

Abstract
Much research now indicates that vagal nerve stimulation results in a systemic reduction in inflammatory cytokine production and an increase in anti-inflammatory cell populations that originates from the spleen. Termed the ‘cholinergic anti-inflammatory pathway’, therapeutic activation of this innate physiological response holds enormous promise for the treatment of inflammatory disease. Much controversy remains however, regarding the underlying physiological pathways mediating this response. This controversy is anchored in the fact that the vagal nerve itself does not innervate the spleen. Recent research from our own laboratory indicating that oral intake of sodium bicarbonate stimulates splenic anti-inflammatory pathways, and that this effect may require transmission of signals to the spleen through the mesothelium, provide new insight into the physiological pathways mediating the cholinergic anti-inflammatory pathway. In this review, we examine proposed models of the cholinergic anti-inflammatory pathway and attempt to frame our recent results in relation to these hypotheses. Following this discussion, we then provide an alternative model of the cholinergic anti-inflammatory pathway which is consistent both with our recent findings and the published literature. We then discuss experimental approaches that may be useful to delineate these hypotheses. We believe the outcome of these experiments will be critical in identifying the most appropriate methods to harness the therapeutic potential of the cholinergic anti-inflammatory pathway for the treatment of disease and may also shed light on the etiology of other pathologies, such as idiopathic fibrosis.
Graphical abstract
Review Outline:
The cholinergic anti-inflammatory pathway (CAIP) is thought to be a part of a neural immune circuit (termed the ‘inflammatory reflex’) that regulates systemic inflammation primarily via efferent neural signals to the spleen1. First identified approximately 20 years ago, activation of this innate physiological pathway via electrical stimulation of the vagal nerve (VN) has been shown to promote an anti-inflammatory state, and therefore holds enormous promise for the treatment of a host of inflammatory diseases. Importantly, a wealth of animal studies have demonstrated that activation of this pathway protects from a wide range of disease states. The success of these pre-clinical studies has led to the initiation of a number of ongoing clinical trials utilizing electrical stimulation of the VN to treat patients with various forms of inflammatory disease2,3. While holding enormous potential for the treatment of disease, much controversy remains regarding the underlying physiological pathways mediating this response. This controversy is anchored in the fact that the VN itself does not innervate the spleen, so some intermediary pathway must transmit the stimulatory signal to the spleen. Recent studies by our own group provide evidence that oral intake of sodium bicarbonate (NaHCO3) may stimulate splenic anti-inflammatory pathways in both rats and humans4. Further, data from our group indicates that cell to cell connections between the splenic capsular mesothelium and mesothelial cells in the peritoneal cavity may be required for the anti-inflammatory response to oral NaHCO3 to be observed4. These findings not only provide a potentially practical method to activate the CAIP that does not require surgical intervention, but may also challenge the currently accepted model of the anti-inflammatory pathway as a neural immune interface. The purpose of this review then, is to: i) frame our recent findings in relation to the published literature investigating the structure of the CAIP; ii) to provide new experimental paradigms that fit with the outcome of this discussion, and finally; iii) to provide an experimental framework to delineate these hypotheses in future. It is hoped that by outlining these arguments, this review will provide a road map to future studies which could be utilized to better understand the underlying physiology of this important pathway.
The inflammatory reflex and the cholinergic anti-inflammatory response in disease
The inflammatory reflex, first proposed by Tracey et al is a parasympathetic neural circuit that is activated in response to inflammation, containing both afferent and efferent arms1,5. Acting as the afferent arm of the inflammatory reflex, afferent VN fibers are able to detect inflammation in the periphery and are activated in response to cytokines, endotoxins, and pathogen associated molecular patterns (PAMPs)6,7. Importantly, increased afferent arm signaling in response to increased levels of peripheral TNF-α has been suggested to result in increases in the motor activity of the efferent vagus via the dorsal vagal complex8-10, which then acts to suppress inflammation, preventing damage mediated by excess cytokine production5. The efferent arm of the inflammatory reflex has been termed ‘the cholinergic anti-inflammatory pathway’. Activation of this arm is thought to reduce pro-inflammatory cytokine production by splenic macrophages and suppress inflammation. While these combined arms are often referred to as a reflex, it has been questioned whether sufficient experimental data support the concept that activation of afferent arm of the inflammatory reflex results in increased efferent vagal activity, and therefore whether the afferent and efferent arms of the inflammatory reflex form a true reflex mechanism or act as independent pathways11. Despite this controversy, there is now little doubt that stimulation of vagal efferent nerves acts to limit systemic inflammatory responses.
Given the ability of the CAIP to modulate systemic inflammation, the therapeutic potential of this pathway has long been apparent. In pioneering studies, Kevin Tracey’s group demonstrated significantly decreased synthesis of the inflammatory cytokine TNF-α in the heart, spleen, and liver and increased survival following efferent vagal nerve stimulation (VNS) in in a murine model of sepsis12,13. The concept that activation of the cholinergic anti-inflammatory pathway is protective in sepsis is supported by the results of Wang et al13 who found that treatment with specific-cholinergic agonists such as nicotine, decreased serum levels of HMGB1 (a molecule that is believed to regulate cytokine expression and production) and also improved survival in experimental models of sepsis. Several approaches for CAIP stimulation have been examined in a variety of animal models, ranging from invasive VNS12 to non-invasive ultrasound techniques14. Importantly, a wealth of animal studies have demonstrated that activation of this pathway protects from a wide range of inflammatory diseases including sepsis, arthritis, lupus and type II diabetes15,16. In human subjects, VNS has been explored for pain management in diseases such as chronic migraine headaches and fibromyalgia17 and VNS is currently FDA approved for seizure disorders and depression in patients aged >12 years old, that are refractory to medication5,17,18. The success of pre-clinical studies toward treating inflammatory disease by activation of the CAIP via VNS has led to the initiation of a number of ongoing clinical trials utilizing electrical stimulation of the VN to treat patients with inflammatory diseases including Crohn’s disease and rheumatoid arthritis2,15. Excitingly, initial reports from clinical trials in which medical devices were surgically implanted around the VN to chronically stimulate this pathway in patients with inflammatory disease have been promising2,15.
To date, activation of the CAIP has been achieved almost exclusively by stimulation of the efferent VN or with administration of cholinergic agonists15. In vitro experiments have demonstrated the ability of acetylcholine (ACh)19 to inhibit TNF-α release in human macrophage cultures after the addition of lipopolysaccharide (LPS)12. Tracey et al established that acetylcholine binding to a specific nicotinic acetylcholine receptor (nAChR) resulted in post-transcriptional inhibition of cytokine synthesis, and inhibition of inflammation through decreased expression of TNF-α protein in macrophages without significantly altering levels of TNF-α messenger RNA1. It has been suggested that acetylcholine inhibits TNF-α production by macrophages via its effect on HMGB117. Other studies have utilized chemical compounds that stimulate the vagal activity in the CNS such as CNI-1493 (a tetravalent guanylhydrazone molecule) to stimulate an anti-inflammatory response18. Muscarinic agonists applied via intracerebroventricular dosing have also been demonstrated to decrease systemic inflammation, however, the vagus nerve (VN) must be intact to observe this effect20. The use of generalized cholinergic receptor agonists like nicotine to stimulate the CAIP are limited due to off-target effects related to global nicotinic ACh receptor activation15. Similarly, the VN innervates a host of organ systems, and generalized electrical stimulation of the cervical VN is also likely to promote unwanted off-target effects, although current clinical trials of VNS therapy in chronic heart failure have demonstrated an ability to minimize non-serious adverse events21. As such, in order to harness the therapeutic potential of the CAIP, much recent research has focused on the use of more specific pharmacologic agonists or ‘learning the (neural) language’3,5,15 of the CAIP, in the hope of stimulating more targeted activation of this pathway and limiting unwanted off-target effects in patients.
Unexpected findings from the kidney
In the following section we will introduce why our group was studying the physiological effects of NaHCO3 and describe the results of studies that we believe may be pertinent to understanding the CAIP. In some instances, we will discuss the methodology of our previous studies in detail, as these details are of importance to the interpretation of these data.
The kidneys contribute to systemic acid/base balance through the regulated excretion of bicarbonate (HCO3−), titratable acids and ammonium. Dysregulation of acid-base status and the development of metabolic acidosis is common in chronic kidney disease (CKD) patients, where severe reductions in renal mass lead to an inability to excrete appropriate amounts of acid in the urine22-24. Metabolic acidosis can result in a number of deleterious sequela including fatigue, bone loss and muscle wasting22. In order to prevent these sequela, it is now common for CKD patients with metabolic acidosis to receive supplementation with oral NaHCO322,25. Interestingly, a number of small clinical trials have indicated that not only does NaHCO3 supplementation limit these sequela, but that it may also slow decline in kidney function in CKD patients26-29. While protective effects of oral NaHCO3 administration on kidney function have previously been reported in rodent models of CKD28, the physiological pathways mediating these beneficial effects remain unclear. The Dahl salt-sensitive rat model is an experimental model of salt-sensitive hypertension and progressive renal injury30. We have recently reported that supplementation of Dahl salt-sensitive rats with 0.1M NaHCO3 in the drinking water markedly reduced tubular cast formation (an index of renal injury) in these animals when fed a high salt diet for two weeks31. While our findings indicate that these reductions in tubular cast formation are likely due to reduced precipitation of acidic proteins in the tubular lumen and unrelated to the inflammatory state of the kidney31, during the course of these studies we investigated the immune profile of kidneys from animals treated with oral NaHCO3 or vehicle (NaCl)4. We believe that our findings in regard to the immune profile of the kidneys following NaHCO3 supplementation (discussed below) may provide important insight into the underlying physiology of the CAIP.
