

Abstract
Gaucher disease (GD) results from mutations in the acid β-glucocerebrosidase (GCase) encoding gene, GBA, which leads to accumulation of glucosylceramides. GD patients and carriers of GD mutations have a significantly higher propensity to develop Parkinson disease (PD) in comparison to the non-GD population. In this study, we used the fruit fly Drosophila melanogaster to show that development of PD in carriers of GD mutations results from the presence of mutant GBA alleles. Drosophila has two GBA orthologs (CG31148 and CG31414), each of which has a minos insertion, which creates C-terminal deletion in the encoded GCase. Flies double heterozygous for the endogenous mutant GBA orthologs presented Unfolded Protein Response (UPR) and developed parkinsonian signs, manifested by death of dopaminergic cells, defective locomotion and a shorter life span. We also established transgenic flies carrying the mutant human N370S, L444P and the 84GG variants. UPR activation and development of parkinsonian signs could be recapitulated in flies expressing these three mutant variants.
UPR and parkinsonian signs could be partially rescued by growing the double heterozygous flies, or flies expressing the N370S or the L444P human mutant GCase variants, in the presence of the pharmacological chaperone ambroxol, which binds and removes mutant GCase from the endoplasmic reticulum (ER). However flies expressing the 84GG mutant, that does not express mature GCase, did not exhibit rescue by ambroxol. Our results strongly suggest that the presence of a mutant GBA allele in dopaminergic cells leads to ER stress and to their death, and contributes to development of PD.
Introduction
Gaucher disease (GD) is a lysosomal storage disease that results from mutation in the GBA gene encoding lysosomal acid β-glucocerebrosidase (GCase) (1). The disease is characterized by accumulation of glucosylceramide mainly in monocyte-derived cells. Due to its heterogeneity, the disease has been divided into three types: the type 1 GD, primarily a non-neurological disease, and Type 2 and 3, two forms associated with a neuronopathic disease (2,3).
More than 300 mutations were identified in the GBA gene. A large fraction of them are missense mutations, though premature termination, splice site mutations, deletions and recombinant alleles have been recognized as well (4). There are several abundant mutations. For example, the N370S mutation is the most prevalent among type 1 GD patients (5), while the L444P mutation is most common among the neuronopathic forms of GD. The majority of patients homozygous for the L444P mutation develop type 3 GD (6). The 84GG mutation is an insertion of a guanine 84 nucleotides downstream from the first initiator methionine of the GBA mRNA, resulting in premature protein termination (7).
As a lysosomal enzyme, GCase in synthesized in the endoplasmic reticulum (ER) on ER-bound polyribosomes. Upon its entry into the ER, it undergoes N-linked glycosylation on four asparagines, after which it is subject to ER quality control. When correctly folded it shuttles to the Golgi compartment for further modifications on its N-linked glycans and finally it traffics to the lysosomes (8–11). Mutant GCase variants undergo ER-associated degradation (ERAD), the degree of which correlates with disease severity (12). ER-retained mutant GCase leads to ER stress and to unfolded protein response (UPR) (13).
In recent years, a large number of independent studies documented association between GD and Parkinson disease (PD). GD patients have significantly higher propensity to develop PD than non-GD population, and a high frequency of GD mutations were found in PD population (14–23). PD is the second most common motor neurodegenerative disorder that usually affects individuals above the age of 60. The hallmark of PD is the loss of dopaminergic cells in the substansia nigra pars compacta, which results in movement disorder with resting tremor, stiffness, postural instability and bradykinesia (24).
The accumulation of misfolded proteins in the brain is a common feature in many neurodegenerative diseases such as Huntington's disease, Alzheimer’s disease and Amyotrophic Lateral Sclerosis (25,26). Accumulation of misfolded proteins has also been documented in PD (27), contributing to ER stress (28) and upregulation of the UPR pathways (29). This raises the possibility that accumulation of misfolded proteins in PD triggers stress-response pathways that induce neurotoxicity and cell death exhibited in brains of afflicted patients.
As ERAD and UPR are well conserved across species, Drosophila has proven an excellent organism to model neuronal degenerative diseases in general and PD in particular (30–33). Deregulation of gene expression can be achieved in the fly by the use of existing mutations, the use of lines containing endogenous genes with an insertion of a transposable element (34), by siRNA or by mutating the gene using the CRISPR-Cas system (35,36). Transgenic expression of heterologous genes is another way to follow the in vivo effect of expression of normal or mutant genes in the fly. Expression of the foreign gene is regulated by the GAL4 transcription factor, under any desired promoter, with a wide or a very narrow expression pattern (37).
In a previous study, we have shown UPR activation in fibroblasts derived from GD patients and in fibroblasts heterozygous for various GD mutations. We, and others, have also demonstrated that in Drosophila melanogaster models for carriers of GD mutations, UPR was activated (13,38) and that locomotion of the flies was impaired (13).
In this study, we extended our analyses to show that aging flies, double heterozygous for the fly GBA variants, presented death of dopaminergic cells, impaired negative geotaxis and a shorter life span compared with normal flies. Transgenic flies expressing the N370S, L444P or the 84GG human mutations also showed UPR and developed parkinsonian signs. With the exception of the 84GG line, in all fly models UPR as well as parkinsonian signs could be partially rescued by the addition of the pharmacological chaperone ambroxol, highlighting the importance of misfolded protein in development of PD.
Results
Parkinsonian signs in GBA double heterozygous flies
Two GBA homologs exist on chromosome 3 of D. melanogaster, designated CG31414 and CG31148, encoding proteins with ∼31% identity and ∼50% similarity to the human GCase. There are two available fly lines, each carrying a minos transposable element insertion in one of the fly GBA orthologs (13). In each case the minos insertion causes premature termination of the fly GCase protein. As a result, mutant CG31414 and CG31148 GCases are 129 and 34 amino acids shorter, respectively, than the corresponding wild-type fly proteins. By crossing these two lines to each other we generated double heterozygous flies as a model for the GD carrier state in humans. These flies did not accumulate glucosylceramide, in comparison to positive controls (fibroblasts derived from type 2 GD patients or mouse macrophages, that were treated with the GCase inhibitor conduritol B epoxide [CBE]), as tested on thin-layer chromatography (TLC) (Fig. 1A). In a previous work we have shown, using the artificial substrate 4-methyl umbelliferyl glucopyranoside, that GCase activity in the double heterozygotes is decreased by 30% compared with that in normal flies. In this study, we tested GCase activity using the synthetic GCase substrate C6-NBD-GlcCer. The results showed a 30% decrease in activity of the GCase in the double heterozygotes compared with that of normal flies (Fig. 1B), which is the expected decrease in activity expected in carriers of GD mutations. We have previously shown that there is UPR activation in these double heterozygous flies and age-dependent decrease in their locomotive ability (13). In this study, we directly tested death of dopaminergic cells by quantifying the amount of tyrosine hydroxylase (TH), as a marker of dopaminergic cells, in heads of aging flies. To do so, we tested the amount of TH on western blots of head lysates or quantified the fluorescence intensity as well as the number of dopaminergic cells that interacted with anti-TH antibodies in fixed, stained brains. As shown in Figure 1C and D there was a significant decrease in the amount of TH in heads of double heterozygous flies already at day 12 post-eclosion, in comparison to control flies. This difference was more pronounced at day 22 post-eclosion, clearly indicating death of dopaminergic cells. Fluorescence intensity of TH was analyzed in the posterior region of the brain, where ∼70 dopaminergic cells occupy very distinct areas (39) (Fig. 1E). As shown (Fig. 1F and G), fluorescence intensity significantly decreased in double heterozygous brains already at day 12 post-eclosion, strongly indicating that dopaminergic cells gradually lose their ability to synthesize TH before they die. TH positive cells were also counted in the protocerebral posterior lateral region 1 (PPL1) and protocerebral posterior medial regions 1, 2 and 3 (PPM1/2, PPM3) (Fig. 1D) at 2, 12 and 22 days post-eclosion (Fig. 2A–C). At days 2 (Fig. 2A) and 12 (Fig. 2B) post-eclosion there was no significant difference in the number of TH positive cells between the double heterozygous flies and the control flies. However, at day 22 post-eclosion (Fig. 2C), a significant decrease in the number of TH positive cells was noted in all the analyzed regions in the double heterozygous flies compared with the control counterparts. Taking into consideration that dying cells express low TH levels, cell numbers were not much different between control and double heterozygous flies at day 12, but the amount of TH on gels and its intensity were significantly different at day 12 between control and double heterozygotes. We have shown in the past a significant decrease in the locomotive ability of aging double heterozygous flies. The survival of the double heterozygous flies was significantly lower than that of the control flies. Although control flies showed 50% survival at day 60, the same percentage of survival for double heterozygotes was found at day 22 (Fig. 2D).
Figure 1.
