

Abstract
Urinary chemistries vary widely in both health and disease and are affected by diet, volume status, medications, and disease states. When properly examined, these tests provide important insight into the mechanism and therapy of various clinical disorders that are first detected by abnormalities in plasma chemistries. These tests cannot be interpreted in isolation, but instead require knowledge of key clinical information, such as medications, physical examination, and plasma chemistries, to include kidney function. When used appropriately and with knowledge of limitations, urine chemistries can provide important insight into the pathophysiology and treatment of a wide variety of disorders.
Keywords: Urologic Diseases, Physical Examination, Urinary Tract Physiological Phenomena, Diagnostic Tests, Routine, Diet
Introduction
Urine chemistries can provide valuable insight into a wide range of clinical conditions. These tests are often underutilized because of the difficulty many physicians find in their interpretation. Whereas a basic metabolic profile obtained from a blood sample has well defined normal values, there are no such values for urine chemistries. Urinary excretion of electrolytes vary widely as the kidney adjusts the rate of excretion to match dietary intake and endogenous production. The excretion of a dilute or concentrated urine or administration of a drug can markedly alter the concentration of urine electrolytes, potentially misleading the clinician as to the absolute quantity in the urine over a given amount of time. This review will be limited in scope and will concentrate on discussing the clinical use and interpretation of urine chemistries to include sodium, potassium, chloride, pH, creatinine, urea, and osmolality. Urine chemistries are best interpreted with knowledge of the clinical setting at the time they are obtained. These tests can be diagnostic and offer mechanistic insight in the evaluation of AKI, volume status, disorders of plasma sodium and potassium, and acid-base disorders.
Urine Sodium
Assessment of Effective Circulating Volume
Under normal conditions, sodium excretion by the kidney equals dietary intake minus the small amount lost in sweat and feces, and typically ranges from 40 to 220 mEq/d. The ability to match dietary intake with excretion enables extracellular fluid volume to be maintained within a narrow range. When effective circulatory volume is reduced, activation of effector mechanisms such as sympathetic nerves and the renin-angiotensin-aldosterone system causes avid sodium retention in the kidneys, lowering urine sodium concentration to values <15 mEq/L. On the other hand, volume expansion suppresses effector mechanisms and stimulates release of atrial natriuretic peptide, leading to a reduction in sodium reabsorption, causing urinary sodium concentration to be high. Thus, the urine sodium concentration is an indirect measure of volume status and reflects the integrity of the kidney to regulate that status.
There are specific circumstances where measurement of urine sodium no longer accurately reflects volume status. The urine sodium concentration is dependent on the amount of free water in the urine and is therefore influenced by the rate of water reabsorption in the kidneys. Under conditions of a water diuresis, the urine sodium concentration may be reduced even though daily excretion is high, falsely suggesting the presence of a low volume state. Similarly, a concentrated urine can increase urine sodium concentration even though the total amount of sodium is low, potentially masking the presence of volume contraction and falsely suggesting euvolemia or volume expansion. The fractional excretion of sodium (FENa) accounts for the effect of water reabsorption in the kidney on urine sodium concentration:
This formula expresses the percentage of filtered sodium excreted in the urine and provides a measure of sodium handling that is independent of urinary concentration.
Differentiation of Prerenal Azotemia versus Acute Tubular Necrosis
Measurement of urine sodium concentration and FENa are frequently utilized to determine whether AKI is prerenal and correctable by restoring intravascular volume, or whether it is secondary to acute tubular necrosis, where administration of fluids might be harmful by causing volume overload (1). A random urine sodium concentration of <15 mEq/L and a FENa of <1% suggest the presence of a volume-responsive component to a reduced GFR. In patients with established acute tubular necrosis, loss of tubular function will prevent maximal sodium retention even when extracellular fluid volume depletion is present. These tests remain useful when evaluating a change in kidney function in patients with previously stable CKD; however, the response to decreased kidney perfusion is delayed and the reduction in urine sodium will be less maximal as compared with normal kidneys (2) (Supplemental Material, Case 1).
There are situations where the urine sodium concentration and FENa inadequately distinguish between azotemia due to kidney parenchymal injury from volume responsive azotemia (3). These conditions are characterized by vasoconstriction in the kidney causing a reduction in GFR and filtered load of sodium in the setting where tubular function remains relatively intact (4–12) (Figure 1). The biochemical profile in the urine can change in a time dependent manner from a prerenal picture to that of acute tubular necrosis. Variability in timing of measurements likely explains disparities in the literature as to sensitivity and specificity of these tests in patients with AKI (13–15). The sensitivity and specificity of the FENa is greatest when applied to patients with oliguria and a reduced GFR (discussed further in Supplemental Material, Cases 1 and 2).