Immune cells participate in both the induction and resolution of inflammation. In addition to serious side-effects, many of the common, currently available therapies for inflammatory disease suppress or inhibit key components of the immune response, thereby limiting pro-inflammatory responses. Unfortunately, this strategy leaves patients at greater risk of infection or malignancy5. In order to improve treatment for inflammatory diseases, much recent research has been directed toward identifying approaches to modulate the activation state of our immune systems towards an anti-inflammatory or ‘regulatory’ environment, without inhibiting the ability of the ‘pro-inflammatory side’ of the immune system to respond to threats5. Macrophages represent a key target in strategies aimed at altering the polarization state of the immune system. Macrophages form an important part of the innate immune response, being critically involved in both the initiation and resolution of inflammatory responses. Either via antigen presentation or via products released by other activated macrophages, macrophages can also recruit and activate cells involved in the adaptive immune response, such as T and B lymphocytes32. Therefore, macrophages form an important link between the innate and adaptive immune systems. Significant plasticity is a hallmark of monocyte-derived cells and it is widely understood that several pathways, including the circulation of factors such as chemokines or microbial-derived molecular products, result in changes in the structural and functional appearance of macrophages33,34. Macrophage polarization can be generally divided between the transformation into either M1 or M2 cells. IFN-γ either alone or in combination with TNF-α or bacterial LPS, contribute to ‘classical’ M1 polarization, while IL-4 and IL-13 contribute to ‘alternative’ M2 polarization through simple inhibition of macrophage activation to the M1 phenotype35. While M1 macrophages commonly participate in inflammatory reactions to noxious stimuli, M2 macrophages have effector functions similar to those of Th2 lymphocytes. Several studies of M2 macrophages have revealed that these cells have important immunoregulatory functions36, as well as roles in tissue remodeling and repair37. Importantly, macrophage polarization is likely to be critical in mediating the end effects of activation of the CAIP to suppress systemic inflammation.
In our studies, following high salt feeding in Dahl salt-sensitive rats in which the drinking water was replaced with 0.1M NaHCO3 solution, T-regulatory cells and M2 macrophages in the kidney were increased while M1 macrophages were decreased, when compared to rats drinking equimolar NaCl solution4. We initially hypothesized that these differences in the renal inflammatory profile were secondary to differences in renal injury. To test this hypothesis, we investigated the effect of just 3 days of NaHCO3 or NaCl drinking on Dahl salt-sensitive rats fed a low salt diet. In these studies, salt load and blood pressure elevations are minimized and differences in renal injury have less time to develop. Surprisingly, despite no differences in renal injury or cast formation being observed at 3 days post-treatment, the immune profile in low salt fed NaHCO3 treated rats remained polarized toward a regulatory profile4. The results of these studies prompted us to perform similar studies in Sprague Dawley rats that do not display significant elevations in blood pressure or develop renal injury when salt-intake is increased. In these animals, NaHCO3 treatment dose dependently shifted the renal immune cell profile away from an inflammatory, and toward an anti-inflammatory phenotype4. In fact, a shift toward a regulatory polarization could be observed with as low as 0.01M NaHCO3 (10% of the concentration of solutions given in initial studies and made equimolar to the vehicle treatment group with 0.09M NaCl) in the drinking water. Given the robust effect of relatively low concentrations of oral NaHCO3 to promote an anti-inflammatory immune cell profile in the kidney, as well as latter studies indicating that ingestion of a single dose of NaHCO3 promoted a similar anti-inflammatory shift in the profile of circulating leukocytes in the venous blood of healthy human subjects4, we decided to further investigate this phenomenon.
The effect of NaHCO3 to promote an anti-inflammatory response in the kidney was found to be dependent on the spleen. Flow cytometry analysis of spleens excised from rats drinking NaHCO3 indicated that changes in the immune cell profile in splenic tissue mirrored those occurring in the kidney4. As the spleen may act as a source of immune cells that infiltrate the kidney parenchyma, we investigated whether removal of the spleen abolished the anti-inflammatory effect of oral NaHCO3 on the renal immune cell profile. In studies in anesthetized Dahl salt-sensitive rats, we first performed an abdominal incision along the Linea Alba before using an index finger to locate the spleen on the left flank and draw it to midline. The spleen was then raised slightly by gently cupping it between two fingers such that the splenic hilum could be visualized and ligated with a 3.0 silk tie. Once ligated, the splenic vessels were cut and the spleen removed. The wound was then closed and the animals allowed to recover for 7 days before beginning the experimental protocol. As the experimental protocol, in which rats were fed high salt along with either NaCl or NaHCO3 in the drinking water, was 3 weeks in length, these surgeries were performed 4 weeks before the kidney was harvested for flow cytometry. As we found that surgical removal of the spleen completely abolished the anti-inflammatory effect of NaHCO3 drinking on the renal immune cell profile4, these studies identified the spleen as critical in the anti-inflammatory response to NaHCO3, and the likely source of infiltrating, polarized immune cells in the kidney.
While on the surface, the results of the above-mentioned studies, in which a prior splenectomy was found to abolish the anti-inflammatory effect of NaHCO3 drinking on the kidney, would appear relatively straightforward in their interpretation, we found that the renal anti-inflammatory response to NaHCO3 was also abolished in our surgical control group4. Our surgical control group consisted of sham splenectomy animals that were treated identically to the splenectomy group except upon raising the spleen out of the wound, the splenic hilum was not ligated and the spleen was carefully returned to its original position on the left flank. The loss of the renal anti-inflammatory response was unlikely to be due to a generalized response to surgery, as we had not previously observed loss of these responses in rats that had telemetry devices implanted using an identical incision and a more involved surgical procedure (albeit, one that did not involve manipulating the spleen). Furthermore, the spleen itself appeared well perfused and was grossly intact at tissue harvest in animals that underwent sham surgery. Therefore, to determine whether moving the spleen to midline during the surgical procedure was responsible for abolishing the anti-inflammatory response to oral NaHCO3, we designed a follow up study in which a laparotomy alone was performed as a surgical control group and the spleen left untouched. This study also contained an intervention group in which we moved the spleen to midline as per the surgical sham group for the original study, but we did not excise the spleen. The results of this study confirmed that simple surgical manipulation of the spleen to midline during the sham splenectomy procedure was sufficient to abolish the anti-inflammatory response to NaHCO34.
Our data indicating that gentle surgical manipulation of the spleen was sufficient to abolish the anti-inflammatory response to NaHCO3 was consistent with reports in mice that surgical clearing of the splenic poles had enhanced systemic inflammation, purportedly by selective parasympathetic denervation of vagal efferents that enter at the splenic poles38. In our experience in rats, only light connective tissue appears to connect the superior margins of the spleen to the inferior aspects of the stomach, while very little if any connective tissue is visible to the naked eye at the inferior border of the spleen or anterior pole. Despite a general consensus that the spleen receives little if any direct vagal innervation, given our observations, we decided to investigate the hypothesis that ‘by surgically manipulating the spleen we had broken vagal efferent fibers that entered not via the splenic hilum, but rather at the splenic poles’.
Surprisingly, data from our studies aimed at investigating the possibility of vagal innervation of the splenic poles, indicated that rather than by nerves, physiologic signals originating at distal anatomical sites may be transmitted to the spleen via signaling between mesothelial cells. We reasoned that if vagal efferent fibers innervated the poles of the rat spleen, we should be able to observe histological evidence of such innervation. On examining trichrome stained sections of the poles of the spleen, we found thin tissue connections attached to the spleen, mostly along the inferior border of the spleen (Figure 1)4. These connections appeared to be made up of collagen filaments surrounded by a single layer of cells, were small enough to be invisible to the naked eye, and appeared sufficiently fragile such that they could easily be broken through surgical manipulation of the spleen without notice. The source of these connections remains undetermined and, due to the length of these connections, cannot easily be identified in thin histological sections. To determine whether these connections contained neurons, we examined these connections with transmission electron microscopy as well as with immunohistochemical staining for the pan neuronal marker PGP9.5, the sympathetic marker tyrosine hydroxylase, and parasympathetic marker acetylcholine esterase. While these connections stained strongly positive for the panneuronal marker PGP9.5, electron microscopy as well as immunostaining against mesothelin, revealed that these connections did not contain neural tissue but instead consisted of mesothelial cells that formed a continuous layer with those on the splenic surface (Figure 2). While the majority of mesothelial cells are negative for PGP9.5, mesothelial cells adjacent to where these connections joined the spleen also stained strongly positive for PGP9.5 (the only other location we have identified PGP9.5 positive mesothelial cells is on the cardiac atria (Figure 2), although such studies have been far from exhaustive). These mesothelial cell-lined connections were found to be negative for the sympathetic marker tyrosine hydroxylase, but stained positive for the parasympathetic marker acetylcholine esterase4. We recognized in our study that staining does not necessarily imply functionality, but the expression of neuronal markers on non-neuronal tissue (e.g. mesothelial cells) provided the impetus for further characterization and study of these cells. Importantly, manipulation of the spleen to midline during surgery promoted proliferation and hypertrophy of the capsular mesothelial cells and prolonged thickening of the underlying fibrous collagen layer. Further, either prior manipulation of the spleen or transection of the VN below the diaphragm (being careful not to manipulate the spleen), promoted loss of acetylcholine esterase staining in these PGP9.5 positive mesothelial cells and prevented the increase in splenic mass we previously observed in response to oral NaHCO34. These data, along with our finding that disruption of these connections by manipulation of the spleen was sufficient to abolish the antiinflammatory response to NaHCO3, are consistent with a model in which signaling through mesothelial cells may transmit anti-inflammatory signals to the spleen4.
