

Abstract
Several studies have suggested that arrestin-mediated signaling by GPCRs requires G protein activation; however, in this issue of Science Signaling, Luttrell et al. documented arrestin-dependent activation of ERK1/2 by a number of GPCRs. These studies do not contradict each other, but illustrate the complexity of cellular signaling that cannot and should not be reduced to simplistic models.
Heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) are the most numerous signaling proteins in animals, and they are targeted by about one-third of clinically used drugs. Arrestin proteins bind to active phosphorylated GPCRs, precluding their coupling to G proteins and organizing various signaling pathways. In this issue of Science Signaling, Luttrell et al. (1) demonstrated the role of nonvisual arrestins (β-arrestin1 and β-arrestin2, also known as arrestin-2 and −3, respectively) in signaling initiated by several GPCRs. The elimination of both arrestins by CRISPR (clustered regularly interspaced short palindromic repeats)/ Cas9–mediated knockout or small interfering RNA (siRNA)–mediated knockdown and their reintroduction in several independent lines of knockout cells had variable effects on signaling (1). Notable differences between lines with genetic knockout clearly showed that the elimination of important proteins led to the rewiring of signaling networks that enabled the cells to survive. This is the main caveat of using genetic knockout cells. The key caveat of using siRNA-mediated knock-down in which mRNA, rather than protein, is targeted, is its incompleteness: If the protein is short-lived, then its expression can be reduced by ~90%, whereas in other cases, knockdown is no more than 50 to 60% successful. Rescue experiments that reinstate proteins targeted by knockout or knockdown are necessary to ascribe observed changes to their elimination or reduction.
Luttrell et al. (1) focused on the phosphorylation (and thus activation) of extracellular signal–regulated kinase 1 and 2 (ERK1/2), as well as monitored G protein activation and GPCR internalization, processes that are sup-pressed and enhanced, respectively, by arrestins. In some cell lines, genetic knockout of arrestins reduced ERK1/2 activation, where-as in others, it increased ERK1/2 activation or had no apparent effect (1). As could be expected, where arrestins play a role, their overexpression and either knockout or knock-down yielded opposite effects. Disparate effects of these means of reducing arrestin protein abundance are hardly surprising: Numerous pathways connect GPCRs with ERK1/2 activation through several types of G proteins and arrestins (2). Because arrestins suppress G protein–mediated signaling (3, 4), the net result of the knockout or knockdown of arrestins depends on the relative contribution of different pathways in particular cell lines.
The key question in arrestin-mediated signaling is whether G protein activation is necessary. Several studies have addressed this question by focusing on GPCR-dependent ERK1/2 activation, which is the most studied arrestin-regulated signaling pathway. It was demonstrated using CRISPR/Cas9 and TALEN (transcriptionactivator–likeeffectornucleases)– mediated gene knockout that arrestin-mediated ERK1/2 activation requires functional G proteins (5, 6), whereas GPCRs could activate ERK1/2 through G proteins with-out arrestins (5–7). Obviously, these findings do not disprove arrestin-mediated ERK1/2 activation, which has been well established by numerous studies, including that of Luttrell et al. (1). Even the most “G protein–centric” study presents data documenting the role of arrestins in ERK1/2 activation, albeit in the supplement (5). New evidence, however, suggests that G protein activation is necessary for arrestin-mediated ERK1/2 activation (5, 6).
The mitogen-activated protein kinase (MAPK) cascades consist of MAP3Ks, MAP2Ks, and MAPKs, where MAP3Ks and MAP2Ks activate their downstream kinases by phosphorylation (8). Because the cell has numerous kinases at each level, signaling is organized by scaffold proteins that bring the kinases of the same cascade into close proximity with each other (Fig. 1). Thus, the activation of the most downstream MAPKs reflects two distinct processes: the strength of the original “push” (MAP3K activation) and the efficiency of signal transduction in the cascade deter-mined by the scaffolds. The mechanisms of activation of different MAP3Ks are complex and kinase-specific, involving diverse proteins and both dephosphorylation and phosphorylation events (Fig. 1) (8). Apparently, to be productive, scaffolds, including arrestins, must bind to an already activated MAP3K. Un-fortunately, neither the classical studies in which arrestin-mediated signaling was dis-covered nor later ones looked at the activation of relevant MAP3Ks. It is possible that G protein activation is required for arrestin-dependent ERK1/2 phosphorylation because G proteins activate the MAP3K c-Raf. The activation of c-Raf involves the small guanosine triphosphatase (GTPase) Ras, which can be activated by GPCRs through G proteins (Fig. 1), as described previously (7), and by other inputs from receptor tyrosine kinases (RTKs) or integrins.