Figure 1.

Causes of AKI where urine sodium concentration and fractional excretion of sodium may be initially reduced, only to later increase. In these conditions, the GFR is reduced because of activation of systemic and/or intrakidney vasoconstrictors causing a reduction in filtered load of sodium at a time when tubular function is relatively intact. As the insult persists, tubular injury becomes more widespread, resulting in an increase in urine sodium concentration and fractional excretion of sodium.
In patients with severe congestive heart failure, advanced cirrhosis of the liver, and extensive burns, a reduction in the filtered load of sodium brought about by intense neurohumoral activation can cause sodium retention in the kidneys to be of such a degree that urine sodium concentration and FENa remain low even when tubular necrosis is present, as manifested by granular casts and tubular cells in the urine (16–19). Despite volume expansion, a low urinary sodium concentration is typically present early in acute GN when tubular function is intact and the filtered load of sodium is reduced because of the decrease in glomerular surface area available for filtration (11).
Active diuretic use is another situation where the urine sodium concentration and FENa may not accurately reflect effective circulatory volume. In congestive heart failure and cirrhosis, the urine sodium concentration and the FENa may be increased because of administration of diuretics and yet effective circulatory volume is reduced (17). In such patients, calculation of the fractional excretion of urea (FEUrea) can be helpful:
Under conditions of volume depletion, proximal reabsorption of water and urea increases in association with decreased kidney perfusion and increased filtration fraction. Thiazide and loop diuretics act distal to the proximal tubule and therefore leave urea reabsorption unaffected. As a result, the FEUrea is reduced even though urine sodium concentration and FENa are increased. An FEUrea <35% suggests low effective volume (20,21). A low FEUrea is also present in adrenal insufficiency where impaired distal reabsorption of sodium due to mineralocorticoid deficiency leads to salt wasting and decreased effective circulatory volume (Supplemental Material, Case 2).
The utility of the FEUrea to assess effective circulatory volume is lost when proximal reabsorption of salt and water is impaired (22). This situation occurs after either administration of acetazolamide, an osmotic diuresis due to administration of mannitol, glycosuria as in uncontrolled diabetes, or increased urea excretion resulting from high protein intake or catabolism. Proximal tubule salt reabsorption may also be impaired in patients with cerebral salt wasting (23).
The FEUrea has also been used to discriminate between volume responsive azotemia and tubular necrosis in the absence of diuretic use. How this test compares to the FENa for this purpose is difficult to determine because studies included patients who were highly selected and excluded those with interstitial nephritis, GN, urinary obstruction, and exposure to radiocontrast material (19,20). Another potential limitation of the FEUrea is its use in elderly patients and those with sepsis. Studies in experimental models show downregulation of urea transporters in the nephron with endotoxemia and aging. This effect would tend to increase the FEUrea in septic and elderly patients even when volume depletion was the only cause of azotemia (24,25).
Conditions in which volume contraction coexists with a nonreabsorbable anion also cause urine sodium concentration and FENa to be increased. As discussed below, urinary chloride concentration is typically low in this setting.
Urinary Chloride
Urinary excretion of chloride mirrors sodium excretion in response to dietary intake. In a manner similar to sodium, the urinary concentration of chloride and fractional excretion of chloride (FECL) can be used as indirect markers of effective circulatory volume. Despite these similarities, there are situations when the directional change in urinary concentration may differ, such that reliance solely on one or the other can lead to an erroneous assessment of effective volume status. These differences are particularly evident when volume disturbances are accompanied by acid-base disorders (26). For this reason, both urine sodium and chloride should be obtained when using urine electrolytes to assess volume or acid-base status.
A high urine sodium and low chloride concentration in the setting of volume depletion suggests the presence of a nonreabsorbable anion. The nature of the anion can be distinguished using the urine pH and clinical context. A urine pH of 7 or 8 indicates significant bicarbonaturia, as with active vomiting or nasogastric suction (27–29). The extrarenal generation of metabolic alkalosis leads to excretion of bicarbonate into the urine, which obligates a component of filtered sodium to accompany the base whereas urine chloride remains low in response to neurohumoral activation resulting from volume contraction. A urine sodium-to-chloride ratio of >1.6 is typical in such patients (26). A urine pH <6 suggests another nonreabsorbable anion is responsible, such as ketoanions, or drugs such as ticarcillin disodium clavulanate, piperacillin tazobactam, or carbenicillin disodium. When given in the setting of low effective volume, these antibiotics couple increased delivery of sodium to increased aldosterone levels in the distal nephron, leading to generation of metabolic alkalosis of a kidney origin, accounting for the low urine pH (Figure 2). In metabolic alkalosis, the urine chloride concentration can be used as a marker as to whether the alkalosis is responsive or resistant to administration of a chloride-containing solution (30,31). A low urine chloride indicates a chloride responsive metabolic alkalosis such as vomiting, chloride wasting diarrhea, remote use of diuretics, and posthypercapneic metabolic alkalosis. By contrast, a high urine chloride concentration indicates a chloride resistant alkalosis, as seen in genetic disorders such as Bartter and Gitelman syndrome and hypertensive disorders caused by increased mineralocorticoids or mineralocorticoid-like effect.