Figure 1. Cartoon demonstrating the location of PGP9.5 positive mesothelial lined connections to the rat spleen.

Solid ovals, PGP9.5 positive mesothelial cells. Open ovals, PGP9.5 negative mesothelial cells as shown in Figure 2.
Figure 2. Mesothelin and PGP9.5 localization in the spleen and heart.

Panel A) Anti-mesothelin staining of the rat cardiac atria. Mesothelial cells cover the entirety of the outer surface of the rat cardiac atria as indicated by positive staining for the mesothelial marker mesothelin (dark brown staining). Original magnification 5X, bar = 400μM. Panel B) Mesothelial cells cover the surface of the spleen and are continuous with mesothelial cells that line collagen connections that make contact with the spleen along its most anterior axis. Original magnification 5X. Panel C) Some of the mesothelial cells lining the cardiac atria stain positive for the pan-neuronal marker PGP9.5 (dark brown staining). Original magnification 5X. Panel D) higher power image of PGP9.5 positive mesothelial cells on the rat cardiac atria. Original magnification 40X, bar = 50μM. Panel E) Low power image of rat spleen stained for PGP9.5. Mesothelial cells of the diaphragmatic surface (white arrow) of the spleen are positive for PGP9.5 while those on the visceral surface are negative. Original magnification 5X.
In another serendipitous finding, we observed that splenic capsular mesothelial cells overlie a dense network of nerves. In an attempt to investigate potential cell-cell signaling between mesothelial cells on the splenic capsule, we loaded cells on the surface of the spleen with the Ca2+-sensitive dye Fluo-4 by immersion of the freshly excised spleen in a solution containing this fluorescent indicator. When imaging the diaphragmatic surface of the splenic capsule (Figure 1) using this dye, we observed a dense network of what appeared to be neural tissue, directly below the splenic capsule4. The cells in this network were confirmed as neurons, both by their ultrastructural features using transmission electron microscopy4, and by their activation following electrical field stimulation4. Importantly, to the best of our knowledge, the network of nerves we observed is far denser that that previously reported to be present in the splenic capsule. Previous reports indicate that nerves observed in the splenic capsule consist of primarily PGP9.5 positive, tyrosine hydroxylase positive neurons that closely associate with the vasculature39,40. We confirmed the presence of these tyrosine hydroxylase positive nerves in the splenic capsule in paraffin embedded sections, the density of which was consistent with previous reports4. This network of tyrosine hydroxylase positive neurons however, was far less dense than that observed using Fluo-4 loaded spleens in which the surface of the splenic capsule was directly imaged (a technique not utilized by previous studies). While the most positive areas of PGP9.5 staining followed a pattern that mirrored the expression of tyrosine hydroxylase, PGP9.5 signal was not limited to tyrosine hydroxylase positive areas. Furthermore, almost the entirety of the splenic capsule stained lightly positive for acetylcholine esterase4. Importantly, our surgical manipulation procedure did not appear to disrupt sympathetic innervation of the spleen, as evidenced by a similar density of tyrosine hydroxylase positive nerve staining in sham and surgically manipulated spleens (supplemental data, Ray et al4). Based on these observations, we concluded that the splenic capsule contains a dense network of nerves or a neural plexus that lies directly beneath collagen surface of the splenic capsule, only a fraction of which are positive for tyrosine hydroxylase, and which stain negative or only lightly for neuronal cell markers.
Alternative models and experimental paradigms to investigate the architecture of the cholinergic anti-inflammatory pathway.
How do our new observations fit with current models of the CAIP? While it is possible that VNS and NaHCO3 activate separate anti-inflammatory pathways, evidence that both responses require the spleen and that the cholinergic receptor antagonist methyllycaconitine was able to block the anti-inflammatory response to NaHCO3 suggest both responses share a common mechanism4. Therefore, in the remainder of the text we will examine our recent findings in relation to previously proposed models of the CAIP. A summary of all models included in this review with supporting and refuting evidence can be found in Table 1.
Table 1. Review of cholinergic anti-inflammatory pathway (CAIP) models.
This table summarizes both supporting and refuting evidence for each proposed pathway of the cholinergic anti-inflammatory pathway discussed throughout the review.
| Model | For | Against |
|---|---|---|
| Di-synaptic | • Splenic nerve denervation or pharmacological depletion of noradrenaline within sympathetic nerve terminals blocked the anti-inflammatory response to VNS | • Inability to validate vagal-sympathetic synapses in the celiac ganglion • Electrical stimulation of the efferent vagus nerve reported to have no direct effect on splenic nerve activity |
| Direct Parasympathetic Splenic Innervation | • Selective surgical clearing of the splenic poles increases systemic inflammation & produces inflammatory splenic immune profile • Neuronal tracers injected into the spleen label the dorsal motor nuclei of the vagus & lesioning parasympathetic nerves at both tips of the spleen abolishes labeling |
• Dearth of histological evidence of vagal innervation in rodent spleen • High risk of artefactual labeling using retrograde tracers from inadvertent spread of the tracer to non-splenic sites |
| Sympathetic Activation & Signaling | • ChAT+CD4+T cells are required for the anti-inflammatory response to VNS • Absence of a valid neuronal pathway capable of transmitting VN mediated signals to the spleen |
• Localization of the required neuronal/T cell interface remains unknown |
| Mesothelial transmission of signals to the spleen | • Presence of thin mesothelial lined collagen connections that join with the capsular mesothelium • Evidence these mesothelial cells express neuronal markers and contain ultrastructural elements resembling signaling components of nerves • Evidence that clearing of connective tissue around the spleen or disruption of these connections inhibits the splenic anti-inflammatory response to NaHCO3 • Evidence that capsular mesothelial cells respond to disruption of mesothelial connections to the spleen (proliferation and collagen deposition) • Evidence of a dense network of nerves below the splenic capsule, providing a potential interface between the splenic capsular mesothelium and splanchnic anti-inflammatory pathway |
• Surgical interventions may damage the spleen leaving it unable to respond to stimuli • NaHCO3 may act by a distinct pathway separate from the CAIP to promote an anti-inflammatory response • Nature and anatomical origin of signals sensed by the spleen and transmitted via the mesothelium to the spleen remains unknown |
The di-synaptic model:
The most widely reported current model of the CAIP is that which has been proposed by Kevin Tracey’s research group at the Feinberg Institute for Medical Research in Manhasset, New York. Tracey et al were the first to discover that stimulation of the vagus nerve resulted in protection against LPS induced endotoxemia12,18 and treatment with a TNF-α monoclonal antibody was protective against the circulatory collapse caused by injection of a lethal dose of Escherichia coli41. From ensuing studies they proposed the CAIP as a ‘reflex arc’ wherein noxious stimuli in the periphery stimulate the afferent arm of the vagus nerve to transmit a signal to the nucleus tractus solitarius where it synapses, to activate efferent vagal activity which mediate the CAIP1,5.
Initial models of the CAIP had proposed a direct connection between the nervous and immune systems, where acetylcholine from vagal efferent fibers was released in proximity to splenic macrophages1,5,12, stimulating the anti-inflammatory response. This model was supported by evidence that exposure of cultured macrophages to nicotine or acetylcholine, agonists of cholinergic receptors (the latter released by post-synaptic VN terminals), resulted in decreased levels of TNF-α, IL-1, and IL-18 through nicotinic, alpha-bungarotoxin sensitive acetylcholine receptors (nAChR)12,42. Importantly, the anti-inflammatory effect of VNS is lost in mice lacking α7nAChR’s, indicating this receptor subtype is critical in mediating the CAIP12. The finding that the spleen was a critical component of the anti-inflammatory response to VNS necessitated an updated model. In 2006 Huston et al demonstrated that VNS failed to inhibit TNF-α production in splenectomized animals during lethal endotoxemia43. These studies indicated that the systemic anti-inflammatory response to VNS requires the spleen. These studies were problematic to initial models of the CAIP as most experimental evidence indicated the spleen receives little, if any vagal innervation44. Instead, efferent innervation of the spleen consists of noradrenergic sympathetic fibers that approach the splenic parenchyma along the splenic nerve via the splenic hilum45. Subsequent studies demonstrating that sympathetic denervation of the spleen, or depletion of noradrenaline within sympathetic nerve terminals, blocked the anti-inflammatory response, along with evidence that some vagal preganglionic neurons terminate in the celiac ganglion (where much of the postganglionic sympathetic nerve supply to the spleen derives)7,46, appeared to solve the problem of how vagal efferent signals are transmitted to the spleen and have led to the current di-synaptic model of the CAIP5. In this di-synaptic model, preganglionic efferent VNs synapse with post-ganglionic sympathetic nerves that then transmit the vagal derived signal to the spleen. Importantly, macrophages express both nicotinic and β-adrenergic receptors45,47, both of which have been demonstrated to suppress TNF-α following activation12. Therefore, it is possible that either noradrenaline or acetylcholine acts as the final chemical signal initiating the anti-inflammatory response in splenic macrophages.