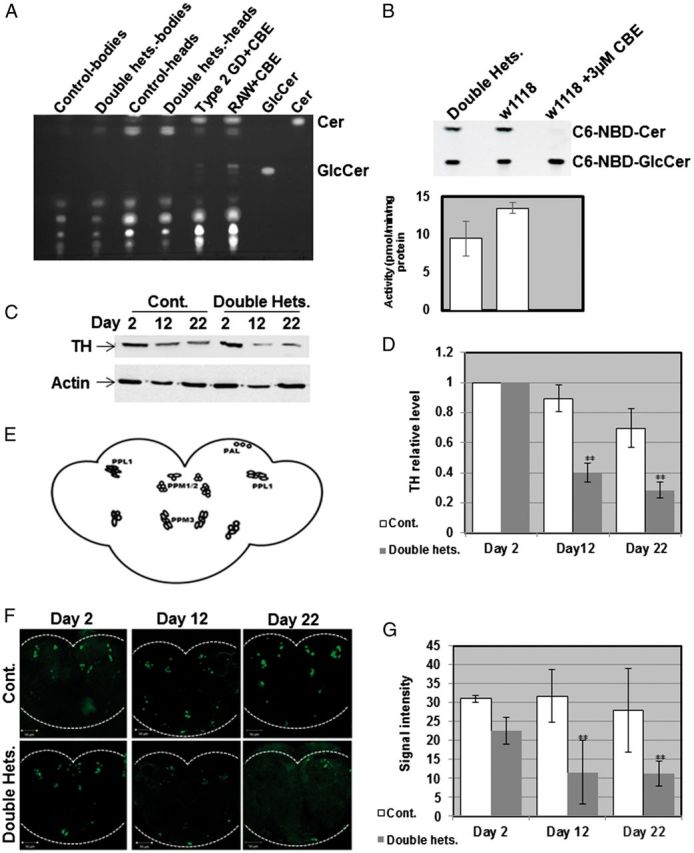
Decrease in amount of TH in flies double heterozygous for the fly GBA homologs. (A) TLC plate showing no GlcCer accumulation in heads and bodies of 22-day-old flies in comparison to its accumulation in type 2 GD fibroblasts or in RAW 264.7 mouse macrophages, both treated with 200 μM CBE for 10 days. (B) Activity assay using the synthetic fluorescent GCase substrate C6-NBD showing activity level of GCase in double heterozygous flies in comparison to control flies. For a positive control, control flies were treated with 3 μM of the GCase inhibitor CBE. (C) Protein lysates, prepared from heads of 10 w1118 (cont.) or double heterozygous flies (double hets.) at days 2, 12 and 22 post-eclosion, were subjected to western blotting. The corresponding blots were interacted with anti TH antibody. As a loading control, the blots were interacted with anti-actin antibody. (D) Intensities of the corresponding bands were quantified by densitometry and the value obtained for control flies was considered 1. Results represent the mean ± SEM of four independent experiments. (E) Schematic representation of dopaminergic neuronal clusters in the posterior region of the Drosophila brain. (F) Representative confocal images of brains that were isolated from w1118 (cont.) or double heterozygous (double hets.) flies after staining with anti-TH antibody, at days 2, 12 and 22 post-eclosion. G. Quantification of signal intensity obtained from brains at 2, 12 and 22 days post-eclosion. Results represent the mean ± SEM of 15 brains (n = 15). **P < 0.01.
Figure 2.

Degeneration of dopaminergic neurons and decreased survival in flies double heterozygous for the fly GBA homologs. Quantitative representation of the average number of cells in dopaminergic clusters in the brains at days 2 (A), 12 (B) or 22 (C) post-eclosion. Results represent the mean ± SEM of 15 brains. (D) Kaplan Meier curve showing the overall survival rates of control and double heterozygote flies tested on 100 flies. Flies were grown as 10 flies per vial and were transferred to fresh food every other day. ***P < 0.005.
Taken together our results point to development of parkinsonian signs in aging flies that are double heterozygous in their two GBA genes, CG31148 and CG31414.
Parkinsonian signs in transgenic flies expressing human mutant GCase
We decided to test the effect of mutant GCase on development of parkinsonian signs in transgenic flies expressing the human GCase variants, and which otherwise have and express the normal set of two endogenous fly GBA genes. The GAL4-UAS bipartite system was used to drive expression of the normal, the N370S and the L444P human GCase variants in the dopaminergic cells, using the Ddc-GAL4 driver (40). We have shown in the past that these human GCase mutant variants present different degrees of ER retention, detected by their endo-H sensitivity, and increased UPR (13). The expression level of the different transgenes was re-tested and was consistent with ERAD of the mutant GCase variants, as previously shown (Fig. 5A in Maor et al., 2013). Thus, the L444P mutant GCase underwent extensive ERAD, in comparison to the N370S mutant variant, in accordance with the severity associated with these two mutations in humans, and as shown in GD patients (12) (Fig. 3A). Therefore, the amount of mature lysosomal L444P GCase was the lowest (Fig. 3A). Dopaminergic cells were stained with anti-TH antibodies and anti-myc antibody to confirm expression of the transgenes in dopaminergic cells (Fig. 3B).
Figure 5.
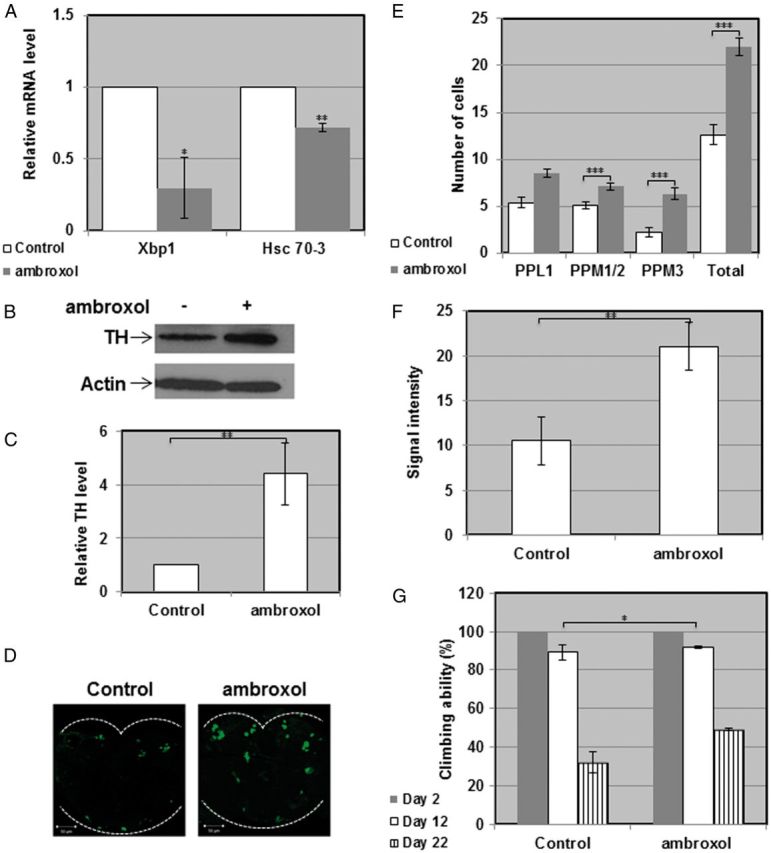
Rescue of the parkinsonian phenotype in flies double heterozygous for the fly GBA homologs. (A) RNA was isolated from ambroxol treated or untreated double heterozygous flies, and the cDNA prepared from it was subjected to quantitative RT-PCR with primers specific for Drosophila Hsc-70-3 or for the spliced form of Drosophila Xbp1. The results (three different experiments) were quantified, and the values obtained for untreated flies were considered 1. RP49 was used as a normalizing control. (B) Protein lysates prepared from heads of ten double heterozygous flies, treated or untreated with 1 mM ambroxol for 22 days, were subjected to western blotting and interaction with anti-TH antibody. As a loading control, the blots were interacted with anti-actin antibody. (C) Intensities of the corresponding bands were quantified by densitometry and the value obtained for untreated flies was considered 1. Results represent the mean ± SEM of four independent experiments. (D) Representative confocal images of adult brains from treated and untreated double heterozygous flies, stained with anti-TH antibody at day 22 post-eclosion. (E) Quantitative representation of the average number of cells in dopaminergic clusters. Results represent the mean ± SEM of 15 brains. (F) Quantification of signal intensity obtained from the brains described in (D). Results represent the mean ± SEM of 15 brains. (G) Five vials, each containing 10 double heterozygous flies were analyzed for locomotion behavior at days 2, 12 and 22 post-eclosion. *P < 0.05; **P < 0.01; ***P < 0.05.
Figure 3.

Parkinsonian signs in transgenic flies expressing mutant versions of human GCase. (A) Protein lysates were prepared from 10 flies expressing the human UAS-mycHisWTGCase or human UAS-mycHisN370SGCase, driven by daughterless (Da) - GAL4. Samples, containing 100 μg of protein, were subjected to overnight Endo-H digestion, after which they were electrophoresed through SDS-PAGE and the corresponding blot was interacted with anti-GCase and anti-actin antibodies. lys-lysosomal, endo-H resistant, fraction. (B) Confocal images of flies expressing mycHis WT, N370S or L444P human GCase transgenes under the Ddc- GAL4 driver. Brains were stained at day 2 using anti-TH antibody (green) and anti-myc-antibody (red). The images show co-expression of TH and GCase in one confocal plane. (C) Protein lysates were prepared from heads of 10 control flies, or 10 flies expressing normal human GCase variants, at days 2, 12 and 22 days post-eclosion, and were subjected to western blotting. The corresponding blots were interacted with anti-TH antibody. As a loading control, the blots were interacted with anti-actin antibody. (D) Intensities of the corresponding bands were quantified by densitometry and the value obtained for control flies was considered 1. Results represent the mean ± SEM of four independent experiments. **P < 0.01.