Figure 2.

Urine chemistry profile in metabolic alkalosis. (A) Loop and thiazide diuretics and their genetic equivalent (Bartter and Gitelman syndrome, respectively) cause contraction of effective circulatory volume and activation of the renin-angiotensin-aldosterone axis. Increased distal delivery of sodium coupled with increased mineralocorticoid levels causes increased potassium secretion and an increased rate of hydrogen ion secretion in the distal nephron, leading to the development of hypokalemic metabolic alkalosis (27). The fractional excretion (FE) of urea is reduced to <35% because proximal reabsorption of urea is stimulated and unaffected by the downstream impairment in sodium chloride transport. Metabolic alkalosis due to remote use of diuretics is sensitive to chloride-containing solutions but resistant in genetic disorders. (B) Antibiotics such as carbenicillin, ticarcillin, and piperacillin given in the setting of decreased effective volume act as nonreabsorbable anions, causing increased delivery of sodium to the distal nephron, resulting in development of hypokalemia and a chloride-responsive from of metabolic alkalosis. The urine pH (UpH) in this setting is acidic because of augmented hydrogen ion secretion. (C) Active vomiting or nasogastric suction generates a chloride-sensitive form of metabolic alkalosis. Bicarbonate acts as a nonreabsorbable anion, causing increased distal sodium delivery and development of potassium wasting (29). (D) A primary increase in mineralocorticoid levels (Conn syndrome) or effect (Liddle syndrome) leads to a chloride resistant form of metabolic alkalosis accompanied by kidney potassium wasting and hypertension. Increased distal delivery of sodium is due to inhibition of proximal reabsorption brought about by effective volume expansion.
A high urine chloride and low sodium concentration in the setting of volume depletion suggests the presence of another cation in the urine. As discussed below, this situation most commonly occurs in diarrhea where development of hypokalemia and metabolic acidosis lead to high rates of ammonium excretion, obligating the excretion of chloride despite depletion of intravascular volume (Figure 3). A urine sodium-to-chloride ratio of <0.76 is typical in such patients (26).
Figure 3.

The urine chloride concentration can be used to distinguish between a chloride responsive and resistant metabolic alkalosis. In a normal gap metabolic acidosis due to diarrhea, a high urine chloride is the result of increased excretion of NH4Cl. Urine chloride is high in kidney tubular acidosis due to acidosis-induced decreased reabsorption of NaCl in the proximal tubule. In the indicated causes of anion gap metabolic acidosis, a high urine chloride is the result of increased excretion of NH4Cl. In these settings, the urine sodium is typically higher than the chloride due to the excretion of sodium and potassium acid salts.
Urine Potassium
Urinary potassium excretion by the kidney can vary from 10–15 mEq/d to as high as 400 mEq/d, depending on dietary intake. Determining the urine potassium concentration in a patient with dyskalemia can help determine whether the kidney is responding appropriately or is responsible for the disorder. A random urine potassium concentration of 5–15 mEq/L is consistent with an extrarenal etiology of hypokalemia, whereas values >40 mEq/L favor a kidney cause of the disorder. These cutoffs to include intermediate values require the clinical context and sometimes the response to therapy for proper interpretation. In general, hypokalemia due to a nonkidney disorder will be more easily corrected, assuming the underlying disturbance is no longer present, whereas ongoing losses in the urine make correction of hypokalemia more difficult. As with urine sodium concentration, a limitation of a random value is the degree of urinary concentration. A urine potassium concentration of 40 mEq/L may be an appropriate response in a patient with hypokalemia with a maximally concentrated urine due to decreased water intake or if obtained in the setting of decreased effective volume. Although decreased volume stimulates aldosterone production, the absolute amount of potassium in the urine remains relatively low because of decreased delivery of sodium and water to the distal nephron. By the same token, a random urine value of <15 mEq/L may represent potassium wasting by the kidneys if obtained in the setting of a water diuresis.