Identification of choline acetyltransferase (ChAT)+ T lymphocytes as another critical component of the anti-inflammatory response led to further refinement of this model of the CAIP. Choline acetyltransferase is the enzyme that catalyzes the synthesis of acetylcholine (ACh)48 and is present in both B and T lymphocytes49. ACh content is greater in CD4+ T lymphocytes compared to either CD8+ T lymphocytes or B lymphocytes50, and reflects increased ChAT function in these cells51. In 2011 Rosas-Ballina et al reported that in nude mice that lack functional T cells, stimulation of the vagus nerve had no anti-inflammatory effect, but found that the anti-inflammatory effect could be partially restored through adoptive transfer of a subset of splenic ChAT+ CD4+ T cells52. Given adrenergic fibers present in the spleen have been identified in close proximity to areas high in ChAT+ CD4+ T cells52,53 (although recently published data refute this finding54), it has been proposed that this subset of T cells serve as a critical link between neuronal input to the spleen and stimulation of the anti-inflammatory response in macrophages.
In summary, in the current di-synaptic model of the CAIP, signals traverse down the efferent arm of the VN, which synapses with sympathetic neuronal cells in the celiac ganglion. The sympathetic signals from the celiac ganglion are transmitted through sympathetic axonal projections to the spleen via the splenic nerve and activate β-adrenergic receptor expressing, ChAT+ CD4+ T-lymphocytes in the spleen. Once active, these CD4+ T-lymphocytes produce acetylcholine, the chemical stimulus that activates α7nAChR expressing macrophages. These activated macrophages serve as the effector cell of the CAIP, resulting in decreased production of TNF-α and an anti-inflammatory state1,55 (Figure 3).
Figure 3. Di-synaptic model of the cholinergic anti-inflammatory pathway.

Efferent parasympathetic vagal nerves (green lines) synapse with post-synaptic sympathetic fibers (blue lines) in the celiac ganglion. Nerve impulses in these post-synaptic sympathetic nerves, that enter the spleen via the splenic hilum, promote release of noradrenaline (blue circles) from nerve terminals in the splenic parenchyma which are in close proximity to Choline acetyl transferase (ChAT) positive T-lymphocytes. Stimulation of adrenergic receptors of ChAT positive T-lymphocytes by noradrenaline promotes acetylcholine (green circles) production by these cells. Increased splenic acetylcholine levels activates nicotinic acetylcholine receptors on splenic macrophages limiting the production of tumor necrosis factor α, or other pro-inflammatory cytokines, and promotes a systemic anti-inflammatory state.
Critique of this model:
Most lymphoid tissues receive input from the autonomic nervous system exclusively in the form of sympathetic innervation and efferent sympathetic activity has been shown to modulate the function of immune cells, which express adrenoreceptors54. Thus, the concept that efferent signals transmitted to the spleen via sympathetic nerves mediates the CAIP, is in agreement with the known action of the sympathetic nervous system at other sites. In contrast, the actions of the parasympathetic and sympathetic nervous systems generally have opposing actions on target tissues. Therefore, the concept that preganglionic efferent vagal neurons synapse with postganglionic sympathetic neurons to stimulate an anti-inflammatory response in the spleen in the di-synaptic model of the CAIP, represents a biologically unique interaction where these two systems work together rather than in opposition. Perhaps unsurprisingly then, the validity of this di-synaptic model has previously been questioned. Data that conflicts with this model include studies by Robin McAllen’s group, which directly examined the proposed synaptic communication of parasympathetic and sympathetic signaling pathways within in the celiac ganglion56. These authors concluded that there was little evidence of vagal-sympathetic connections in the celiac ganglion and that electrical stimulation of the efferent vagus nerve had no direct effect on splenic nerve activity56. Instead, it has been suggested that increases in splenic nerve activity observed following vagal activation are due to stimulation of vagal afferent activation and reflex activation of the sympathetic nerves, a pathway which has also been suggested to suppress inflammation (termed the sympathetic splanchnic anti-inflammatory pathway)11. A di-synaptic model, where signaling to the spleen occurs via the splenic nerve, is also unable to explain data from our group and from Kooijman et al indicating that movement of the spleen (which did not appear to disrupt sympathetic innervation of the spleen via the splenic nerve)4 or clearing of the splenic poles38 promotes an inflammatory profile within the spleen.
How do our data fit with the di-synaptic model?
Both VNS and ingestion of NaHCO3 appear to promote an anti-inflammatory response that requires the spleen. In our study we did not directly examine whether vagal signaling was required to mediate the anti-inflammatory response to NaHCO3. Therefore, we cannot exclude the possibility the ingestion of NaHCO3 stimulated afferent vagal activity which may promote a splenic anti-inflammatory response via reflex activation of either efferent sympathetic or VNs. As vagal dennervation affects the function of the GI-tract including acid secretion57,58, the results of such studies may be difficult to interpret (discussed later). None of these possibilities however would explain loss of the anti-inflammatory response following disruption of mesothelial connections to the spleen. In relation to controversy regarding how vagal efferent signals reach the spleen, our data indicating that mesothelial cells may transmit anti-inflammatory signals to the spleen provides a potential alternative pathway for vagal efferent signals to reach the spleen that does not require a di-synaptic connection. Rather, parasympathetic, vagal efferent signals may directly or indirectly stimulate signaling through the mesothelium, initiating the anti-inflammatory response independent of a requirement for activation of sympathetic efferent nerve impulses via the splenic nerve.
Key studies needed to delineate this hypothesis:
A relatively straightforward experiment to test the alternate hypothesis indicated by our findings that ‘mesothelial cells act as the intermediate pathway between efferent VNS and splenic anti-inflammatory responses’ would be to determine whether mesothelial connections to the spleen are required to observe the anti-inflammatory response to efferent VNS in animals in which the cervical vagus has been transected. The studies published by Bratton et al already provide strong evidence against a di-synaptic model in mediating the anti-inflammatory action of efferent VNS56.
Direct parasympathetic innervation of the spleen:
Kooijman et al have previously reported that either selective sympathetic denervation of the spleen (by transecting the splenic nerve) or selective parasympathetic denervation of the spleen (by surgical clearing of the splenic poles) increased systemic inflammation when compared to sham surgical controls in mice38. The parasympathetic denervation procedure assumes that parasympathetic nerve fibers innervate the spleen at the poles, a feature that would support a model of the CAIP in which vagal efferent fibers directly innervate the splenic parenchyma (Figure 4). Key data supporting this model include evidence that selective surgical clearing of the splenic poles had a marked inflammatory effect on the splenic immune profile38, and that neuronal tracers injected into the spleen label the dorsal motor nuclei of the vagus. This labelling is lost by lesioning putative parasympathetic nerves at both tips of the spleen38.
Figure 4. Direct parasympathetic innervation model of the cholinergic anti-inflammatory pathway.

Vagal parasympathetic fibers (green lines) innervate the spleen at the splenic poles. Activation of efferent parasympathetic fibers directly promotes acetylcholine release within the splenic parenchyma. This pathway presumably also requires Choline acetyl transferase (ChAT) positive T-lymphocytes and splenic macrophages however specific signaling pathways within the spleen have not been detailed for this model.
Critique of this model:
Most published reports directly examining the issue of parasympathetic innervation to the rodent spleen have found little evidence of significant vagal innervation and it remains generally accepted that the VNs do not directly innervate the spleen11,38,40,44,53,55,56,59,60. Furthermore, Anderson et al suggest that the results of studies using retrograde tracers must be interpreted cautiously, due to the possibility of artefactual labeling from inadvertent spread of the tracer to non-splenic sites where vagal innervation is present38.
How do our data fit with a direct vagal innervation of the spleen model?
As discussed, our data indicating that fragile collagen connections lined by mesothelial cells may transmit antiinflammatory signals to the spleen, provides a potential alternative pathway for efferent anti-inflammatory signals to reach the spleen. Our data are highly consistent with the results of Kooijman et al in that we both report significant effects of surgical interventions that may disrupt these connections on the inflammatory profile of the spleen4,38. It should be noted that we were unaware of the results of Kooijman, or any controversy regarding the physiological pathways through which the CIAP is mediated at the time we observed that surgical manipulation of the spleen negated the anti-inflammatory response to NaHCO3 ingestion. Given the similarities between our results and that of Kooijman however, our data could be viewed as independent verification, in a second species (rats rather than mice), that surgical interventions that do not disrupt the splenic hilum, but damage connective tissue to the body of the spleen, alter splenic immune responses. Opposing their conclusion of direct parasympathetic innervation at the splenic poles however, in rats we found no histological evidence of neuronal innervation of the splenic poles using the pan-neuronal marker PGP9.5. Rather, in all histological sections studied, PGP9.5 labeling outside the sub-capsular splenic parenchyma was limited to mesothelial cells4. We have since found evidence of similar mesothelial connections along the inferior axis of the spleen in mice (Figure 5). Furthermore, these mesothelial cells were found to contain neuronal like cellular organelles and disrupting these mesothelial cell-lined connections promoted hypertrophy and hyperplasia of capsular mesothelial cells, as well as progressive capsular fibrosis in both rats and mice (Figure 5)4. These data indicating that the splenic capsular mesothelium responds to disruption of these connections, provides further evidence that biological signaling is occurring between these cells. Coupled with the dearth of evidence of significant neuronal innervation from previous investigations, these findings lead us to favor a model of the CAIP in which anti-inflammatory signals are transmitted to the spleen via the mesothelium.