To follow death of dopaminergic cells, TH was quantified in brain lysates on western blots. The results indicated a significant decrease in the amount of TH in brains of transgenic flies expressing mutant human GCase, at day 22 post-eclosion, in comparison to its amount in flies expressing normal human GCase or in control flies expressing only the Ddc-GAL4 driver (Fig. 3C and D).
Quantification of the amount of TH fluorescence emitted from dopaminergic cells (Fig. 4A) showed limited death of dopaminergic cells at days 2 and 12 post-eclosion for all tested lines (Fig. 4B). Extent of death of dopaminergic cells in the flies expressing the mutant variant forms in comparison to the normal human GCase was significantly higher at day 22 post-eclosion (Fig. 4B). The number of dopaminergic neurons was tested in the PPL1, PPM1/2 and PPM3 regions of brains of the transgenic flies. There was no significant difference in the number of dopaminergic cells between flies expressing the transgenic normal or the mutant (N370S or L444P) GCase variants at days 2 (Fig 4C) or 12 (Fig 4D) post-eclosion. At day 22 post-eclosion, there was a significant decrease in the number of TH positive cells in all tested areas of the brains of transgenic flies expressing mutant human GCase variants but not in brains of flies expressing the normal human GCase (Fig. 4E).
Figure 4.
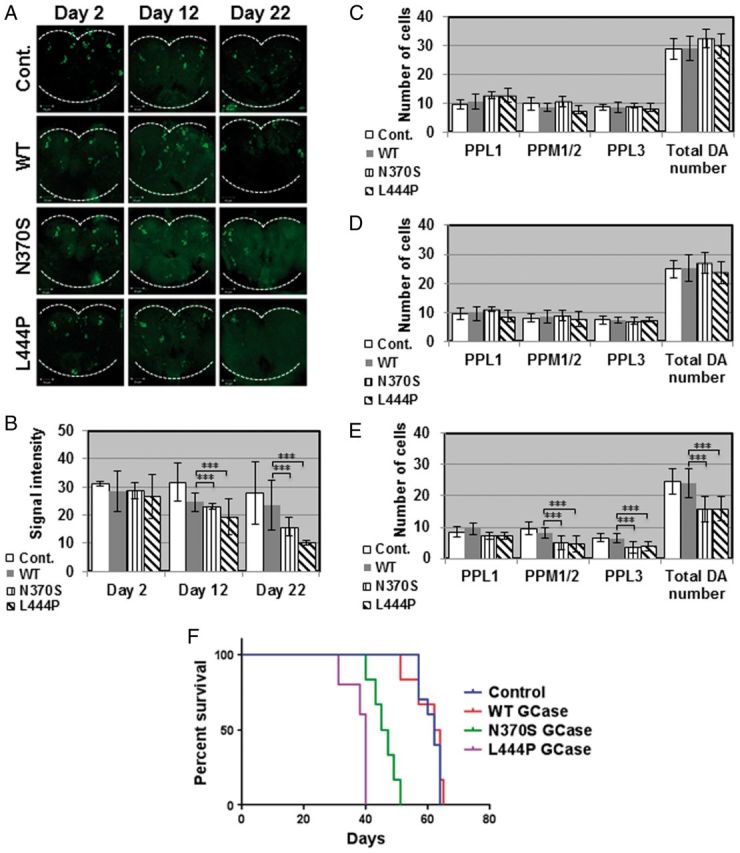
Degeneration of dopaminergic neurons and decreased survival in transgenic flies expressing mutant versions of human GCase. (A) Representative confocal images of brains isolated from control flies (w118, cont.) or flies expressing the different human GCase transgenes, following their staining with anti-TH antibody, at days 2, 12 and 22 post-eclosion. (B) Quantification of signal intensity obtained from the tested brains described in (A). Quantitative representation of the average number of cells in dopaminergic clusters at days 2 (C), 12 (D) or 22 (E) post-eclosion. Results represent the mean ± SEM of 15 brains. (F) Kaplan Meier curve showing the overall survival rates of control (Ddc-GAL4), or flies expressing wild-type, N370S or L444P human GCase flies tested on 100 flies. Flies were grown as 10 flies per vial and were transferred to fresh food. ***P < 0.005.
We have already shown in the past that aging flies expressing the human N370S or the L444P mutant variants present decreased locomotor activity, as tested by climbing assays (13). In the present study we tested the effect of expression of mutant human GCase in dopaminergic cells on fly survival. The results (Fig. 4F) strongly indicated that flies expressing the N370S or the L444P mutant GCase variants in their dopaminergic neurons presented premature death in comparison with flies expressing the normal human GCase.
To summarize, the results strongly indicate the development of parkinsonian signs in transgenic flies expressing the human mutant GCase variants N370S and L444P.
Rescue of parkinsonian signs in GBA double heterozygous and GCase transgenic flies by ambroxol
Our results suggest that the parkinsonian signs, presented by the GBA double heterozygous flies as well as by the transgenic flies, expressing human mutant N370S or L444P, resulted from ER retention of mutant GCase, which activated UPR. If this is really so, removing mutant GCase from the ER should rescue, at least partially, the phenotype. Such removal of mutant misfolded GCase molecules from the ER can be achieved by pharmacological chaperones (41–44). Pharmacological chaperones are small molecules that bind to mutant proteins in the ER and stabilize their native folding, thus enabling them to leave the ER (45,46). As small molecules, pharmacological chaperones can cross the blood brain barrier, thus be applicable for neurodegenerative diseases (47–49). One such GCase chaperone is ambroxol, previously used to increase amount and lysosomal activity of mutant GCase in GD-derived skin fibroblasts (38,42,50,51). Growth of GBA double heterozygous flies on 1 mM ambroxol containing food resulted in decreased levels of UPR parameters: Hsc-70-3 mRNA level and splicing of Xbp-1 mRNA compared with untreated flies (Fig. 5A). The same was true for ambroxol treated transgenic flies, expressing the N370S or the L444P mutant GCase variants in comparison to flies expressing the normal human GCase (Fig. 6A). The amount of TH (tested by western blotting, by its fluorescence intensity and by the number of TH containing cells in the posterior region of the brain) increased in the treated double heterozygous flies (Fig. 5B–F) as well as in treated transgenic flies expressing mutant, but not normal human GCase compared with the corresponding untreated flies (Fig. 6B–F). It also led to partial rescue of the defective locomotion of the flies compared with untreated counterparts (Fig. 5G and 6G), strongly indicating that removal of mutant misfolded GCase from the ER alleviated development of PD-like disease in the fly.
Figure 6.
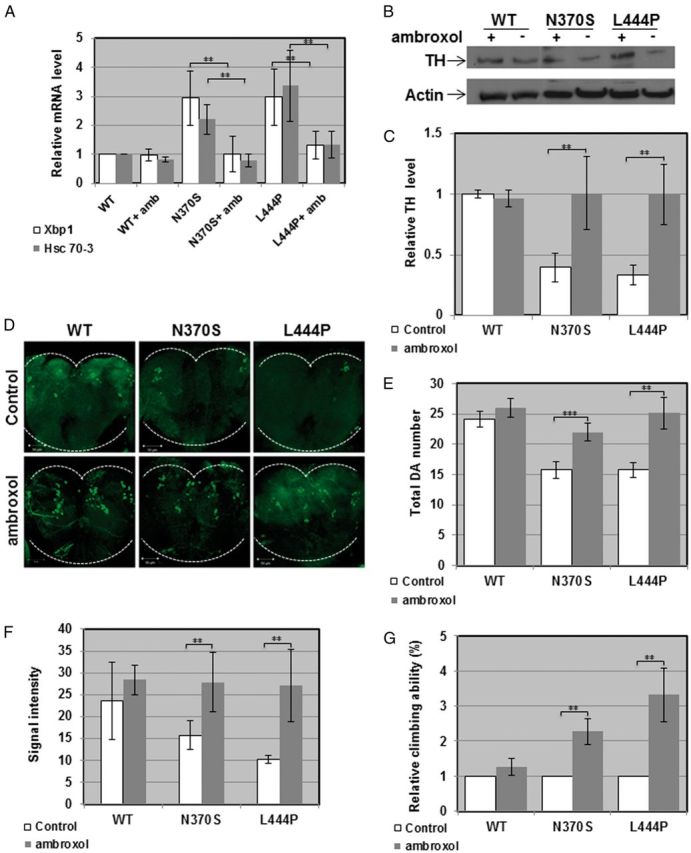
Rescue of parkinsonian phenotype in transgenic flies expressing mutant human GCase. (A) RNA was isolated from ambroxol treated or untreated transgenic flies, expressing the normal or the N370S or L444P human GCase variants, and the cDNA prepared from it was subjected to quantitative RT-PCR with primers specific for Drosophila Hsc-70-3 or for the spliced form of Drosophila Xbp1. The results (three different experiments) were quantified, and the values obtained for untreated flies were considered 1. RP49 was used as a normalizing control. (B) Lysates prepared from heads of transgenic flies, treated or untreated with 1mM ambroxol, were analyzed by western blotting, as described in the legend to Figure 5. (C) Intensities of the corresponding bands were quantified by densitometry and the value obtained for untreated flies was considered 1. Results represent the mean ± SEM of four independent experiments. (D) Representative confocal images of adult brains of transgenic flies, expressing the human normal (mycHisWTGCase), the N370S (mycHisN370SGCase) or the L444P (mycHisL444PGCase) variants, untreated or ambroxol treated, following their staining with anti-TH antibody at day 22 post-eclosion. (E) Quantitative representation of the average number of cells in dopaminergic clusters. Results represent the mean ± SEM of 15 brains. (F) Quantification of TH signal intensity (Signal intensity) obtained from tested brains at day 22 post-eclosion. Results represent the mean ± SEM of 15 brains. (G) Flies expressing wild-type, N370S or L444P human GCase transgenes in their dopaminergic cells were treated with 1mM ambroxol and analyzed for climbing ability. **P < 0.01.