The transtubular potassium gradient (TTKG) is a method designed to overcome the limitations of a random urine potassium concentration in the evaluation of a patient with dyskalemia:
The formula estimates the ratio of potassium in the lumen of the cortical collecting duct to that in the peritubular capillaries at a point where tubular fluid is isotonic relative to plasma (32,33). In a patient with hypokalemia, a value of <3 is consistent with an appropriate kidney response to the disorder whereas a value >7 indicates potassium wasting by the kidneys. Mineralocorticoid activity and TTKG correlate positively, and in a patient with hyperkalemia, a value of <6 generally indicates an inappropriate response by the collecting duct, although studies vary as to the precise cutoff value. This calculation requires the urine sodium concentration to be at least 25 mEq/L and urine osmolality to be equal to or greater than the plasma osmolality.
Recognizing that urea and sodium are reabsorbed in the downstream medulla has led some to question the utility of this calculation because the validity of this formulae assumes absorption of osmoles distal to the collecting duct is negligible. For this reason, a urine potassium-to-creatinine ratio is used to assess potassium handling by the kidneys (34). This formula takes advantage of the near constant rate of urinary secretion of creatinine and therefore the ratio corrects for variability in urine concentration. A ratio of <13 mEq potassium/g creatinine (<2.5 mEq potassium/mmol creatinine) is considered an appropriate response to gastrointestinal potassium loss, remote use of diuretics, decreased dietary intake, and potassium shift into cells. Higher values suggest an inappropriate response of the kidney (Figure 4).
Figure 4.

A flow diagram for the approach to patients with hypokalemia on the basis of the potassium-to-creatinine ratio in the urine. RTA, renal tubular acidosis.
Urine Chemistries in the Evaluation of Metabolic Acidosis
Analysis of urine chemistries can provide information as to the amount of ammonium excretion by the kidney and therefore are useful in distinguishing between renal and extrarenal causes of a nonanion gap hyperchloremic metabolic acidosis. Metabolic acidosis of a kidney origin is characterized by low ammonium excretion rates, whereas ammonium excretion is elevated in metabolic acidosis of an extrarenal origin. Direct measurement of ammonium in the urine is the most appropriate way to make this distinction, but if this assay is unavailable, then urinary ammonium excretion can be indirectly assessed by measuring the urinary anion gap (UAG):
The UAG is normally positive, ranging from 30 to 50 mmol/L, because of the urinary excretion of unmeasured anions such as phosphate, sulfate, and organic anions. In the setting of metabolic acidosis, ammonia production by a normal kidney increases from baseline values of 30–40 to >200 mmol/d. Ammonium is excreted into the urine with chloride and the increased concentration of ammonium chloride will cause the UAG to become negative, indicating the acidosis is of an extrarenal origin (35,36) (Figure 5). A positive UAG indicates an impairment in ammonium production and identifies the kidney as the cause of the acidosis. An increased plasma chloride and low bicarbonate concentration also develops as a compensatory response in chronic respiratory alkalosis. In this setting, the UAG is positive because urinary ammonium excretion is appropriately low (37).
Figure 5.

Urine chemistry profile in metabolic acidosis of kidney and nonkidney origin. (A) In diarrhea, stool loss of potential bicarbonate, sodium chloride, and potassium lead to a normal gap hyperchloremic metabolic acidosis, hypokalemia, and extracellular fluid volume contraction. Acidemia and hypokalemia stimulate kidney ammoniagenesis allowing for increased amounts of distal hydrogen ion secretion to occur. Ammonium is excreted coupled to chloride, accounting for the development of a negative urinary anion gap and increased osmolal gap. The urine pH (UpH) is not maximally acidic despite robust distal hydrogen ion secretion because the free hydrogen ion concentration is reduced due to the buffering effect of urinary ammonium. (B) Shown are various examples of overproduction aciduria. The excretion of the sodium or potassium salts of these acids (NaA) into the urine represent the indirect loss of bicarbonate from the body and cause the urine anion gap to remain positive despite large amounts of ammonium excretion. Urine sodium and potassium concentration will be greater than the urine chloride concentration because chloride is retained in response to volume contraction. Although some ammonium is excreted as NH4Cl, a large amount of ammonium is excreted coupled to the anion salts of the acids. The urine osmolal gap is increased due to the large amount of ammonium in the urine indicating the acidosis is of extrakidney origin. (C) Distal (type 1) and proximal (type 2) renal tubular acidosis (RTA) are characterized by hypokalemic hyperchloremic normal anion gap metabolic acidosis. Distal (type 1) RTA results from defects in hydrogen ion secretion in the distal nephron interfering with bicarbonate regeneration. The urine pH is always alkaline (pH 6–7) and urine potassium is increased due to coupling of increased distal sodium delivery with increased aldosterone resulting from acidosis-induced decreased sodium reabsorption in the proximal tubule (36). Impairment in activity of the H+-K+-ATPase can also contribute to kidney K+ wasting. Proximal (type 2) RTA is the result of impaired bicarbonate reclamation in the proximal tubule due to a decrease in the tubular maximum for reabsorption. When the plasma bicarbonate exceeds the tubular maximum, the urine pH will be alkaline and urine sodium and potassium will be increased whereas urine chloride is low . Once the plasma bicarbonate concentration falls to the reduced tubular maximum, the urine pH will become acidic and the degree of potassium wasting will decrease. Type 4 RTA is characterized by hyperkalemic normal anion gap metabolic acidosis. When the disorder results from a disturbance in the renin-angiotensin-aldosterone axis, the urine pH is acidic, whereas when it results from tubular dysfunction, the urine pH is more alkaline. In all types of RTA, urine ammonium excretion is reduced, reflected by a positive urine anion gap and no increase in the urine osmolal gap.