Fig. 1. Multiple pathways lead to MAPK activation.
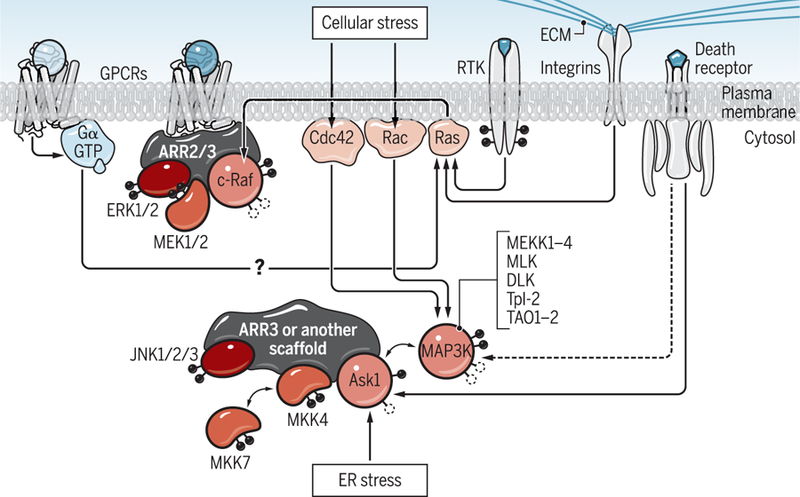
MAPK cascades consist of three kinases, MAP3K, MAP2K, and MAPK, which sequentially activate their downstream targets by phosphorylation. Arrestins have not been reported to participate in MAP3K activation, which initiates the signaling in MAPK cascades. G proteins can be activated by GPCR-dependent and GPCR-independent mechanisms. MAP3Ks are activated by RTKs, integrins, and several G protein–dependent mechanisms. Only after MAP3K activation can various scaffolds, including free and GPCR-bound arrestins, help to transform this first push into the activation of the most downstream MAPKs that are experimentally monitored in most studies of biased signaling. Note that JNK1/2/3 pathways are activated not only by ASK1 (which is not only scaffolded by arrestin-3) but also by other MAP3Ks, including MEKK1 to MEKK4, MLK, DLK, Tpl-2, and TAO1-2, with the help of other scaffolds. Note that c-Raf (cellular rapidly accelerated fibrosarcoma), ASK1 (apoptosis signal–regulated kinase 1), and MEKK1 to MEKK4, MLK, DLK, Tpl-2, and TAO1-2 are MAP3Ks; MEK1/2 and MKK4/7 are MAP2Ks; and Ras, Rac, and Cdc42 are small (single subunit) G proteins. Black circles indicate phosphorylation. Empty circles represent the dephosphorylation that is necessary for activation. ECM, extracellular matrix; PM, plasma membrane; ARR2/3, arrestin-2 or -3.
Cultured cells in all reported experiments were serum-starved, which precludes RTK signaling, and were plated on supports that do not activate integrins. In contrast, cells in vivo have access to growth factors, and growth factor receptors (RTKs) are the main activators of MAP3Ks (8). Therefore, in cultured cells, G proteins could be the only available activators of c-Raf, which is a pre-requisite for arrestin-dependent scaffolding of the ERK cascade. In a more natural setting with multiple sources of c-Raf activation, arrestin-mediated ERK activation might be G protein–independent.
Arrestin-dependent phosphorylation of ERK1/2 requires the binding of arrestins to active GPCRs (9). Arrestin-3, but not the highly homologous arrestin-2, facilitates the activation of c-Jun N-terminal kinase 3 (JNK3) independently of GPCRs. Arrestin-3 mutants that do not bind to GPCRs promote JNK3 activation (9). When the extent of JNK3 and ERK1/2 phosphorylation was monitored in the same cells expressing endogenous β2-adrenergic receptors treated with activating agonist, inactivating inverse agonist, or vehicle, it was shown that, whereas ERK phosphorylation reflects the receptor state, JNK3 phosphorylation only depends on the arrestin-3 variant expressed (9). Moreover, a short arrestin-3–derived peptide lacking receptor-binding elements facilitates JNK3 phosphorylation in cells (10). The JNK pathway is initiated by diverse signals, including death receptors, cellular stress (through the small GTPases Rac and Cdc42), and endoplasmic reticulum (ER) stress (Fig.1). A possible role of G proteins in the arrestin-3–dependent activation of JNK3 has never been tested. If MAP3Ks are activated through mechanisms that do not involve GPCRs, then the arrestin-3– dependent activation of JNK would likely be G protein–independent.
