenching Buffer (1M Tris·HCl, pH 8.0) for 15 minutes. The cross-linked samples were then subjected to five rounds of buffer exchange in to Mass Spectrometry Buffer A (436 mM ammonium acetate, 13.5 mM ammonium bicarbonate, pH 7.5, and 5% glycerol) using 10,000 MWCO (molecular weight cut-off) Amicon Ultra-0.5 Centrifugal Filter Units and concentrated to 20 μM final Cas1 concentration. For non-cross-linked samples, Cas1 and pseudo-full-site substrate samples were complexed and purified as described above on a Superdex 200 (16/60) column. The major peak was concentrated to an A280 of 3.0 using Nanodrop and subjected to five rounds of buffer exchange in to Mass Spectrometry Buffer B (242 mM ammonium acetate, 7.5 mM ammonium bicarbonate, pH 7.5, and 5% glycerol). Native mass spectrometry measurements were performed using a Synapt G2-Si mass spectrometer equipped with a nanoelectrospray ionization source, as described previously (Liu et al., 2017).
Quantification and Statistical Analysis
Integration assays (protospacer length and overhang length)
Fluorescent bands were visualized by Typhoon FLA gel imaging scanner and quantified using ImageQuantTL. The fraction integrated was calculated as the ratio of the integration product fluorescence intensity to the total intensity of the free fluorescent protospacer and the integration product. When examining integration by open-circle formation, gel bands were visualized by ChemiDoc MP (BioRad) and quantified using Image Lab (BioRad). The fraction integrated was calculated as the ratio of the open-circle integration product band intensity to the total intensity of both the open-circle band and the supercoiled plasmid band. Statistical analyses were performed using Prism GraphPad. Data were presented as the mean ± SD (error bars) of three independent experiments.
Dynamic light scattering
Sizes were presented as the mean ± SD of three runs with 70 measurements per run collected for each sample.
Full-site selection
Numbers of integration events were presented as the mean ± SD (error bars) of three independent experiments with 50 sequenced colonies for each experiment.
Supplementary Material
Highlights.
A mini-integrase comprising Cas1 alone catalyzes integration of short DNA fragments
V-C Cas1 has an intrinsic mechanism to orient protospacers during integration
Cas1 tetramerizes for integration, supporting a shortened internal ruler mechanism
Minimal integrase may represent the function of the ancestral Cas1
ACKNOWLEDGMENTS
A.V.W. is supported by a US National Science Foundation Graduate Fellowship. J.Y.W is supported by the NIH T32 066698 training grant and the Berkeley Graduate Fellowship Program. This work was funded by US National Science Foundation grant number 1244557. This work used a mass spectrometer that was purchased using NIH funding (grant 1S10OD020062–01) and the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. This work was partly conducted by the US Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, under contract number DE-AC02–05CH11231. J.A.D. is an investigator of the Howard Hughes Medical Institute. We thank the Francis lab for use of their Agilent 1260 Infinity Multi-detector system and Malvern Zetasizer Nano instrument and M.J. Lobba for help in data collection with these instruments. We thank G.J. Knott for input on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
UC Regents has filed patents related to this work on which D.B., J.F.B., L.B.H., and J.A.D are inventors. L.B.H. is a co-founder of Mammoth Biosciences. J.F.B. is a co-founder of Metagenomi. J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Intellia Therapeutics, Scribe Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribous Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Synthego, Metagenomi, Mammoth Biosciences, and Inari. J.A.D is a member of the board of directors at Johnson & Johnson, and has sponsored research projects by Pfizer, Inc. and Biogen.