The UAG can be misleading when other unmeasured ions are excreted. For example, increased urinary excretion of sodium keto acid salts in diabetic and alcoholic ketoacidosis and urinary excretion of sodium hippurate and sodium benzoate in toluene exposure can keep the UAG positive despite an appropriate increase in urinary ammonium excretion (38,39). A similar effect can occur in D-lactic acidosis. The stereospecificity of the sodium-L-lactate cotransporter in the luminal membrane of the proximal tubule results in less efficient reabsorption of filtered D-lactate as compared with L-lactate, causing excretion of D-lactate into the urine as a sodium or potassium salt. Increased urinary excretion will also be missed when ammonium is excreted with an anion other than chloride, such as β-hydroxybutyrate or hippurate (Figure 5). In these settings, calculation of the urine osmolal gap is used as an indirect measure of ammonium excretion (40). The urine osmolal gap is the difference between the measured and the calculated urine osmolality:
 |
The urine osmolal gap normally ranges from approximately 10 to 100 mOsmol/kg. Because ammonium salts are generally the only other major urinary solute that contribute importantly to the urine osmolality, values appreciably >100 mOsmol/kg reflect increased excretion of ammonium salts. There are other osmotically active solutes that can increase the urinary osmolal gap in the absence of increased ammonium excretion. These include mannitol and alcohols such as ethanol, methanol, and ethylene glycol. Although ethanol is metabolized rapidly, urinary excretion of methanol and ethylene glycol, particularly during treatment with fomepizole, will significantly increase the urine osmolal gap. An increased plasma osmolal gap and clinical setting are important clues to the presence of toxic alcohol ingestion.
Urine pH does not reliably differentiate acidosis due to kidney disease from that of extrarenal origin (Figure 5). A small amount of distal H+ secretion can lower urine pH to values <5 in the setting of decreased ammoniagenesis. Despite the acid urine pH, net acid excretion is low because of low ammonium excretion. Similarly, an alkaline urine does not necessarily imply a kidney acidification defect. When ammonia production is stimulated, distal H+ secretion can be robust and yet the urine remains relatively alkaline because of the buffering effects of ammonia. A severe reduction in distal sodium delivery, as in advanced cirrhosis, can limit H+ secretion and also cause the urine to be relatively alkaline (Supplemental Material, Case 3).
Urine Osmolality
Measurement of urine osmolality along with urine chemistries can provide important diagnostic information in approaching a patient with disorders in water balance. Because urine osmolality can normally range from <100 to approximately 1200 mOsm/kg, one measures urine osmolality to determine if the value is in the expected range for the given clinical situation. A maximally dilute urine in a patient with hypotonic hyponatremia is found in primary polydipsia where ingestion of large quantities of water (>20 L/d in the setting of normal kidney function) overwhelm the normal excretory capacity of the kidney. This threshold decreases as kidney function declines. A similar value is found with more moderate fluid intake when combined with extremely limited solute intake, a condition often referred to as “beer potomania” syndrome (39). Another example would be a “tea and toast” diet sometimes present in an elderly patient. Low urinary solute excretion limits water excretion by the kidneys because solute excretion determines the upper limits for the volume of water loss by the kidneys.
More commonly, the urine osmolality in hypotonic hyponatremia is some value >200 mOsm/kg resulting from the action of vasopressin to decrease free water excretion. Urine electrolytes can be used to assess volume status and determine whether the cause of increased vasopressin levels is secondary to a circulatory disturbance causing unloading of baroreceptors, or is baroreceptor independent as occurs in the syndrome of inappropriate antidiuretic hormone release (41).