Figure 5. Capsular fibrosis and capsular cell proliferation following surgical manipulation of the spleen in the mouse.

We have previously reported capsular fibrosis and mesothelial cell hypertrophy and hyperplasia on the surface of the rat spleen following surgical manipulation that disrupted thin mesothelial lined connections to the splenic capsule. While, due to the thinner capsule, fibrosis is not easily observed 3 weeks post-surgery in the mouse spleen, capsular fibrosis is evident in trichrome stained sections of the mouse spleen (A) when compared to spleens that were not manipulated during surgery (B) (original magnification of both images 5X). Panel C shows capsular thickening and cell proliferation (arrows) on the surface of the surgically manipulated mouse spleen, similar to that previously reported in the rat. Original magnification 40X.
Key studies needed to delineate this hypothesis:
A caveat that should be considered in relation to our own data and that of Kooijman et al, is that surgical interventions in which the spleen is manipulated or connective tissue adjacent to the spleen is cleared, may have caused damage to the spleen. Such injury to the spleen may limit normal splenic functions, preventing the spleen from responding to anti-inflammatory signals from other sources. This could then lead us to mistakenly conclude that the immune response was modulated by severing inputs that are normally received via the body, rather than the hilum, of the spleen. We do not favor this hypothesis for two reasons. Firstly, we were careful to minimize the physical impact on the splenic parenchyma during our surgical procedure and feel it is unlikely we caused significant trauma. Certainly, we do not feel that we would have caused sufficient trauma to continually and completely inhibit splenic function for over 4 weeks post-surgery, when our immunological measurements were made. Further, we confirmed by immunohistochemical staining for tyrosine hydroxylase that we had not inadvertently sympathetically denervated the spleen4. Secondly, Kooijman et al found that proliferation of immune cells within the spleen was increased, rather than inhibited following surgical clearing of the splenic poles. This finding at least argues against generalized paralysis of splenic immune responses. Nevertheless, the use of alternative approaches to disrupt cell to cell signaling in mesothelial cells that do not require surgical intervention may help address this issue, as well as delineate whether vagal or mesothelial signaling may mediate these effects. Such approaches could include Cre recombinase mediated deletion of key proteins involved in mesothelial signaling. With regard to this approach, mice with promotor-driven Cre recombinase positioned specifically in proximity to mesothelial promotors such as Wilm’s Tumor 1 (WT-1) are available61. Importantly, WT-1 is not known to be expressed in peripheral nerves or the immune system62. Therefore, use of this promotor would allow us to exclude the major alternative hypothesis. A limitation of this approach is our current lack of knowledge of signaling between mesothelial cells, and therefore our ability of identify appropriate genetic targets involved in mesothelial cell-cell signaling. One potential target is the α7nAChR, which has been reported to mediate anti-inflammatory responses in cultured mesothelium60.
Stimulation of splenic sympathetic nerve terminals by circulating ChAT-positive T cells:
In challenging the di-synaptic model of the CAIP in their excellent review “The cholinergic anti-inflammatory pathway: A critical review”, Martelli, McKinley and McAllen suggest that rather than a di-synaptic connection between vagal efferents and sympathetic neurons, a non-neuronal mechanism must transmit vagal signals to the spleen11. The authors acknowledge a critical role of sympathetic nerves in the CAIP, but suggest that this role does not involve transmission of action potentials to the spleen via the splenic nerve11. Instead, two possibilities are proposed. In the first, acetylcholine release from infiltrating ChAT+ CD4+ T cells promotes noradrenaline release from adrenergic nerve terminals expressing α7nAChR’s within the spleen in the absence of an action potential. This then promotes the anti-inflammatory response by stimulating β-adrenergic receptors on splenic macrophages. The second proposed mechanism hypothesizes that acetylcholine released from infiltrating ChAT+ CD4+ T cells directly stimulates action potentials in adrenergic nerve terminals. Both pathways propose β-adrenergic receptors on splenic macrophages rather than α7nAChR’s as the final cell-cell signaling component initiating anti-inflammatory response. Critically, both pathways also propose that non-neuronal signaling, rather than a di-synaptic model, is responsible for transmitting signals resulting from vagal efferent stimulation to the spleen11 (Figure 6).
Figure 6. A model of the cholinergic anti-inflammatory pathway initiated by stimulation of splenic sympathetic nerve terminals by circulating ChAT-positive T cells.

Vagal parasympathetic fibers (green lines) release acetylcholine (green circles) in proximity to Choline acetyl transferase (ChAT) positive T-lymphocytes at some as yet unidentified site outside the spleen. These activated ChAT positive T-lymphocytes then infiltrate the spleen via the circulation and release acetylcholine in proximity to sympathetic nerve terminals (blue line). This promotes one of either depolarization of these nerve terminals and release of noradrenaline (blue circles) or direct release of noradrenaline in the absence of terminal depolarization. This noradrenaline then stimulates β-receptors on splenic macrophages, limiting the production of tumor necrosis factor α, or other pro-inflammatory cytokines, and promoting a systemic anti-inflammatory state
Critique of this model:
While these authors speculate that ChAT+ CD4+ T cells act as the intermediate signal carrier in this pathway, the authors acknowledge that the localization of the required neuronal/T cell interface remains unknown11.
How do our data fit with a stimulation of splenic sympathetic nerve terminals by circulating ChAT-positive T cells model?
Through their own experimental results Bratton et al had excluded the possibility that sympathetic nerves transmitted vagal efferent signals to the spleen56. In their work, speculation that ChAT+ CD4+ T cells may act as the intermediate signal carrier was based on the absence of a known alternative signal carrier56. Our finding that mesothelial connections may transmit signals to the spleen is consistent with the hypothesis of Martelli et al, except that rather than ChAT+ CD4+ T cells acting as the signal carrier to the spleen, signals are transmitted to the spleen via cell-cell signaling between mesothelial cells. Mesothelial cells line the body's serous cavities and internal organs63. Interestingly, most mesothelial cells form monolayers of predominantly flattened, squamous-like cells devoid of significant mitochondria or Golgi apparatus. Mesothelial cells on some surfaces however, including that of the spleen, appear cuboidal and have abundant mitochondria and rough endoplasmic reticulum (RER), suggesting a more metabolically active state63. Traditionally, the mesothelium was thought only to provide a non-adhesive, protective surface that allows the body’s internal surfaces to rub against each other without injury. More recently however, it has become clear that the mesothelium is involved in a number of other important functions, including the transport and movement of fluid across the serosal cavities, synthesis of pro-inflammatory cytokines and growth factors, release of factors to promote both the deposition and clearance of fibrin, and antigen presentation63. Much evidence indicates that that cells outside the parasympathetic, primarily cholinergic neuronal network also synthesize, contain, and release ACh64. Importantly, mesothelial cells have previously been reported to express both ChAT and nicotinic receptors as well as other components required for ACh signaling63. Therefore, mesothelial cells appear to contain the necessary cellular machinery to act as an initiating source of acetylcholine that could activate splenic anti-inflammatory pathways. Taken together, our data suggest a previously unrecognized function of the mesothelium to potentially mediate the splenic cholinergic anti-inflammatory response via transmission of signals (potentially via ACh) to the splenic capsule.
In addition to identifying the mesothelium as a potential signal carrier required to initiate the CAIP, our data may also provide a location of interface between mesothelial cells and the splenic nerves. Given the close anatomical association between mesothelial cells and the dense network of nerves we observed underlying the splenic capsule, we speculate that mesothelial cells on the capsular surface may act via paracrine signaling to alter the release of neurotransmitters from this underlying neural network. These neurotransmitters may then act on immune cells to modulate the inflammatory response. Assuming these capsular nerves arise from the splenic nerve, such a pathway would be consistent with the requirement to have both a patent splenic nerve as well as intact mesothelial connections to the splenic capsule in order to stimulate the CAIP20. Localization of the splenic capsule as an integral site in the CAIP is consistent with this region being an important immunomodulatory area65. Interestingly, most of the nerves we observed in the splenic capsule were negative for tyrosine hydroxylase, suggesting that most of the observed capsular nerves may not release noradrenaline. The significance of this observation in regard to the CAIP remains unclear. Further studies are required to confirm nerves underlying the splenic capsule arise from the splenic nerve. Browning et al has suggested that the anti-inflammatory actions of VNS on the GI tract may be mediated indirectly via its effect on the enteric nervous system7. Given the paucity of data regarding the origin of this dense network of nerves below the capsule, until the source of these capsular nerves is confirmed, it remains possible that similar intrinsic neural networks may be present in the spleen.
Key studies needed to delineate this hypothesis:
A series of studies are required to test whether cholinergic signaling between mesothelial cells or infiltration of ChAT+ CD4+ T cells initiates the cholinergic anti-inflammatory response, and whether these signals may interact with nerves underlying the splenic capsule. Given the proposed role of ChAT+ CD4+ T cells in mediating downstream signaling involved in activation of the CAIP52, it may be easier to exclude a role of mesothelium in transmitting anti-inflammatory signals to the spleen than that of this T cell subset, as depletion of ChAT+ CD4+ T cells may inhibit the CAIP at multiple points. However, specific depletion of ChAT in mesothelial cells utilizing targeted Cre recombinase could delineate the requirement of cholinergic signaling through mesothelial cells. As per the discussion of direct vagal innervation, chief among experiments that may help delineate these hypotheses would involve determining whether genetic deletion of ChAT or other key signaling components in mesothelial cells inhibits splenic cholinergic anti-inflammatory responses to efferent vagal stimulation. As this hypothesis suggests that mesothelial cells transmit anti-inflammatory signals to the spleen in response to efferent VNS, and that activation of sympathetic nerve terminals underlying the splenic capsule are required to activate the anti-inflammatory response, physical or genetic disruption of signaling along these mesothelial connections would be predicted to abolish the anti-inflammatory response to efferent VNS. Splenic anti-inflammatory responses to either direct electrical stimulation of the splenic nerve or reflex activation of the splenic nerve secondary to vagal afferent signaling however, would be predicted to remain. As the network of nerves underlying the capsule can easily be loaded with and visualized using Ca2+-sensitive fluorescent dies4, it may also be possible to investigate potential mesothelial/neural interactions on the splenic surface using in situ imaging techniques.