In MAPK signaling, arrestins act as scaffolds (3, 4), but they are not the only scaffolds for any MAPK cascade. Thus, where the strength of the first push is the key and other scaffolds effectively facilitate signaling, the role of arrestins in ERK or JNK activation would be relatively small. In contrast, in situations in which arrestins serve as the key scaffolds, the translation of MAP3K activation into MAPK phosphorylation would depend on arrestin abundance. The cell is often thought of as a sack of water; however, it is highly structured, more like a city, with streets, intersections, and even construction zones obstructing traffic. MAPK activity in different compartments exerts distinct bio-logical effects. The localization of a scaffold determines where active MAPKs are generated, thereby affecting the biological consequence of their activation. MAPKs phosphorylate various transcription factors in the nucleus, regulating gene expression, and cytoplasmic substrates, with totally different consequences. Arrestins are predominantly cytoplasmic proteins; receptor-bound arrestins localize their partners even more restrictively to the plasma membrane and endosomes.
To summarize, the apparent controversy regarding arrestin-mediated signaling carries several take-home messages. First, just like we need two legs to walk properly, the cell needs both G proteins and arrestins to adequately respond to stimuli. Second, we need to elucidate the molecular mechanisms of the activation of MAP3Ks and MAP2Ks and the relative roles of G proteins, RTKs, and other inputs at these stages, rather than focus solely on the most downstream MAPKs, such as ERKs and JNKs. Third, the mechanisms underlying MAPK activation by different GPCRs likely differ. Fourth, both genetic ablation of signaling proteins by gene editing and classical knockdown methods have caveats. Fifth, genetically altered knockout cells are not necessarily the best model to screen for G protein– or arrestin-biased GPCR-targeting drugs. Cells most closely resembling the intended target cells in the body should be used for this purpose. The presence of serum mimics the situation in vivo, whereas serum starvation generates artificial conditions, excluding the contribution of RTKs, the main regulators of MAPKs. Thus, we need to study cell signaling in its natural complexity (Fig. 1), avoiding oversimplifications and unwarranted generalizations.
REFERENCES
- 1.Luttrell LM, Wang J, Plouffe B, Smith JS,Yamani L, Kaur S, Jean-Charles P-Y, Gauthier C, Lee M-H, Pani B, Kim J, Ahn S, Rajagopal S,Reiter E, Bouvier M, Shenoy SK, Laporte SA,Rockman HA, Lefkowitz RJ, Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal 11, eaat7650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luttrell LM, ‘Location, location, location’: Activation and targeting of MAP kinases by G protein-coupled receptors. J. Mol. Endocrinol 30, 117–126 (2003). [DOI] [PubMed] [Google Scholar]
- 3.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK, β-arrestins and cell signaling. Annu. Rev. Physiol 69, 483–510 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Peterson YK, Luttrell LM, The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol. Rev 69, 256–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Rüttiger N, Ziegler N, Benkel T, Schmitt NK, Ishida S, Müller I, Reher R, Kawakami K, Inoue A, Rick U, Kühl T, Imhof D, Aoki J, König GM, Hoffmann C, Gomeza J, Wess J, Kostenis E, Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun 9, 341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, Milligan G, Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem 291, 27147–27159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng K Kawakami J. Aoki, Messer K, Inoue R. Sunahara, von Zastrow M, Gutkind JS, Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal 10, eaal3395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrington TP, Johnson GL, Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol 11, 211–218 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Breitman M, Kook S, Gimenez LE, Lizama BN, Palazzo MC, Gurevich EV, Gurevich VV, Silent scaffolds: Inhibition of c-Jun N-terminal kinase 3 activity in the cell by dominant-negative arrestin-3 mutant. J. Biol. Chem 287, 19653–19664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan X, Stoy H, Kaoud TS, Perry NA, Chen Q, Vucak G, Perez A, Els-Heindl S, Slagis JV, Iverson TM, Beck-Sickinger AG, Gurevich EV, Dalby KN, Gurevich VV, Peptide mini-scaffold facilitates JNK3 activation in cells. Sci. Rep 6, 21025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]