The urine osmolality should be maximally concentrated (>1000 mOsm/kg H2O) in patients with hypernatremia who have intact hypothalamic and kidney function due to increased plasma vasopressin levels. A lower maximum is seen in elderly patients because of age-related reductions in urine concentrating ability. If urine osmolality is <300 mOsm/kg, then either central or nephrogenic diabetes insipidus is present. These disorders are distinguished by examining the change in urine osmolality and urine volume after the administration of exogenous vasopressin. Urine osmolality will increase and urine volume decrease in central diabetes insipidus, whereas no response will occur in the nephrogenic form. Patients can present with incomplete forms of central and nephrogenic diabetes insipidus making the response to exogenous vasopressin less straightforward.
Urine osmolality can distinguish between a water diuresis and osmotic diuresis in patients with polyuria and polydipsia (42,43). A value <100 mOsm/kg indicates a water diuresis as seen in diabetes insipidus or primary polydipsia whereas a value between 300 and 600 mOsm/kg indicates an osmotic diuresis. Intermediate values between 100 and 300 mOsm/kg suggest a mixed polyuria as with a partial central or nephrogenic diabetes insipidus or simultaneous water and solute intake. Daily osmolar excretion averages 600–900 mOsm when ingesting a normal diet, and is largely accounted for by urea, sodium, potassium, and ammonium salts. Another osmole is contributing to the polyuria when the product of urine volume and urine osmolality exceeds this range. Glycosuria as in uncontrolled diabetes, increased urea generation due to high protein nutritional supplementation, salt-containing intravenous fluids, or administration of mannitol can all lead to an osmotic diuresis (Figure 6). Polyuria in the absence of polydipsia can be seen in patients with hyponatremia undergoing a brisk aquaresis when the defect in water excretion is rapidly reversed as with discontinuation of thiazides, or after cortisol or thyroid hormone replacement.
Figure 6.

Urine chemistry tests for the diagnostic evaluation of a patient with polyuria. Polyuria is generally said to be present when urine output is >3 L/d and can be due to either a water diuresis or an osmotic diuresis. A urine osmolality (Uosm) <100 mOsm/kg indicates a water diuresis. Restricting free water intake will cause a decrease in urine volume and an increase in urine osmolality in psychogenic polydipsia, whereas no effect is seen in diabetes insipidus. An increase in urine osmolality after the administration of desmopressin is found in central diabetes insipidus whereas no response is found in the nephrogenic form. A urine osmolality >300 mOsm/kg is observed in osmotic diuresis. Values between 100 and 300 mOsm/kg favor a mixed polyuria as with simultaneous water and solute intake or partial central or nephrogenic diabetes insipidus. Patients with CKD are unable to maximally dilute the urine and can also fall within this range when challenged with a water load. Administration of NaCl is responsible when the majority of osmoles can be accounted for by two times the sum of Na+ and K+ concentration in the urine. An osmotic diuresis due either to glucose, urea, or mannitol is present when there is a large gap between the urine osmolality and the urine electrolyte concentration. Even though urine osmolality is often greater than plasma values, hypernatremia can develop during an osmotic diuresis because the plasma Na+ concentration is determined by the relative loss of water and electrolytes and not total solutes. Calculation of electrolyte free water excretion predicts the change in plasma Na+ concentration in this setting. The graph demonstrates that with progressive osmotic diuresis the urine osmolality will progressively decrease to values that are isosmolar with plasma because high tubular flow rates prevent osmotic equilibrium with medullary interstitial fluid even when arginine vasopressin is not limiting (44).
Patients undergoing an osmotic diuresis may have a urine osmolality greater than the plasma osmolality and still be in negative water balance (44,45). Free water loss is present when the concentration of sodium and potassium in the urine is less than the plasma concentration. Calculation of electrolyte free water clearance is useful in predicting the directional change in plasma sodium concentration:
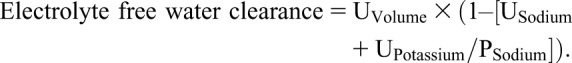 |
A positive value indicates loss of free water from the body and predicts an increase in the plasma sodium concentration over time (46,47). A negative value suggests water intake would tend to lower the plasma sodium concentration after taking into account insensible water losses (Supplemental Material, Case 4).
Conclusions
Urinary chemistries vary widely in both health and disease and are affected by diet, volume status, medications, and disease states. When properly examined these tests provide important insight into the mechanism and therapy of various clinical disorders that are first detected by abnormalities in plasma chemistries. These tests cannot be interpreted in isolation, but instead require knowledge of key clinical information, such as medications, physical examination, and plasma chemistries, to include kidney function. When used appropriately and with knowledge of limitations, urine chemistries can provide important insight into the pathophysiology and treatment of a wide variety of disorders.