Activation of UPR pathways and development of parkinsonian signs in 84GG transgenic flies
The 84GG mutation is an insertion of a guanine after the 84th nucleotide of the human GBA mRNA (7), starting from the first ATG (52,53). This mutation leads to shift of frame and premature termination 18 amino acids downstream from the insertion and to a putative 48 amino acids peptide. The amount of GCase mRNA in individuals carrying this mutation is normal, but no protein product has been detected (7). No homozygous newborns for this mutation have been reported thus far. Flies expressing the human 84GG transgene under the Da-GAL4 driver contained an 84GG mRNA product (Fig. 7A). They presented elevation in transcription of the chaperone Hsc-70-3, in splicing of the transcription factor Xbp-1 (Fig. 7B) and in phosphorylation of the initiation factor eIF2α (Fig. 7C and D). Like human carriers of these mutations, flies transgenic for the 84GG mutations presented activation of the UPR pathways.
Figure 7.
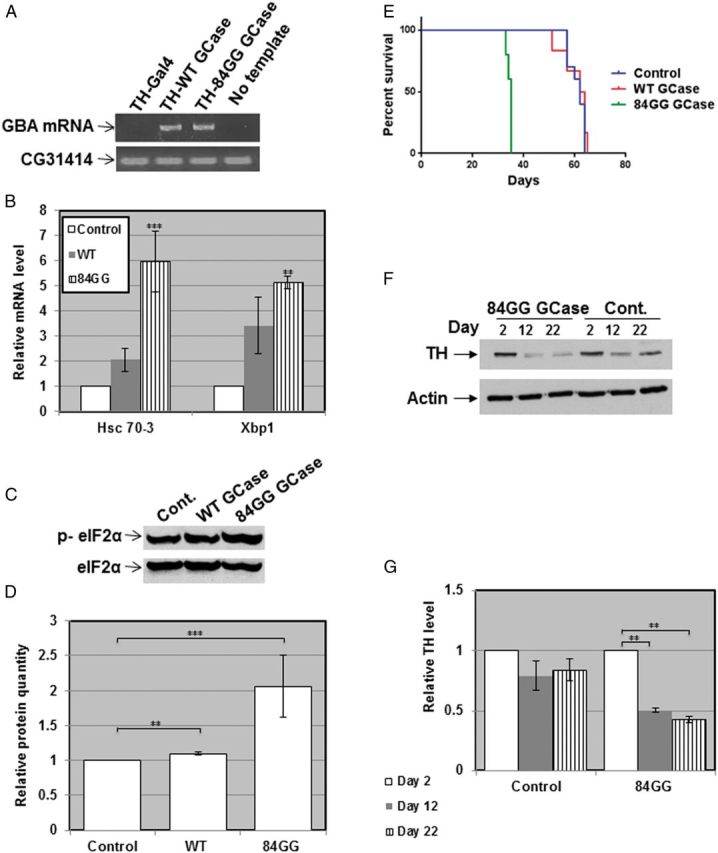
Activation of the UPR pathways and development of parkinsonian signs in 84GG transgenic flies. (A) RNA was isolated from Ddc-GAL4 or from flies expressing the human wild-type or the 84GG GBA transgenes under the Ddc-GAL4 driver. cDNA was prepared and subjected to PCR using primers specific for an 1200 bp fragment of the human GBA mRNA. Primers for the CG31414 Drosophila gene were used as a DNA loading control. Products were separated through a 1% agarose gel. (B) RNA was isolated from the above-mentioned flies, and the cDNA prepared from it was subjected to quantitative RT-PCR with the appropriate primers. RP49 was used as a normalizing control. The results (three different experiments) were quantified and the values obtained for flies expressing normal human GCase were considered 1. (C) Protein lysates, prepared from Da-GAL4 (Cont.), or flies expressing WT and 84GG human GCase using the Da-GAL4 driver, were subjected to western blotting and interaction with anti-phosphorylated eIF2α antibodies (p-eIF2α). As a loading control, the blot was interacted with anti-IF2α antibodies. (D) The results (three different experiments) were quantified and the values obtained for control flies were considered 1. (E) Kaplan Meier curve showing the overall survival rates of control (Ddc-GAL4), or flies expressing wild-type, or 84GG human GCase tested on 100 flies. Flies were grown as 10 flies per vials and were transferred to fresh food every other day. (F) Protein lysates were prepared from heads of 10 transgenic flies with the genotypes: Ddc-GAL4; wild-type GCase (cont.) or Ddc-GAL4; 84GG GCase, at days 2, 12 and 22 post-eclosion. They were subjected to western blotting and the corresponding blots were interacted with anti-TH antibody. As a loading control, the blots were interacted with anti-actin antibody. (G) Intensities of the corresponding bands were quantified by densitometry and the value obtained for control flies was considered 1. Results represent the mean ± SEM of four independent experiments. **P < 0.01.
The 84GG transgenic flies, expressing the transgene under the Ddc-GAL4 driver developed parkinsonian signs. Their survival was significantly shorter than that of wild-type flies or flies expressing the normal human GCase (Fig. 7E). Likewise, there was a significant decrease in the amount of TH in the 84GG expressing flies compared with flies expressing the normal human GCase, as tested by western blotting (Fig. 7F).
In the 84GG expressing flies UPR and parkinsonian signs cannot be attributed to retention of misfolded GCase. Therefore, ambroxol should not rescue their defective phenotype. Indeed, ambroxol did not change the tested UPR parameters: Hsc-70-3 mRNA and Xbp1 splicing (Fig. 8A) in these flies. More so, there was no change in the number of dopaminergic cells in these flies as inferred from examining the TH levels in their brains by western blotting (Fig. 8B and C), by counting the number of dopaminergic cells in the posterior region of their brain (Fig. 8D and E) and by monitoring the intensity of fluorescence emitted by TH containing cells in their brain (Fig. 8F). The results of all these experiments strongly indicated death of TH containing, dopaminergic cells. As expected, growing the 84GG transgenic flies on ambroxol containing food did not affect their defective locomotion (Fig. 8G). These results imply that there is no mature 84GG protein that ambroxol can bind and remove from the ER. Therefore, there is no alleviation of UPR in the 84GG transgenic flies and no improvement in PD-like signs by a pharmacological chaperone.
Figure 8.
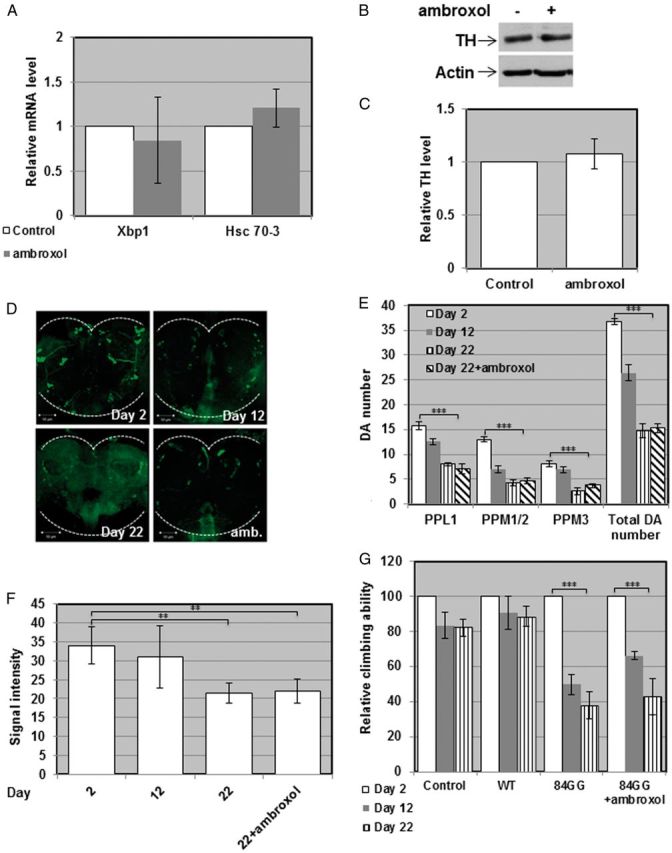
Development of parkinsonian signs in 84GG transgenic flies. (A) Transcription levels of Drosophila Hsc-70-3 or of the spliced form of Drosophila Xbp1 in ambroxol treated or untreated flies, expressing the human 84GG GBA transgene, were analyzed by quantitative RT PCR as described in the legend to Figure 5. The results (three different experiments) were quantified, and the values obtained for untreated flies were considered 1. RP49 was used as a normalizing control. (B) Protein lysates prepared from heads of ten Ddc-GAL4 flies or flies expressing the 84GG human GBA transgene, treated or untreated with 1 mM ambroxol for 22 days, were subjected to western blotting and interaction with anti-TH antibody. As a loading control, the blots were interacted with anti-actin antibody. (C) Intensities of the corresponding bands were quantified by densitometry and the value obtained for untreated flies was considered 1. Results represent the mean ± SEM of four independent experiments. (D) Representative confocal images of the posterior region of brains isolated from Ddc-GAL4 flies or flies expressing the human 84GG GBA transgene, stained with anti-TH antibody at days 2, 12, 22 post-eclosion and treatment with 1 mM ambroxol. (E) Quantitative representation of the average number of cells in dopaminergic clusters. Results represent the mean ± SEM of 15 brains. (F) Quantification of TH signal intensity obtained from tested brains at days 2, 12, 22 post-eclosion and treatment with 1 mM ambroxol. Results represent the mean ± SEM of 15 brains. (G) Five vials, each containing 10 control flies or control, or flies expressing the wild-type or the 84GG human GBA transgenes were analyzed for locomotion behavior. Climbing ability was tested at days 2, 12 and 22 post-eclosion. For each fly line, locomotion at day 2 was considered 100%. **P < .01, ***P < 0.005.