A new model of the cholinergic anti-inflammatory pathway incorporating the mesothelium:
Based on the above discussion, we propose a novel model the of CAIP wherein anti-inflammatory signals are transmitted to the splenic parenchyma via paracrine signaling between PGP9.5 positive mesothelial cells that form continuous layers with those that cover the splenic capsule. These signals reach the splenic capsule via thin mesothelial lined collagen connections that join with the capsular mesothelium, primarily at the inferior border of the spleen (note; these data come from qualitative assessment of histological sections and further mapping of these connections is required to confirm the location of these connections). We propose that release of ACh or other mediators from capsular mesothelial cells, interacts with a dense network of nerves that may derive from the splenic nerve, and underlie the splenic capsule. Either via stimulation of action potentials or via direct stimulation of neurotransmitter release from nerve terminals in this network, anti-inflammatory signals may be propagated along the length of the splenic parenchyma underlying the capsule. Release of neurotransmitters from these nerves then initiates signaling pathways requiring (at a minimum) both ChAT+ CD4+ T cells and splenic macrophages, promoting a systemic anti-inflammatory response (Figure 7).
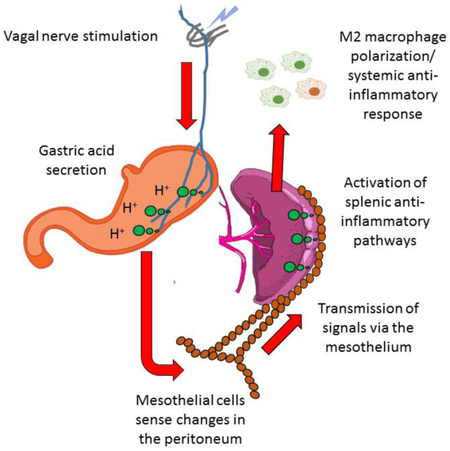
Figure 7. A model of the cholinergic anti-inflammatory pathway as a gastro-intestinal - splenic axis, mediated by non-neuronal signaling through the mesothelium.

Changes in venous blood draining the GI-tract or within the peritoneal environment such as increased levels of gastric hormones or direct input from efferent vagal nerves, stimulates signaling between PGP9.5 positive mesothelial cells (red ovals). These signals reach the spleen via paracrine signaling along thin mesothelial cell lined collagen connections that adjoin the anterior edge and inferior border of the spleen and in turn activate PGP9.5 positive mesothelial cells on the capsular surface. These mesothelial cells release acetylcholine (green circles) or some other substance which stimulates release of noradrenaline or other mediators from a web of nerves underlying the capsule, derived from the sympathetic splenic nerves. This noradrenaline then stimulates β-receptors on splenic macrophages, limiting the production of tumor necrosis factor α, or other pro-inflammatory cytokines, and promoting a systemic anti-inflammatory state.
Critique of this model:
A key component of this newly proposed model is that non-neuronal singling between mesothelial cells transmits anti-inflammatory signals to the spleen. While many cells are capable of cholinergic signaling64, the concept that signaling between mesothelial cells can conduct biological signals between distal anatomical sites would represent a new method of biological signaling, not previously observed, comparable (at least in its function to transmit biological signals between distal sites) to hormonal or neural signaling. Further, it is unknown whether ACh or other signaling molecules are produced in mesothelial cells in quantities high enough to effectively penetrate the splenic parenchyma and activate pathways involved in mediating the CAIP (note; our data, in which we were able to rapidly immersion load neuronal cells underlying the splenic capsule with Ca2+ sensitive dyes however, indicates that the collagen surface of the capsule may be quite permeable to small molecules). In any case, our hypothesis should be viewed critically and requires vigorous testing. Other critiques of this model include those already mentioned, such as the possibility that surgical interventions may damage the spleen rendering it unable to respond to stimuli, or that NaHCO3 acts by a distinct pathway different to that of the CAIP to promote an anti-inflammatory response. The nature and anatomical origin of signals potentially transmitted via the mesothelium also remains unclear.
How do other data fit with this new model?
As discussed above, this newly proposed model fits with key data underlying the CAIP including:
Evidence that stimulation of efferent VNs activates a systemic anti-inflammatory response5
That this response requires an intact splenic nerve20
As well as data that has challenged a direct parasympathetic or di-synaptic model of the CAIP including:
That efferent VNs do not innervate the spleen44
That efferent vagal signals are unlikely to be transmitted to the spleen via the splenic nerve56
That clearing of the splenic poles promotes a pro-inflammatory response38
A critical component of this newly proposed model which we have not yet addressed is ‘how could efferent vagal signals promote signaling within the mesothelium?’ In support of a direct connection between efferent vagal pathways and the mesothelium, Mihara et al recently reported that within the ileum, nerve terminals are found in close proximity to the mesothelium and that these cells are likely capable of responding to ACh via nicotinic receptors60. As such, efferent vagal signals may directly stimulate signaling in mesothelial cells which could then modulate immune responses in the spleen and GI tract. Teleologically, if vagal efferent nerves evolved in part to promote a systemic anti-inflammatory response that required the spleen, why not simply have direct vagal innervation of the spleen? Why (as our data suggest) rely on indirect transmission through mesothelial cells to promote a response? A potential answer to this question is that the CAIP did not evolve simply to respond to vagal efferent signals, but rather, evolved as a sensory pathway which responds to signals arising from the GI tract that are effected by VNS. We speculate that vagal efferent nerves do not drive the CAIP through a direct vagal-mesothelial signaling interface, but rather mesothelial cells in the peritoneum sense changes in the peritoneal cavity and respond by sending signals to the spleen to modulate the systemic inflammatory environment. As such, we propose that the CAIP may not represent a neural-immune axis but would be more appropriately described as GI-immune axis. Much evidence now supports a ‘splanchnic anti-inflammatory pathway’66 in which efferent sympathetic nerve impulses to the spleen promote an anti-inflammatory response. In line with the proposal of Martelli et al11, rather than developing an entirely new system to support a GI-immune axis, we suggest that this GI-immune axis evolved to piggyback on the existing machinery of the splanchnic anti-inflammatory pathway. Such a relationship would explain the requirement for a patent sympathetic nerve in mediating CAIP, absent a requirement of signaling through the sympathetic nerves to mediate this response.
Certainly, the idea that the CAIP represents a GI-immune axis rather than a neural-immune axis cannot be easily excluded from the available data. While the VN innervates a wide array of organs in the human body, in regard to the CAIP its connections to the abdominal viscera are of greatest interest. Efferent branches of the VN from the dorsal motor nucleus combine to form preganglionic fibers that course to cholinergic ganglia. Within these cholinergic ganglia, postganglionic neurons send projections to target, effector organs, supplying thoracic and abdominal viscera via several prevertebral and autonomic plexi67,68. Stimulation of VN activity decreases heart rate via vagal innervation of the cardiac atria and stimulates gastric, hepatic, and pancreatic glandular secretion via vagal innervation to the gastrointestinal tract and pancreas7,67. VN activity also increases gastrointestinal peristalsis67. In regard to the GI tract, the stomach and upper GI tract are most densely innervated by vagal parasympathetic nerves69. Given the well-known actions of efferent VN activity on the gastro-intestinal tract, it is somewhat surprising that little effort appears to have been made to exclude the secondary effects of vagal efferent stimulation on gastro-intestinal (GI)19 physiology toward mediating the CAIP. This is despite a practical inability to dissociate vagal efferent signals to the GI tract from those that mediate the CAIP; since there is no direct vagal innervation of the spleen, studies utilizing vagal denervation have transected the vagal trunk below the diaphragm, thereby disrupting vagal signaling to the liver, stomach, pancreases, gall bladder and mesentery by removing normal preganglionic cholinergic drive. Mapping of the neuroanatomical pathways and deciphering the ‘language’ of the CAIP has also failed to rule out this possibility, as the neuroanatomical pathways and stimulation frequencies found to drive the CAIP3,70 are remarkably similar to those that are known to stimulate gastric acid secretion57,71. Lining the entirety of the peritoneal cavity, the mesothelium is ideally situated to sense and respond to changes in GI function and/or secretion or absorption from the GI tract as well as potential threats from invading pathogens in this anatomically high-risk location. Our observation that oral ingestion of NaHCO3 promotes splenic anti-inflammatory responses is highly suggestive of a model in which GI physiology, rather than neural signals, mediates the CAIP. Evidence that both high fat feeding72 and the ‘hunger hormone’ Ghrelin stimulate the CAIP73 is also suggestive of a pathway that responds to primarily GI rather than neuronal signals, albeit these two stimuli are reported to have largely opposing actions on both vagal activation and acid secretion71,74,75.