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.10330818/-/DCSupplemental.
Case 1. Use of the fractional excretion of sodium in a patient with pre-existing chronic kidney disease.
Case 2. Distinguishing between prerenal azotemia and acute tubular necrosis in the setting of diuretic therapy.
Case 3. Distinguishing whether metabolic acidosis is due to kidney disease or not.
Case 4. Use of urine chemistries in the approach to a polyuric patient with hypernatremia and a patient with hyponatremia.
References
- 1.Miller TR, Anderson RJ, Linas SL, Henrich WL, Berns AS, Gabow PA, Schrier RW: Urinary diagnostic indices in acute renal failure: A prospective study. Ann Intern Med 89: 47–50, 1978 [DOI] [PubMed] [Google Scholar]
- 2.Nguyen MT, Maynard SE, Kimmel PL: Misapplications of commonly used kidney equations: Renal physiology in practice. Clin J Am Soc Nephrol 4: 528–534, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zarich S, Fang LS, Diamond JR: Fractional excretion of sodium. Exceptions to its diagnostic value. Arch Intern Med 145: 108–112, 1985 [PubMed] [Google Scholar]
- 4.Steiner RW: Interpreting the fractional excretion of sodium. Am J Med 77: 699–702, 1984 [DOI] [PubMed] [Google Scholar]
- 5.Vanmassenhove J, Glorieux G, Hoste E, Dhondt A, Vanholder R, Van Biesen W: Urinary output and fractional excretion of sodium and urea as indicators of transient versus intrinsic acute kidney injury during early sepsis. Crit Care 17: R234, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang LS, Sirota RA, Ebert TH, Lichtenstein NS: Low fractional excretion of sodium with contrast media-induced acute renal failure. Arch Intern Med 140: 531–533, 1980 [PubMed] [Google Scholar]
- 7.Palmer BF: Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 43: 516–533, 1995 [PubMed] [Google Scholar]
- 8.Steiner RW: Low fractional excretion of sodium in myoglobinuric renal failure. Arch Intern Med 142: 1216–1217, 1982 [Google Scholar]
- 9.Hoffman LM, Suki WN: Obstructive uropathy mimicking volume depletion. JAMA 236: 2096–2097, 1976 [PubMed] [Google Scholar]
- 10.Pimentel JL Jr., Martinez-Maldonado M, Wilcox JN, Wang S, Luo C: Regulation of renin-angiotensin system in unilateral ureteral obstruction. Kidney Int 44: 390–400, 1993 [DOI] [PubMed] [Google Scholar]
- 11.Wen SF, Wagnild JP: Acute effect of nephrotoxic serum on renal sodium transport in the dog. Kidney Int 9: 243–251, 1976 [DOI] [PubMed] [Google Scholar]
- 12.Hong CD, Kapoor BS, First MR, Pollak VE, Alexander JW: Fractional excretion of sodium after renal transplantation. Kidney Int 16: 167–178, 1979 [DOI] [PubMed] [Google Scholar]
- 13.Pahwa AK, Sperati CJ: Urinary fractional excretion indices in the evaluation of acute kidney injury. J Hosp Med 11: 77–80, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Bagshaw SM, Langenberg C, Bellomo R: Urinary biochemistry and microscopy in septic acute renal failure: A systematic review. Am J Kidney Dis 48: 695–705, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Schrier RW: Urinary indices and microscopy in sepsis-related acute renal failure. Am J Kidney Dis 48: 838–841, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Diamond JR, Yoburn DC: Nonoliguric acute renal failure associated with a low fractional excretion of sodium. Ann Intern Med 96: 597–600, 1982 [DOI] [PubMed] [Google Scholar]
- 17.Palmer BF: Pathogenesis of ascites and renal salt retention in cirrhosis. J Investig Med 47: 183–202, 1999 [PubMed] [Google Scholar]
- 18.Belcher JM, Parikh CR, Garcia-Tsao G: Acute kidney injury in patients with cirrhosis: Perils and promise. Clin Gastroenterol Hepatol 11: 1550–1558, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alsaad AA, Wadei HM: Fractional excretion of sodium in hepatorenal syndrome: Clinical and pathological correlation. World J Hepatol 8: 1497–1501, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvounis CP, Nisar S, Guro-Razuman S: Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int 62: 2223–2229, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB: The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract 114: c145–c150, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Schrier RW: Diagnostic value of urinary sodium, chloride, urea, and flow. J Am Soc Nephrol 22: 1610–1613, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer BF: Hyponatremia in patients with central nervous system disease: SIADH versus