Discussion
In this study, we show that flies carrying mutations in the two Drosophila GBA genes: CG31148 and CG31414 (namely double heterozygous flies) or transgenic flies expressing the human mutant N370S, the L444P or the 84GG mutations, developed parkinsonian signs. We also show that the parkinsonian phenotype could be alleviated in flies carrying misfolded mutant GCase (the N370S or for the L444P mutant variants) by growing them in the presence of the pharmacological chaperone ambroxol. These results strongly imply that presence of mutant GCase in the ER plays a major role in development of Parkinson’s-like disease in the fly. Another group has shown that expression of the mutant human R120W or the RecNci variants in the fly eye leads to ER stress, UPR activation and degeneration of the eye (38). Both studies imply that ER stress caused by mutant GCase leads to neurodegeneration.
In this study, we also tested the effect of the 84GG GBA allele that does not express a mature GCase, on UPR activation and development of parkinsonian signs. It is of note that the mutant 84GG allele has been widely observed among PD patients (54,55). Moreover, we have documented UPR activation in skin fibroblasts derived from an 84GG-carrying individual (13). The 84GG expressing flies exhibited activation of UPR and development of parkinsonian signs. Treatment with ambroxol did not improve UPR parameters or the course of Parkinson’s-like disease. The question remains what drives this UPR activation. We hypothesize that a small, 48 amino acids peptide, translated from the 84GG mRNA, either clogs the translocons (56) or else, enters the ER with or without its signal peptide and activates one of the BiP-bound ER receptors, thus, inducing UPR. There is evidence that small molecules or peptides can activate the UPR. For example, a small peptide that binds IRE1, one of the three ER membranal receptors (57), activated the UPR (58). In another work three chemical compounds were shown to activate UPR via binding to PERK (59). Further studies are needed to solve this enigma.
It has been well documented that GD patients and carriers of GD mutations have a significantly higher propensity to develop PD in comparison to the non-GD population (14–19,22,23,48,60–67). Since carriers of GD mutations develop PD, a mutant GBA allele is a predisposing factor for the development of PD and, therefore, a dominant effect of mutant GBA gene should be considered. Dominance can result from haplo-insufficiency or from gain of function (68). Haplo-insufficiency is a situation where the single normal allele does not yield enough product, leading to development of clinical disease signs. Gain of function refers to a gene product with a deleterious cellular effect (68). Since there is no accumulation of glucosylceramide in brains of carriers of GD mutations (69), dominant deleterious effect of the mutant GBA gene product is a possible cause for development of PD in the carriers. In the present work we show that presence of endogenous mutant misfolded GCase or heterologously expressed transgenic mutant GCase variants activate the UPR and lead to development of parkinsonian signs in D. melanogaster. Such effects of mutant GCase on development of Parkinson’s-like disease has not been shown in the different mice models that exist for GD. There are several mouse models for GD, ranging from knockout to knockin models. GBA knockout mice have a complete loss of GBA expression. They accumulate the glucosylceramide substrate but do not have misfolded GCase (70–73), and therefore are not suitable as models to study the contribution of mutant GCase to the development of PD. On the other hand, none of the GBA knockin animals mimic the human phenotype. Thus, while most N370S homozygous patients present with a mild disease (5,74), mice with the same genotype die soon after birth due to a severe skin lesion (75). On the other hand, patients homozygous for the L444P mutation develop Type 3 GD, whereas the parallel animals do not accumulate glucosylceramide in their brain (76). Mouse models for GD, and mice carrying a mutant GBA allele, have been reported to develop synucleinopathies, but none of the knockout or knockin models has ever developed death of dopaminergic cells or exhibited decline of motoric skills (48,77–81). The fact that none of the GD mice models phenocopy the human disease makes it reasonable to use Drosophila models to study the development of parkinsonian signs in carriers of GD mutations. As mentioned earlier, Drosophila has proven as an excellent tool to study human neurodegenerative diseases and protein misfolding diseases in general (31,32,33) and PD in particular (40,82–86).
A growing number of publications discuss a possible contribution of factors like substrate accumulation (48,80), altered lipid metabolism or lysosomal dysfunction (87) to the development of PD. Substrate accumulation has never been recorded in carriers of GD mutations (69) or in mice heterozygous for a null or a knockin GBA mutation (48,77), nor in Type 1 GD patients. In this study, we show that GBA double heterozygous mutant flies do not accumulate the glucosylceramide substrate (Fig. 1A). A minor accumulation of GlcCer has been reported in neurons differentiated from inducible pluripotent stem cells heterozygous for GBA mutations (88).
Altered lipid metabolism has also been discussed as a possible mechanism to initiate development of PD in carriers of GD mutations (69). Concerning lysosomal dysfunction, we assume that it results from aggravated autophagy, already reported in cells carrying GD mutations (89,90). Dysregulated autophagy is a well-documented outcome of ER stress (91), which occurs in carriers of GD mutations due to ER retention of mutant GCase.
A key factor contributing to PD is α-synuclein. One of the major characteristics of PD is the presence of insoluble oligomeric and fibrillar α-synuclein-positive inclusions known as Lewy bodies and Lewy neurites in neurons in the substantia nigra pars compacta (92) (for review, see:(93)) Alpha-synuclein is a membrane associated 19 kDa protein, found in neuronal synapses (94). In brain autopsies from PD patients, who were GD patients or carriers of GBA mutations, there was accumulation of α-synuclein aggregates in the substantia nigra (21,95,96). Furthermore, mutant GCase contributes to aggregation of α-synuclein (48,77,80,95–99). In α-synuclein-KO mice, transgenic for the human A53T mutant α-synuclein and heterozygous for the L444P mutant allele of GBA there was a significant increase in α-synuclein half-life, compared with mice expressing the human A53T mutant α-synuclein only, highlighting the effect mutant GCase on α-synuclein (81).
D. melanogaster does not contain α-synuclein expressing gene. Therefore, the contribution of α-synuclein to development of PD has traditionally been studied in transgenic flies expressing either the normal or the A53T mutant α-synuclein variants. The results demonstrated that overexpression of the normal or mutant α-synuclein leads to development of Parkinson’s-like disease in the fly, manifested by death of dopaminergic cells, decreased mobility, and shorter life span (13,30,31,40,84,85,100).
To summarize our results strongly argue that presence of mutant misfolded GCase in dopaminergic cells of D. melanogaster leads to development of parkinsonian signs. This phenotype can be partially rescued by treatment with the pharmacological chaperone ambroxol, again, underscoring the role of mutant GCase in development of PD.
Materials and Methods
Fly strains and maintenance
All experiments were performed in isogenized w118 background (which was also used as a control) obtained from the Bloomington Drosophila Stock Center, Indiana University, Bloomington. Strains harboring a minos transposable element in CG31414 [Mi{ET1}CG31414] or in CG31148 [Mi{ET1}CG31148] (Nos. 23602 and 23435, respectively) were obtained from Bloomington Stock Center. Transgenic flies, harboring pUASTmycHisGCase, pUASTmycHisN370SGCase, pUASTmycHisL444PGCase on the second chromosome, or pUASTmycHis84GGGCase on the first chromosome, were established by Bestgene (Chino Hills, CA, USA). Da-GAL4 driver line (No. 55849) and Ddc-GAL4 driver line (No. 7009) were from Bloomington Stock Center. Strains were maintained on standard cornmeal-molasses medium at 25 °C.
Cell lines
RAW 264.7 mouse macrophages (ATCC, No. TIB-71) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Beit Haemek, Israel). Human GD-derived skin fibroblasts (NIGMS, No. GM1260) were grown in DMEM supplemented with 20% FBS. All cells were grown at 37 °C in the presence of 5% CO2.
Survival assay
For each fly strain, 10 vials, each containing 10 flies, were maintained on food from day one post-eclosion. Fresh food was supplied every other day and deaths were recorded. Results are presented as percentage of initial number of flies in each vial. Kaplan-Meier was used to plot survival using the graphpad prism 6 software.
Climbing assay
Five vials, each containing 10 flies were knocked to the bottom and allowed 15 s to climb 5 cm up the vial, containing standard cornmeal molasses medium. The percentage of flies passing the 5 cm line was recorded. Results are presented as percentage of all flies in the vial. Climbing ability was tested at days 2, 12 and 22 post-eclosion.