REFERENCES
- 1.Arslan Z, Hermanns V, Wurm R, Wagner R, and Pul Ü (2014). Detection and characterization of spacer integration intermediates in type IE CRISPR–Cas system. Nucleic Acids Res 42, 7884–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 3.Beguin P, Charpin N, Koonin EV, Forterre P, and Krupovic M (2016). Casposon integration shows strong target site preference and recapitulates protospacer integration by CRISPR-Cas systems. Nucleic Acids Res 44, 10367–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolotin A, Quinquis B, Sorokin A, and Ehrlich SD (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- 5.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC, Doudna JA and Banfield JF (2017). New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen IMA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, Ratner A, Huang J, Andersen E, Huntemann M, and Varghese N (2016). IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res 45, D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, and Semenova E (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun 3, 945. [DOI] [PubMed] [Google Scholar]
- 10.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P and Moineau S (2008). Phage response to CRISPRencoded resistance in Streptococcus thermophilus. J. Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Díez-Villaseñor C, Guzmán NM, Almendros C, García-Martínez J, and Mojica FJ (2013). CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR-Cas IE variants of Escherichia coli. RNA Biol 10, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fagerlund RD, Wilkinson ME, Klykov O, Barendregt A, Pearce FG, Kieper SN, Maxwell HW, Capolupo A, Heck AJ, Krause KL, and Bostina M (2017). Spacer capture and integration by a type IF Cas1–Cas2–3 CRISPR adaptation complex. Proc. Natl. Acad. Sci. U. S. A 114, E5122–E5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- 14.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, and Terns MP (2009). RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hickman AB and Dyda F (2015). The casposon-encoded Cas1 protein from Aciduliprofundum boonei is a DNA integrase that generates target site duplications. Nucleic Acids Res 43, 10576–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krupovic M and Koonin EV (2016). Self-synthesizing transposons: unexpected key players in the evolution of viruses and defense systems. Curr. Opin. Microbiol 31, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krupovic M, Béguin P, and Koonin EV (2017). Casposons: mobile genetic elements that gave rise to the CRISPR-Cas adaptation machinery. Curr. Opin. Microbiol 38, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krupovic M, Makarova KS, Forterre P, Prangishvili D, and Koonin EV (2014). Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu TY, Iavarone AT, and Doudna JA (2017). RNA and DNA targeting by a reconstituted Thermus thermophilus Type III-A CRISPR-Cas system. PloS One 12, e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarova KS, Wolf YI, and Koonin EV (2018). Classification and nomenclature of CRISPR-Cas systems: where from here?. The CRISPR Journal, 1(5), 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- 24.Mevel M, Neveu C, Goncalves C, Yaouanc JJ, Pichon C, Jaffrès PA, and Midoux P (2008). Novel neutral imidazole-lipophosphoramides for transfection assays. Chem Commun 27, 3124–3126. [DOI] [PubMed] [Google Scholar]
- 25.Mojica FJ, Díez-Villasenor C, Garcia-Martinez J, and Almendros C (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740. [DOI] [PubMed] [Google Scholar]
- 26.Mojica FJM, Díez-Villaseñor C, García-Martínez J, and Soria E (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol 60, 174–182. [DOI] [PubMed] [Google Scholar]
- 27.Nivala J, Shipman SL, and Church GM (2018). Spontaneous CRISPR loci generation in vivo by non-canonical spacer integration. Nat. Microbiol 3, 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuñez JK, Bai L, Harrington LB, Hinder TL, and Doudna JA (2016). CRISPR immunological memory requires a host factor for specificity. Mol. Cell, 62(6), 824–833. [DOI] [PubMed] [Google Scholar]
- 29.Nuñez JK, Harrington LB, Kranzusch PJ, Engelman AN, and Doudna JA (2015). Foreign DNA capture during CRISPR–Cas adaptive immunity. Nature 527, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, and Doudna JA (2014). Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol 21, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nuñez JK, Lee AS, Engelman A, and Doudna JA (2015). Integrase-mediated spacer acquisition during CRISPR–Cas adaptive immunity. Nature 519, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pourcel C, Salvignol G, and Vergnaud G (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. [DOI] [PubMed] [Google Scholar]
- 33.Rollie C, Graham S, Rouillon C, and White MF (2018). Prespacer processing and specific integration in a Type IA CRISPR system. Nucleic Acids Res 46, 1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sashital DG, Wiedenheft B, and Doudna JA (2012). Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell 46, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, et al. (2015). Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell 60, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sternberg SH, Richter H, Charpentier E, and Qimron U (2016). Adaptation in CRISPR-Cas systems. Mol. Cell 61, 797–808. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Li J, Zhao H, Sheng G, Wang M, Yin M, and Wang Y (2015). Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR-Cas systems. Cell 163, 840–853. [DOI] [PubMed] [Google Scholar]
- 38.Wei Y, Chesne MT, Terns RM, and Terns MP (2015). Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res 43, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright AV and Doudna JA (2016). Protecting genome integrity during CRISPR immune adaptation. Nat. Struct. Mol. Biol 23, 876. [DOI] [PubMed] [Google Scholar]
- 40.Wright AV, Liu JJ, Knott GJ, Doxzen KW, Nogales E, and Doudna JA (2017) Structures of the CRISPR genome integration complex. Science 357, 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright AV, Nuñez JK, and Doudna JA (2016). Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164, 29–44. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Y, Ng S, Nam KH, and Ke A (2017). How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature 550, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoganand KNR, Sivathanu R, Nimkar S, and Anand B (2017). Asymmetric positioning of Cas1–2 complex and Integration Host Factor induced DNA bending guide the unidirectional homing of protospacer in CRISPR-Cas type I-E system. Nucleic Acids Res 45, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yosef I, Goren MG, and Qimron U (2012). Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40, 5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.