CSW. Trends Endocrinol Metab 14: 182–187, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Schmidt C, Höcherl K, Bucher M: Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol 292: F1479–F1489, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Trinh-Trang-Tan MM, Geelen G, Teillet L, Corman B: Urea transporter expression in aging kidney and brain during dehydration. Am J Physiol Regul Integr Comp Physiol 285: R1355–R1365, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Wu KL, Cheng CJ, Sung CC, Tseng MH, Hsu YJ, Yang SS, Chau T, Lin SH: Identification of the causes for chronic hypokalemia: Importance of urinary sodium and chloride excretion. Am J Med 130: 846–855, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Palmer BF, Alpern RJ: Metabolic alkalosis. J Am Soc Nephrol 8: 1462–1469, 1997 [DOI] [PubMed] [Google Scholar]
- 28.Palmer BF, Naderi AS: Metabolic complications associated with use of thiazide diuretics. J Am Soc Hypertens 1: 381–392, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Carlisle EJ, Donnelly SM, Ethier JH, Quaggin SE, Kaiser UB, Vasuvattakul S, Kamel KS, Halperin ML: Modulation of the secretion of potassium by accompanying anions in humans. Kidney Int 39: 1206–1212, 1991 [DOI] [PubMed] [Google Scholar]
- 30.Luke RG, Galla JH: It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 23: 204–207, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrington JT, Cohen JJ: Measurement of urinary electrolytes - indications and limitations. N Engl J Med 293: 1241–1243, 1975 [DOI] [PubMed] [Google Scholar]
- 32.Choi MJ, Ziyadeh FN: The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 19: 424–426, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Kamel KS, Ethier JH, Richardson RM, Bear RA, Halperin ML: Urine electrolytes and osmolality: When and how to use them. Am J Nephrol 10: 89–102, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Lin SH, Lin YF, Chen DT, Chu P, Hsu CW, Halperin ML: Laboratory tests to determine the cause of hypokalemia and paralysis. Arch Intern Med 164: 1561–1566, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Batlle DC, Hizon M, Cohen E, Gutterman C, Gupta R: The use of the urinary anion gap in the diagnosis of hyperchloremic metabolic acidosis. N Engl J Med 318: 594–599, 1988 [DOI] [PubMed] [Google Scholar]
- 36.Aronson PS, Giebisch G: Effects of pH on potassium: New explanations for old observations. J Am Soc Nephrol 22: 1981–1989, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batlle D, Chin-Theodorou J, Tucker BM: Metabolic acidosis or respiratory alkalosis? Evaluation of a low plasma bicarbonate using the urine anion gap. Am J Kidney Dis 70: 440–444, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer BF, Clegg DJ: Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 373: 548–559, 2015 [DOI] [PubMed] [Google Scholar]
- 39.Palmer BF, Clegg DJ: Electrolyte disturbances in patients with chronic alcohol-use disorder. N Engl J Med 377: 1368–1377, 2017 [DOI] [PubMed] [Google Scholar]
- 40.Carlisle EJ, Donnelly SM, Vasuvattakul S, Kamel KS, Tobe S, Halperin ML: Glue-sniffing and distal renal tubular acidosis: Sticking to the facts. J Am Soc Nephrol 1: 1019–1027, 1991 [DOI] [PubMed] [Google Scholar]
- 41.Palmer BF: Diagnostic approach and management of inpatient hyponatremia. J Hosp Med 5[Suppl 3]: S1–S7, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Berl T: Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol 19: 1076–1078, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Bhasin B, Velez JC: Evaluation of polyuria: The roles of solute loading and water diuresis. Am J Kidney Dis 67: 507–511, 2016 [DOI] [PubMed] [Google Scholar]
- 44.Brodsky WA, Rapoport S, West CD: The mechanism of glycosuric diuresis in diabetic man. J Clin Invest 29: 1021–1032, 1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gennari FJ, Kassirer JP: Osmotic diuresis. N Engl J Med 291: 714–720, 1974 [DOI] [PubMed] [Google Scholar]
- 46.Popli S, Tzamaloukas AH, Ing TS: Osmotic diuresis-induced hypernatremia: Better explained by solute-free water clearance or electrolyte-free water clearance? Int Urol Nephrol 46: 207–210, 2014 [DOI] [PubMed] [Google Scholar]
- 47.Bodonyi-Kovacs G, Lecker SH: Electrolyte-free water clearance: A key to the diagnosis of hypernatremia in resolving acute renal failure. Clin Exp Nephrol 12: 74–78, 2008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.