Immunofluorescence
Adult brains were dissected in PBS and fixed with 4% paraformaldehyde for 60 min. Following rinsing with PBT (1× PBS supplemented with 0.3% Triton X-100), samples were reacted with anti-TH antibodies (1:80, AB152, Millipore, Billeria, MA, USA) or with anti-myc antibody (1:100, Cell Signaling Technology, Beverly, MA, USA) diluted in BBT (1× PBS supplemented with 0.1% BSA, 0.1% Tween-20 and 250 mM NaCl) for overnight incubation at 4 °C with shaking. Following several washes with PBT, Cy2-conjugated goat anti-rabbit or Cy3-conjugated goat anti-rabbit (1:200, Jackson Immuno Research Laboratories, West Grove, PA, USA) secondary antibodies were added and incubated with shaking for 2 h at room temperature (68). After several washes with PBT the preparations were mounted in Galvanol. Slides were visualized using LSM510 Meta (ZEISS) confocal microscope. For quantitative studies, Z-projections of confocal sections (exposed and processed identically) were analyzed images of a given experiment were exposed and processed identically, unless otherwise detailed. Captured images were analyzed using ImageJ software. The ImageJ software, substrates background staining. Pixel intensity (in arbitrary units) was used to quantify fluorescence in the indicated experiments. Data was statistically evaluated using Student’s t test.
Construction of plasmids
A XbaI-SapI fragment, isolated from pcDNA4, was subcloned between XbaI and SapI restriction sites of pUAST, to create pUASTmycHis. An EcoRI-XhoI fragment, containing the 84GG mutant human GCase cDNA, was cloned in pUASTmycHis, cleaved with the same restriction enzymes, to create pUASTmycHis84GGGCase. Plasmids containing the human mutant N370S or the L444P GCase variants were described elsewhere (13).
RNA preparation
Total RNA was isolated using EZ-RNA kit (Biological Industries, Beit Haemek, Israel), according to the manufacturer's instructions. For RNA extraction form flies, adult flies were frozen in liquid nitrogen and then homogenized in TRI Reagent solution (MRC, Cincinnati, OH, USA). The extraction was performed according to the manufacturer’s recommendations.
RT- PCR
Two micrograms of RNA were reverse transcribed with M-MLV reverse transcriptase (Promega Corporation, Madison, WI, USA), using oligo-dT primer (IDT, Jerusalem, Israel) in a total volume of 20 µl, at 42 °C for 60 min. Reactions were stopped by incubation at 70 °C for 15 min. One to two microliters of the resulting cDNA were amplified by PCR or by quantitative real time PCR.
Quantitative real time PCR
One microliter of cDNA was used for quantitative real time PCR. PCR was performed using ‘power SYBR green QPCR mix reagent kit’ (Applied Biosystems, Foster City, CA, USA) by Rotor-Gene 6000. The reaction mixture contained 50% QPCR mix, 300 nM of forward primer and 300 nM of reverse primer, in a final volume of 10 µl. Thermal cycling conditions were 95 °C (10 min), and 40 cycles of 95 °C (10 s) 60 °C (20 s) and 72 °C (20 s). Relative gene expression was determined by Ct value.
SDS-PAGE and western blotting
Flies (usually 10 flies in each preparation) were homogenized in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS), containing protease inhibitors (10 µg/ml leupeptin, 10 µg/ml aprotinin and 0.1 mM phenylmethylsulfonylfluoride, all from Sigma-Aldrich, Israel). Lysates were incubated on ice for 30 min and centrifuged at 10 000g for 15 min at 4 °C. Samples, containing the same amount of protein, were electrophoresed through 10% SDS/PAGE and electroblotted onto a nitrocellulose membrane (Schleicher and Schuell BioScience, Keene, NH, USA). Membranes were blocked with 5% (w/v) non-fat dried skimmed milk powder and 0.1% Tween 20 in TBS (Tris-buffered saline; 20 mM Tris/HCl, 4 mM Tris-base, 140 mM NaCl and 1 mM EDTA) for 30 min at RT and incubated with the primary antibodies overnight at 4 °C. The membranes were then washed three times with 0.1% Tween 20 in TBS and incubated with the appropriate secondary antibodies for 1 h at room temperature. After washing, membranes were incubated with enhanced chemiluminescence detection reagent (Santa Cruz Biotechnology, Dallas, TX, USA) and analyzed using a luminescent image analyzer (ChemiDoc XRS+, Bio-Rad, Hercules, CA, USA).
Antibodies for western blotting
The following primary antibodies were used in this study: rabbit polyclonal anti-TH antibodies AB152 (Millipore, MA, USA), rabbit polyclonal anti-phospho-eIF2α (Ser51) antibodies, rabbit polyclonal anti-eIF2α antibodies (from Cell Signaling Technology, Beverly, MA, USA), and mouse monoclonal anti-actin antibody (Sigma-Aldrich, Israel).
Secondary antibodies used were: Horseradish peroxidase-conjugated goat anti-mouse antibodies and Horseradish peroxidase-conjugated goat anti-rabbit antibodies (both from Jackson Immuno Research Laboratories, West Grove, PA, USA).
Ambroxol treatment
Ambroxol (Sigma Aldrich, Rehovot, Israel) was added into 10 ml food containing vials, to a final concentration of 1 mM.
Separation of sphingolipids on TLC
Flies were lysed in 300 µl of distilled water. Protein amounts were determined and 900µl chloroform:methanol (2:1) were added. After centrifugation, the lower phase was isolated according to the Folch protocol (101) and dried. Twenty µl of chloroform:methanol (2:1) were added and samples containing 100 μg of protein were separated by TLC (Silica gel 60A; Sigma-Aldrich, St Louis, MO, USA) in CHCl3: MeOH: ammonium hydroxide (65:25:4, by vol.). The TLC plates were developed with Primuline reagent (Sigma-Aldrich, St Louis, MO, USA) and quantified by ChemiDoc XRS (Bio-Rad laboratories, GmbH, Munich).
GCase activity assay
Frozen flies were lysed in McIlvaine’s buffer (0.1 M citric acid, pH 4.2, 0.2 M Na2HPO0, 29:21, vol:vol) and protein concentrations were estimated by the Bio-Rad protein assay (Bio-Rad laboratories, GmbH, Munich). Tissue homogenates containing 100 µg of protein were incubated at 37 °C with 8 µM N-[6-[(7-Nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-Glucosylceramide (C6-NBD-GlcCer) (Avanti Polar Lipids, Alabaster, AL, USA) in a final volume of 50 µl McIlvaine’s buffer for 1 h. Reactions were terminated by addition of three volumes of chloroform:methanol (2:1). Lipids were extracted and the lower phase was separated by TLC as described under ‘Total lipid extraction and loading experimnts’. N-[6-[(7-Nitro-2-1,3-benzoxadiazol-4-yl)amino]caproyl]-Ceramide (C6-NBD-Cer) (Matreya LLC, State college, PA, USA) was identified with an authentic standard using Amersham imager 600 (Sucursal EM Portugal).
CBE treatment
Cells were treated with 200 μM CBE (Sigma-Aldrich, Rehovot, Israel) for 10 days.
Endonuclease H sensitivity
Endo-H sensitivity was tested essentially as described elsewhere (13).
Primers
For Hsc 70-3: F 5′-GCTGGTGTTATTGCCGGTCTGC-3′
R 5′-GATGCCTCGGGATGGTTCCTTGC-3′
For Xbp1 spliced form: F 5′-CCGAACTGAAGCAGCAACAGC-3′
R 5′-GTATACCCTGCGGCAGATCC-3′
For RP49: F 5′-TAAGAAGCGCACAAAGCACT-3′
R 5′-GGGCATCAGATATTGTCCCT-3′
For human 84GG GBA allele: F 5′-CATCCGGGTACCCATGGCCAGCTGTG-3′
R 5′-TCGATCCCAGGAGCCTAGCCGCAGAC-3′
Acknowledgements
Gali Maor is a recipient of a fellowship from the Buchmann Scholarship Fund. This work was supported by research grants from the Israel Science Foundation (1300/13), from Pfizer and from the NNE Research Program administered by TEVA (to M.H.).
Conflict of Interest Statement. None declared.
Funding
Gali Maor is a recipient of a fellowship from the Buchmann Scholarship Fund. This work was supported by research grants from the Israel Science Foundation (grant number 1300/13), from Pfizer and from the NNE Research Program administered by TEVA (to MH).