The issue of whether vagal efferent stimulation directly stimulates splenic anti-inflammatory pathways or promotes splenic anti-inflammatory responses indirectly via its effects on GI physiology, is critical to identifying the most appropriate therapeutic strategies. Evidence from our study indicating that the anti-inflammatory response to NaHCO3 ingestion was blocked by treatment with a proton pump inhibitor4 suggest that gastric acid secretion is a key component of the anti-inflammatory response we observed. If rather than responding to direct cholinergic input from efferent VNs, mesothelial signaling to the spleen was found to sense and respond to gastric acid secretion, ingestion of NaHCO3 may not only be preferred over implantation of electrodes designed to stimulate vagal efferent activity, but may also promote an even greater anti-inflammatory effect. This is because ingestion of NaHCO3 raises the pH of the gastric fluid, and stimulation of acid secretion using this approach would not be limited by feedback inhibition from already low gastric pH76. Further, due to the strong buffering capacity of HCO3− solutions, acid secretion may be stimulated for a significant duration after ingestion of a single dose. A gastric acid secretion-mediated mechanism would be consistent with the robust changes in inflammatory cell polarization we observe after ingestion of NaHCO34, even in the absence of a prior pro-inflammatory stimuli (often a requisite to detect the anti-inflammatory responses to VNS). If the CAIP is mediated by gastric acid secretion, promotion of gastric acid secretion with alkali like NaHCO3, would also represent a more direct stimulus, avoiding potential adverse consequences of generalized VNS on other physiological functions.
On a separate but related topic, if our hypothesis that communication between specialized mesothelial cells transmits the signals that mediate the CAIP is correct, this finding may shed light on the etiology of some forms of idiopathic fibrosis. Splenic capsular fibrosis or perisplenitis (also known colloquially as ‘icing sugar spleen’), is a common incidental finding at autopsy77 (Figure 8). However, the etiology of capsular fibrosis remains unknown77. In trichrome stained sections, the layered arrangement of the thickened capsule with proliferating mesothelial cells at the surface, looks similar to the tissue within the connections themselves (Figure 8). The progressive nature of the fibrosis, with continued capsular thickening over weeks to months following surgery, is also unlike the common fibrotic response to injury or wound healing, where following the initial fibrotic response the scar tissue retracts. We speculate that the fibrotic response we observed following manipulation of the spleen may be triggered by loss of cell-cell signaling between capsular mesothelial cells and those lining the thin connections to the splenic pole. In support of this, it has been reported that splenic capsular fibrosis is most commonly found in patients with a history of peritonitis or portal hypertension77, where mesothelial cells in these connections may have been damaged or lost. While capsular fibrosis is generally considered a benign finding, based on our data, we speculate that this pathology may represent loss of the CAIP and a physiological attempt to reconnect the splenic capsule to damaged or broken mesothelial connections that supply signaling input to the capsular surface. As mesothelial cells have been implicated in idiopathic fibrosis of other organs61, we speculate that in some cases fibrosis may not represent tissue scarring in response to injurious or inflammatory stimuli, but rather, may represent continuing failed attempts at re-establishing lost pathways of cellular communication.
Figure 8. Progressive fibrosis and thickening of the rat splenic capsule is reminiscent of human perisplenitis and may represent a continued and unsuccessful attempt of the capsular mesothelium to reconnect signaling input.

Panel A) normal histology of the rat splenic capsule 8 weeks after surgery in which the spleen was not manipulated. Original magnification 20X. Panel B) significant thickening of the splenic capsule following manipulation of the spleen in the rat 8 weeks post-surgery. Original magnification 20X. Panel C) low power image of normal spleen and connections shown in panel A. Original magnification 5X. Panel D) low power image of thickened splenic capsule and connections in image shown in panel B. Original magnification 5X. Panel E) mesothelial cell lined collagen connection attached to the inferior border of the rat spleen in trichrome stained paraffin section. Original magnification 10X. Panel F) Example of shredding of the capsule often observed in rat with splenic capsular fibrosis, reminiscent of mesothelial lined connections present in the normal spleen. Panel G) Gross histological section of human spleen demonstrating significant splenic capsular fibrosis (perisplenitis). Note the general appearance and location of the fibrosis is similar to that observed in the rat spleen 8 weeks post-surgical manipulation (panel D). Image in panel G is modified from image shown in Monash University museum of pathology (http://museum.med.monash.edu.au/spec/index.cfm?spec=D3D1) with permission.
Key studies needed to delineate this hypothesis:
A critical test of the hypothesis that VNS promotes splenic anti-inflammatory responses indirectly via stimulation of gastric acid secretion would be to determine whether inhibition of acid secretion using proton pump inhibitors or other drug classes that inhibit gastric acid secretion, or following selective denervation of the VN innervating the stomach, inhibit the anti-inflammatory response to efferent VNS. If VNS acts by an independent signaling pathway, pharmacological inhibition of gastric acid secretion should have little effect to inhibit the anti-inflammatory response. If these studies further implicate gastric acid secretion in mediating the CIAP, additional studies would be required to identify the signals associated with gastric acid secretion that are sensed outside the stomach as well as how these may be sensed by the mesothelium. Such signals could include changes in pH of the blood draining the stomach or release of gastric hormones associated with acid secretion.
Summary and conclusions
In summary, the CAIP is thought to be a part of a neural immune circuit (termed the ‘inflammatory reflex’) that regulates systemic inflammation primarily via efferent neural signals to the spleen5. Much controversy still exists however, regarding the physiological pathways that mediate the anti-inflammatory response. Recent data from our laboratory may provide novel insight into the mechanisms that mediate the CAIP. Importantly, these data indicate that rather than a neural-immune circuit, the CAIP may represent a novel gastrointestinal-immune axis, mediated not by neuronal signaling but rather by signaling between mesothelial cells.
Ingestion of NaHCO3 is touted as an effective home remedy for the treatment of at least two inflammatory conditions, cystitis78 and gout arthritis79. More recent evidence from clinical studies indicates that NaHCO3 may also slow the decline in kidney function as well as lessen insulin resistance in CKD patients29. While, all of these diseases are thought to involve an inflammatory component, the beneficial effects of NaHCO3 ingestion are thought to be related to the alkalizing effect of NaHCO3 on the urine or blood (the latter representing a best modest change in healthy individuals). Such mechanisms however, remain largely untested, and a potential anti-inflammatory action of NaHCO3 in mediating any therapeutic benefits cannot be excluded. Given the controversy surrounding the physiological pathways underlying the CIAP, as well as the potential role of NaHCO3 in mediating beneficial effects in inflammatory states, it is the author’s view that further studies are warranted to determine the role of NaHCO3, GI physiology and the mesothelium in mediating the CAIP.
Delineating the hypothetical models proposed to underlie the CAIP will be likely be critical in leveraging the therapeutic potential of this innate physiological pathway. A number of key experiments capable of delineating these hypotheses are listed, some of which are currently underway in our own laboratory. It is hoped that by outlining the arguments underlining current models of the CAIP as well as suggesting an experimental framework to delineate these hypotheses in future, this review will provide a road map to future studies which could be utilized to better understand the underlying physiology of this important pathway. Perhaps a cheap and freely available antacid will rival VNS in treating a wide variety of inflammatory disorders?