References
- 1.Beutler E. (1980) Gaucher’s disease. Compr Ther, 6, 65–68. [PubMed] [Google Scholar]
- 2.Beutler E. (1999) Gaucher disease. Arch. Intern. Med., 159, 881–882. [DOI] [PubMed] [Google Scholar]
- 3.Brady R.O., Kanfer J.N., Shapiro D. (1965) Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher's Disease. Biochem. Biophys. Res. Commun., 18, 221–225. [DOI] [PubMed] [Google Scholar]
- 4.Hruska K.S., LaMarca M.E., Scott C.R., Sidransky E. (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat., 29, 567–583. [DOI] [PubMed] [Google Scholar]
- 5.Tsuji S., Martin B.M., Barranger J.A., Stubblefield B.K., LaMarca M.E., Ginns E.I. (1988) Genetic heterogeneity in type 1 Gaucher disease: multiple genotypes in Ashkenazic and non-Ashkenazic individuals. Proc. Natl. Acad. Sci. U S A, 85, 2349–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuji S., Choudary P.V., Martin B.M., Stubblefield B.K., Mayor J.A., Barranger J.A., Ginns E.I. (1987) A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N. Engl. J. Med., 316, 570–575. [DOI] [PubMed] [Google Scholar]
- 7.Beutler E., Gelbart T., Kuhl W., Sorge J., West C. (1991) Identification of the second common Jewish Gaucher disease mutation makes possible population-based screening for the heterozygous state. Proc. Natl. Acad. Sci. U S A, 88, 10544–10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benyair R., Ron E., Lederkremer G.Z. (2011) Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell. Mol. Biol., 292, 197–280. [DOI] [PubMed] [Google Scholar]
- 9.Beutler E., Kuhl W. (1986) Glucocerebrosidase processing in normal fibroblasts and in fibroblasts from patients with type I, type II, and type III Gaucher disease. Proc. Natl. Acad. Sci. U S A, 83, 7472–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson A.H., Ginns E.I., Barranger J.A. (1985) Biosynthesis of the lysosomal enzyme glucocerebrosidase. J. Biol. Chem., 260, 14319–14324. [PubMed] [Google Scholar]
- 11.Jonsson L.M., Murray G.J., Sorrell S.H., Strijland A., Aerts J.F., Ginns E.I., Barranger J.A., Tager J.M., Schram A.W. (1987) Biosynthesis and maturation of glucocerebrosidase in Gaucher fibroblasts. Eur. J. Biochem., 164, 171–179. [DOI] [PubMed] [Google Scholar]
- 12.Ron I., Horowitz M. (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet., 14, 2387–2398. [DOI] [PubMed] [Google Scholar]
- 13.Maor G., Rencus-Lazar S., Filocamo M., Steller H., Segal D., Horowitz M. (2013) Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet. J. Rare Dis., 8, 140.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neudorfer O., Giladi N., Elstein D., Abrahamov A., Turezkite T., Aghai E., Reches A., Bembi B., Zimran A. (1996) Occurrence of Parkinson's syndrome in type I Gaucher disease. Qjm, 89, 691–694. [DOI] [PubMed] [Google Scholar]
- 15.Tayebi N., Callahan M., Madike V., Stubblefield B.K., Orvisky E., Krasnewich D., Fillano J.J., Sidransky E. (2001) Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol. Genet. Metab., 73, 313–321. [DOI] [PubMed] [Google Scholar]
- 16.Bembi B., Zambito Marsala S., Sidransky E., Ciana G., Carrozzi M., Zorzon M., Martini C., Gioulis M., Pittis M.G., Capus L. (2003) Gaucher’s disease with Parkinson disease: clinical and pathological aspects. Neurology, 61, 99–101. [DOI] [PubMed] [Google Scholar]
- 17.Tayebi N., Walker J., Stubblefield B., Orvisky E., LaMarca M.E., Wong K., Rosenbaum H., Schiffmann R., Bembi B., Sidransky E. (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab., 79, 104–109. [DOI] [PubMed] [Google Scholar]
- 18.Varkonyi J., Rosenbaum H., Baumann N., MacKenzie J.J., Simon Z., Aharon-Peretz J., Walker J.M., Tayebi N., Sidransky E. (2003) Gaucher disease associated with parkinsonism: four further case reports. Am. J. Med. Genet. A, 116A, 348–351. [DOI] [PubMed] [Google Scholar]
- 19.Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. (2004) Mutations in the glucocerebrosidase gene and Parkinson disease in Ashkenazi Jews. N. Engl. J. Med., 351, 1972–1977. [DOI] [PubMed] [Google Scholar]
- 20.Sidransky E. (2005) Gaucher disease and parkinsonism. Mol. Genet. Metab., 84, 302–304. [DOI] [PubMed] [Google Scholar]
- 21.Hruska K.S., Goker-Alpan O., Sidransky E. (2006) Gaucher disease and the synucleinopathies. J. Biomed. Biotechnol., 2006, 78549.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan E.K., Tong J., Fook-Chong S., Yih Y., Wong M.C., Pavanni R., Zhao Y. (2007) Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch. Neurol., 64, 1056–1058. [DOI] [PubMed] [Google Scholar]
- 23.Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A, et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson disease. N. Engl. J. Med., 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shulman J.M., De Jager P.L., Feany M.B. (2011) Parkinson disease: genetics and pathogenesis. Annu. Rev. Pathol., 6, 193–222. [DOI] [PubMed] [Google Scholar]
- 25.Wang M., Ye R., Barron E., Baumeister P., Mao C., Luo S., Fu Y., Luo B., Dubeau L., Hinton D.R, et al. (2010) Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ., 17, 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stutzbach L.D., Xie S.X., Naj A.C., Albin R., Gilman S., Lee V.M., Trojanowski J.Q., Devlin B., Schellenberg G.D. (2013) The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol. Commun., 1, 31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forman M.S., Lee V.M., Trojanowski J.Q. (2003) ‘ Unfolding’ pathways in neurodegenerative disease. Trends Neurosci., 26, 407–410. [DOI] [PubMed] [Google Scholar]
- 28.Mercado G., Valdes P., Hetz C. (2013) An ERcentric view of Parkinson disease. Trends Mol. Med., 19, 165–175. [DOI] [PubMed] [Google Scholar]
- 29.Varma D., Sen D. (2015) Role of the unfolded protein response in the pathogenesis of Parkinson disease. Acta Neurobiol. Exp. (Wars), 75, 1–26. [PubMed] [Google Scholar]
- 30.Lu B., Vogel H. (2009) Drosophila models of neurodegenerative diseases. Annu. Rev. Pathol., 4, 315–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitworth A.J. (2011) Drosophila models of Parkinson disease. Adv. Genet., 73, 1–50. [DOI] [PubMed] [Google Scholar]
- 32.West R.J., Furmston R., Williams C.A., Elliott C.J. (2015) Neurophysiology of Drosophila models of Parkinson disease. Parkinsons Dis., 2015, 381281.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rincon-Limas D.E., Jensen K., Fernandez-Funez P. (2012) Drosophila models of proteinopathies: the little fly that could. Curr. Pharm. Des., 18, 1108–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaminker J.S., Bergman C.M., Kronmiller B., Carlson J., Svirskas R., Patel S., Frise E., Wheeler D.A., Lewis S.E., Rubin G.M, et al. (2002) The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol., 3, RESEARCH0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gokcezade J., Sienski G., Duchek P. (2014) Efficient CRISPR/Cas9 plasmids for rapid and versatile genome editing in Drosophila. G3 (Bethesda), 4, 2279–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bassett A.R., Liu J.L. (2014) CRISPR/Cas9 and genome editing in Drosophila. J. Genet. Genomics, 41, 7–19. [DOI] [PubMed] [Google Scholar]
- 37.Duffy J.B. (2002) GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis, 34, 1–15. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T., Shimoda M., Ito K., Hanai S., Aizawa H., Kato T., Kawasaki K., Yamaguchi T., Ryoo H.D., Goto-Inoue N, et al. (2013) Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PLoS One, 8, e69147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao Z., Davis R.L. (2009) Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: anatomical and physiological heterogeneity. Front. Neural Circuits, 3, 5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feany M.B., Bender W.W. (2000) A Drosophila model of Parkinson disease. Nature, 404, 394–398. [DOI] [PubMed] [Google Scholar]
- 41.Sawkar A.R., Cheng W.C., Beutler E., Wong C.H., Balch W.E., Kelly J.W. (2002) Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. U S A, 99, 15428–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bendikov-Bar I., Maor G., Filocamo M., Horowitz M. (2013) Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis., 50, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimran A., Altarescu G., Elstein D. (2013) Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis., 50, 134–137. [DOI] [PubMed] [Google Scholar]
- 44.McNeill A., Magalhaes J., Shen C., Chau K.Y., Hughes D., Mehta A., Foltynie T., Cooper J.M., Abramov A.Y., Gegg M, et al. (2014) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain, 137, 1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mu T.W., Ong D.S., Wang Y.J., Balch W.E., Yates J.R., 3rd, Segatori L., Kelly J.W. (2008) Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell, 134, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araki K., Nagata K. (2011) Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol., 3, a007526.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalia S.K., Kalia L.V., McLean P.J. (2010) Molecular chaperones as rational drug targets for Parkinson disease therapeutics. CNS Neurol. Disord. Drug Targets, 9, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cullen V., Sardi S.P., Ng J., Xu Y.H., Sun Y., Tomlinson J.J., Kolodziej P., Kahn I., Saftig P., Woulfe J, et al. (2011) Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol., 69, 940–953. [DOI] [PubMed] [Google Scholar]
- 49.Cortez L., Sim V. (2014) The therapeutic potential of chemical chaperones in protein folding diseases. Prion, 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babajani G., Tropak M.B., Mahuran D.J., Kermode A.R. (2012) Pharmacological chaperones facilitate the post-ER transport of recombinant N370S mutant beta-glucocerebrosidase in plant cells: evidence that N370S is a folding mutant. Mol. Genet. Metab., 106, 323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maegawa G.H., Tropak M.B., Buttner J.D., Rigat B.A., Fuller M., Pandit D., Tang L., Kornhaber G.J., Hamuro Y., Clarke J.T, et al. (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem., 284, 23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sorge J.A., West C., Kuhl W., Treger L., Beutler E. (1987) The human glucocerebrosidase gene has two functional ATG initiator codons. Am. J. Hum. Genet., 41, 1016–1024. [PMC free article] [PubMed] [Google Scholar]
- 53.Pasmanik-Chor M., Elroy-Stein O., Aerts H., Agmon V., Gatt S., Horowitz M. (1996) Overexpression of human glucocerebrosidase containing different-sized leaders. Biochem. J., 317, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lesage S., Anheim M., Condroyer C., Pollak P., Durif F., Dupuits C., Viallet F., Lohmann E., Corvol J.C., Honore A, et al. (2011) Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson disease. Hum. Mol. Genet., 20, 202–210. [DOI] [PubMed] [Google Scholar]
- 55.Gan-Or Z., Giladi N., Rozovski U., Shifrin C., Rosner S., Gurevich T., Bar-Shira A., Orr-Urtreger A. (2008) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology, 70, 2277–2283. [DOI] [PubMed] [Google Scholar]
- 56.Ast T., Michaelis S., Schuldiner M. (2016) The Protease Ste24 Clears Clogged Translocons. Cell, 164, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Welihinda A.A., Tirasophon W., Kaufman R.J. (1999) The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr., 7, 293–300. [PMC free article] [PubMed] [Google Scholar]
- 58.Mendez A.S., Alfaro J., Morales-Soto M.A., Dar A.C., McCullagh E., Gotthardt K., Li H., Acosta-Alvear D., Sidrauski C., Korennykh A.V, et al. (2015) Endoplasmic reticulum stress-independent activation of unfolded protein response kinases by a small molecule ATP-mimic. Elife, 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie W., Pariollaud M., Wixted W.E., Chitnis N., Fornwald J., Truong M., Pao C., Liu Y., Ames R.S., Callahan J, et al. (2015) Identification and characterization of PERK activators by phenotypic screening and their effects on NRF2 activation. PLoS One, 10, e0119738.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lwin A., Orvisky E., Goker-Alpan O., LaMarca M.E., Sidransky E. (2004) Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab., 81, 70–73. [DOI] [PubMed] [Google Scholar]
- 61.Aharon-Peretz J., Badarny S., Rosenbaum H., Gershoni-Baruch R. (2005) Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype-genotype correlation. Neurology, 65, 1460–1461. [DOI] [PubMed] [Google Scholar]
- 62.Sato C., Morgan A., Lang A.E., Salehi-Rad S., Kawarai T., Meng Y., Ray P.N., Farrer L.A., St George-Hyslop P., Rogaeva E. (2005) Analysis of the glucocerebrosidase gene in Parkinson disease. Mov. Disord., 20, 367–370. [DOI] [PubMed] [Google Scholar]
- 63.Halperin A., Elstein D., Zimran A. (2006) Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol. Dis., 36, 426–428. [DOI] [PubMed] [Google Scholar]
- 64.Wu Y.R., Chen C.M., Chao C.Y., Ro L.S., Lyu R.K., Chang K.H., Lee-Chen G.J. (2007) Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J. Neurol. Neurosurg. Psychiatry, 78, 977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitsui J., Mizuta I., Toyoda A., Ashida R., Takahashi Y., Goto J., Fukuda Y., Date H., Iwata A., Yamamoto M, et al. (2009) Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch. Neurol., 66, 571–576. [DOI] [PubMed] [Google Scholar]
- 66.Rosenbloom B., Balwani M., Bronstein J.M., Kolodny E., Sathe S., Gwosdow A.R., Taylor J.S., Cole J.A., Zimran A., Weinreb N.J. (2011) The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol. Dis., 46, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beavan M.S., Schapira A.H. (2013) Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann. Med., 45, 511–521. [DOI] [PubMed] [Google Scholar]
- 68.Griffiths A., Miller J., Suzuki D., Lewontin R, Gelbart W. (2000) An Introduction to Genetic Analysis, W. H. Freeman, New York. [Google Scholar]
- 69.Gegg M.E., Sweet L., Wang B.H., Shihabuddin L.S., Sardi S.P., Schapira A.H. (2015) No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord., 30, 1085–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tybulewicz V.L., Tremblay M.L., LaMarca M.E., Willemsen R., Stubblefield B.K., Winfield S., Zablocka B., Sidransky E., Martin B.M., Huang S.P, et al. (1992) Animal model of Gaucher's disease from targeted disruption of the mouse glucocerebrosidase gene. Nature, 357, 407–410. [DOI] [PubMed] [Google Scholar]
- 71.Enquist I.B., Nilsson E., Ooka A., Mansson J.E., Olsson K., Ehinger M., Brady R.O., Richter J., Karlsson S. (2006) Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. U S A, 103, 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mistry P.K., Liu J., Yang M., Nottoli T., McGrath J., Jain D., Zhang K., Keutzer J., Chuang W.L., Mehal W.Z, et al. (2010) Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. U S A, 107, 19473–19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu Y.H., Reboulet R., Quinn B., Huelsken J., Witte D., Grabowski G.A. (2008) Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol. Genet. Metab., 94, 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beutler E., Nguyen N.J., Henneberger M.W., Smolec J.M., McPherson R.A., West C., Gelbart T. (1993) Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am. J. Hum. Genet., 52, 85–88. [PMC free article] [PubMed] [Google Scholar]
- 75.Xu Y.H., Quinn B., Witte D., Grabowski G.A. (2003) Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am. J. Pathol., 163, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizukami H., Mi Y., Wada R., Kono M., Yamashita T., Liu Y., Werth N., Sandhoff R., Sandhoff K., Proia R.L. (2002) Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Invest., 109, 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sardi S.P., Singh P., Cheng S.H., Shihabuddin L.S., Schlossmacher M.G. (2012) Mutant GBA1 expression and synucleinopathy risk: first insights from cellular and mouse models. Neurodegener. Dis., 10, 195–202. [DOI] [PubMed] [Google Scholar]
- 78.Xu Y.H., Sun Y., Ran H., Quinn B., Witte D., Grabowski G.A. (2011) Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab., 102, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sardi S.P., Clarke J., Kinnecom C., Tamsett T.J., Li L., Stanek L.M., Passini M.A., Grabowski G.A., Schlossmacher M.G., Sidman R.L, et al. (2011) CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U S A, 108, 12101–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mazzulli J.R., Xu Y.H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A., Krainc D. (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 146, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fishbein I., Kuo Y.M., Giasson B.I., Nussbaum R.L. (2014) Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain, 137, 3235–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature, 441, 1162–1166. [DOI] [PubMed] [Google Scholar]
- 83.Ng C.H., Mok S.Z., Koh C., Ouyang X., Fivaz M.L., Tan E.K., Dawson V.L., Dawson T.M., Yu F., Lim K.L. (2009) Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci., 29, 11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pesah Y., Pham T., Burgess H., Middlebrooks B., Verstreken P., Zhou Y., Harding M., Bellen H., Mardon G. (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development, 131, 2183–2194. [DOI] [PubMed] [Google Scholar]
- 85.Moore D.J., Dawson V.L., Dawson T.M. (2006) Lessons from Drosophila models of DJ-1 deficiency. Sci. Aging Knowledge Environ., 2006, pe2.. [DOI] [PubMed] [Google Scholar]
- 86.Bonini N.M. (2002) Chaperoning brain degeneration. Proc. Natl. Acad. Sci. U S A, 99(suppl. 4), 16407–16411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McNeill A. (2015) Lysosomal dysfunction in Parkinson disease. Brain, 138, e339.. [DOI] [PubMed] [Google Scholar]
- 88.Schondorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B., Sardi S.P., Valsecchi M., Hoffmann S., Schwarz L.K, et al. (2014) iPSC-derived neurons from GBA1-associated Parkinson disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun., 5, 4028.. [DOI] [PubMed] [Google Scholar]
- 89.Awad O., Sarkar C., Panicker L.M., Miller D., Zeng X., Sgambato J.A., Lipinski M.M., Feldman R.A. (2015) Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet., 24, 5775–5788. [DOI] [PubMed] [Google Scholar]
- 90.Du T.T., Wang L., Duan C.L., Lu L.L., Zhang J.L., Gao G., Qiu X.B., Wang X.M., Yang H. (2015) GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy, 11, 1803–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matus S., Lisbona F., Torres M., Leon C., Thielen P., Hetz C. (2008) The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr. Mol. Med., 8, 157–172. [DOI] [PubMed] [Google Scholar]
- 92.Jenner P., Olanow C.W. (1998) Understanding cell death in Parkinson disease. Ann. Neurol., 44, S72–S84. [DOI] [PubMed] [Google Scholar]
- 93.Puschmann A., Bhidayasiri R., Weiner W.J. (2012) Synucleinopathies from bench to bedside. Parkinsonism Relat. Disord., 18(Suppl. 1), S24–S27. [DOI] [PubMed] [Google Scholar]
- 94.Gasser T. (1998) Genetics of Parkinson disease. Ann. Neurol., 44, S53–S57. [DOI] [PubMed] [Google Scholar]
- 95.Choi J.H., Stubblefield B., Cookson M.R., Goldin E., Velayati A., Tayebi N., Sidransky E. (2011) Aggregation of alpha-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab., 104, 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goker-Alpan O., Stubblefield B.K., Giasson B.I., Sidransky E. (2010) Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol., 120, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clark L.N., Kartsaklis L.A., Wolf Gilbert R., Dorado B., Ross B.M., Kisselev S., Verbitsky M., Mejia-Santana H., Cote L.J., Andrews H, et al. (2009) Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch. Neurol., 66, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Manning-Bog A.B., Schule B., Langston J.W. (2009) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology, 30, 1127–1132. [DOI] [PubMed] [Google Scholar]
- 99.Velayati A., Yu W.H., Sidransky E. (2010) The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr. Neurol. Neurosci. Rep., 10, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Z., Wang X., Yu Y., Li X., Wang T., Jiang H., Ren Q., Jiao Y., Sawa A., Moran T, et al. (2008) A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. U S A, 105, 2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Folch J., Lees M., Sloane Stanley G.H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem., 226, 497–509. [PubMed] [Google Scholar]