Acknowledgements
This work was supported by National Institute of health grants to Paul O’Connor (NIH DK099548 and 1P01HL134604).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853–859. [DOI] [PubMed] [Google Scholar]
- 2.Koopman FA, Chavan SS, Miljko S, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2016;113(29):8284–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwan H, Garzoni L, Liu HL, et al. Vagus Nerve Stimulation for Treatment of Inflammation: Systematic Review of Animal Models and Clinical Studies. Bioelectron Med. 2016;3:1–6. [PMC free article] [PubMed] [Google Scholar]
- 4.Ray SC, Baban B, Tucker MA, et al. Oral NaHCO3 Activates a Splenic Anti-Inflammatory Pathway: Evidence That Cholinergic Signals Are Transmitted via Mesothelial Cells. J Immunol. 2018;200(10):3568–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9(6):418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex--linking immunity and metabolism. Nat Rev Endocrinol. 2012;8(12):743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Browning KN, Verheijden S, Boeckxstaens GE. The Vagus Nerve in Appetite Regulation, Mood, and Intestinal Inflammation. Gastroenterology. 2017;152(4):730–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci. 2000;85(1-3):49–59. [DOI] [PubMed] [Google Scholar]
- 9.Hermann GE, Emch GS, Tovar CA, Rogers RC. c-Fos generation in the dorsal vagal complex after systemic endotoxin is not dependent on the vagus nerve. Am J Physiol Regul Integr Comp Physiol. 2001;280(1):R289–299. [DOI] [PubMed] [Google Scholar]
- 10.Emch GS, Hermann GE, Rogers RC. TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J Physiol Gastrointest Liver Physiol. 2000;279(3):G582–586. [DOI] [PubMed] [Google Scholar]
- 11.Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014;182:65–69. [DOI] [PubMed] [Google Scholar]
- 12.Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–462. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Liao H, Ochani M, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10(11):1216–1221. [DOI] [PubMed] [Google Scholar]
- 14.Gigliotti JC, Huang L, Ye H, et al. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol. 2013;24(9):1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoover DB. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol Ther. 2017;179:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Opland D, Tsai S, et al. Pten deletion in RIP-Cre neurons protects against type 2 diabetes by activating the anti-inflammatory reflex. Nat Med. 2014;20(5):484–492. [DOI] [PubMed] [Google Scholar]
- 17.Johnson RL, Wilson CG. A review of vagus nerve stimulation as a therapeutic intervention. J Inflamm Res. 2018;11:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernik TR, Friedman SG, Ochani M, et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. 2002;195(6):781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czikora I, Alli A, Bao HF, et al. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am J Respir Crit Care Med. 2014;190(5):522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavlov VA, Ochani M, Gallowitsch-Puerta M, et al. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci U S A. 2006;103(13):5219–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Premchand RK, Sharma K, Mittal S, et al. Autonomic regulation therapy via left or right cervical vagus nerve stimulation in patients with chronic heart failure: results of the ANTHEM-HF trial. J Card Fail. 2014;20(11):808–816. [DOI] [PubMed] [Google Scholar]
- 22.Dobre M, Rahman M, Hostetter TH. Current status of bicarbonate in CKD. J Am Soc Nephrol. 2014;26(3):515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz WB, Hall PW 3rd, Hays RM, Relman AS. On the mechanism of acidosis in chronic renal disease. J Clin Invest. 1959;38(1, Part 1):39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wrong O, Davies HE. The excretion of acid in renal disease. Q J Med. 1959;28(110):259–313. [PubMed] [Google Scholar]
- 25.Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int. 2014;86(5):1031–1038. [DOI] [PubMed] [Google Scholar]
- 26.Goraya N, Simoni J, Jo C, Wesson DE. Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int. 2012;81(1):86–93. [DOI] [PubMed] [Google Scholar]
- 27.Ori Y, Zingerman B, Bergman M, Bessler H, Salman H. The effect of sodium bicarbonate on cytokine secretion in CKD patients with metabolic acidosis. Biomed Pharmacother. 2015;71:98–101. [DOI] [PubMed] [Google Scholar]
- 28.Loniewski I, Wesson DE. Bicarbonate therapy for prevention of chronic kidney disease progression. Kidney Int. 2014;85(3):529–535. [DOI] [PubMed] [Google Scholar]
- 29.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol. 2009;20(9):2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori T, Polichnowski A, Glocka P, et al. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol. 2008;19(8):1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray SC, Patel B, Irsik DL, et al. Sodium bicarbonate loading limits tubular cast formation independent of glomerular injury and proteinuria in Dahl salt-sensitive rats. Clin Sci (Lond). 2018;132(11):1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. Journal of Translational Medicine. 2017;15(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23(4):344–346. [DOI] [PubMed] [Google Scholar]
- 34.Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541–566. [DOI] [PubMed] [Google Scholar]
- 35.Gordon S Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. [DOI] [PubMed] [Google Scholar]
- 37.Noel W, Raes G, Hassanzadeh Ghassabeh G, De Baetselier P, Beschin A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004;20(3):126–133. [DOI] [PubMed] [Google Scholar]
- 38.Kooijman S, Meurs I, van Beek L, et al. Splenic autonomic denervation increases inflammatory status but does not aggravate atherosclerotic lesion development. Am J Physiol Heart Circ Physiol. 2015;309(4):H646–654. [DOI] [PubMed] [Google Scholar]
- 39.Elfvin LG, Johansson J, Hoijer AS, Aldskogius H. The innervation of the splenic capsule in the guinea pig: an immunohistochemical and ultrastructural study. J Anat. 1994;185 (Pt 2):267–278. [PMC free article] [PubMed] [Google Scholar]
- 40.Bellinger DL, Felten SY, Collier TJ, Felten DL. Noradrenergic sympathetic innervation of the spleen: IV. Morphometric analysis in adult and aged F344 rats. J Neurosci Res. 1987;18(1):55–63, 126-129. [DOI] [PubMed] [Google Scholar]
- 41.Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330(6149):662–664. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–388. [DOI] [PubMed] [Google Scholar]
- 43.Huston JM, Ochani M, Rosas-Ballina M, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203(7):1623–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bellinger DL, Lorton D, Hamill RW, Felten SY, Felten DL. Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav Immun. 1993;7(3):191–204. [DOI] [PubMed] [Google Scholar]
- 45.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987 2007). Brain Behav Immun. 2007;21(6):736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berthoud HR, Powley TL. Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia. J Auton Nerv Syst. 1993;42(2):153–169. [DOI] [PubMed] [Google Scholar]
- 47.Kohm AP, Sanders VM. Norepinephrine: a messenger from the brain to the immune system. Immunology Today. 2000;21(11):539–542. [DOI] [PubMed] [Google Scholar]
- 48.Tallini YN, Shui B, Greene KS, et al. BAC transgenic mice express enhanced green fluorescent protein in central and peripheral cholinergic neurons. Physiol Genomics. 2006;27(3):391–397. [DOI] [PubMed] [Google Scholar]
- 49.Fujii T, Mashimo M, Moriwaki Y, et al. Expression and Function of the Cholinergic System in Immune Cells. Front Immunol. 2017;8:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rinner I, Kawashima K, Schauenstein K. Rat lymphocytes produce and secrete acetylcholine in dependence of differentiation and activation. J Neuroimmunol. 1998;81(1-2):31–37. [DOI] [PubMed] [Google Scholar]
- 51.Rinner I, Schauenstein K. Detection of choline-acetyltransferase activity in lymphocytes. J Neurosci Res. 1993;35(2):188–191. [DOI] [PubMed] [Google Scholar]
- 52.Rosas-Ballina M, Olofsson PS, Ochani M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felten SY, Olschowka J. Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synapticlike contacts on lymphocytes in the splenic white pulp. J Neurosci Res. 1987;18(1):37–48. [DOI] [PubMed] [Google Scholar]
- 54.Murray K, Godinez DR, Brust-Mascher I, Miller EN, Gareau MG, Reardon C. Neuroanatomy of the spleen: Mapping the relationship between sympathetic neurons and lymphocytes. PLoS One. 2017;12(7):e0182416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murray K, Reardon C. The cholinergic anti-inflammatory pathway revisited. Neurogastroenterol Motil. 2018;30(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol. 2012;97(11):1180–1185. [DOI] [PubMed] [Google Scholar]
- 57.Pevsner L, Grossman MI. The mechanism of vagal stimulation of gastric acid secretion. Gastroenterology. 1955;28(4):493–499; discussion, 531-495. [PubMed] [Google Scholar]
- 58.Dempsey DT. Chapter 26. Stomach In: Brunicardi FC, Andersen DK, Billiar TR, et al. , eds. Schwartz's Principles of Surgery, 9e. New York, NY: The McGraw-Hill Companies; 2010. [Google Scholar]
- 59.Cano G, Sved AF, Rinaman L, Rabin BS, Card JP. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J Comp Neurol. 2001;439(1):1–18. [DOI] [PubMed] [Google Scholar]
- 60.Mihara T, Otsubo W, Horiguchi K, et al. The anti-inflammatory pathway regulated via nicotinic acetylcholine receptors in rat intestinal mesothelial cells. J Vet Med Sci. 2017;79(11):1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zolak JS, Jagirdar R, Surolia R, et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am J Pathol. 2013;182(4):1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walker C, Rutten F, Yuan X, Pass H, Mew DM, Everitt J. Wilms' tumor suppressor gene expression in rat and human mesothelioma. Cancer Res. 1994;54(12):3101–3106. [PubMed] [Google Scholar]
- 63.Mutsaers SE. Mesothelial cells: their structure, function and role in serosal repair. Respirology. 2002;7(3):171–191. [DOI] [PubMed] [Google Scholar]
- 64.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154(8):1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simkins TJ, Fried D, Parikh K, et al. Reduced Noradrenergic Signaling in the Spleen Capsule in the Absence of CB1 and CB2 Cannabinoid Receptors. J Neuroimmune Pharmacol. 2016;11(4):669–679. [DOI] [PubMed] [Google Scholar]
- 66.Martelli D, Farmer DG, Yao ST. The splanchnic anti-inflammatory pathway: could it be the efferent arm of the inflammatory reflex? Exp Physiol. 2016;101(10):1245–1252. [DOI] [PubMed] [Google Scholar]
- 67.Waxman SG. Cranial Nerves and Pathways Clinical Neuroanatomy, 28e. New York, NY: McGraw-Hill Education; 2017. [Google Scholar]
- 68.Waxman SG. The Autonomic Nervous System Clinical Neuroanatomy, 28e. New York, NY: McGraw-Hill Education; 2017. [Google Scholar]
- 69.Browning KN, Travagli RA. Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr Physiol. 2014;4(4):1339–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kressel AM, Tsaava T, Pavlov VA, Chavan SS, Tracey KJ. Cholinergic signaling in the dorsal motor nucleus regulates systemic inflammatory responses via the vagus nerve. The Journal of Immunology. 2018;200(1 Supplement):170.111–170.111. [Google Scholar]
- 71.Berthoud HR, Laughton WB, Powley TL. Vagal stimulation-induced gastric acid secretion in the anesthetized rat. J Auton Nerv Syst. 1986;16(3):193–204. [DOI] [PubMed] [Google Scholar]
- 72.Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med. 2005;202(8):1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mao Y, Tokudome T, Kishimoto I, et al. Endogenous ghrelin attenuates pressure overload-induced cardiac hypertrophy via a cholinergic anti-inflammatory pathway. Hypertension. 2015;65(6):1238–1244. [DOI] [PubMed] [Google Scholar]
- 74.Yakabi K, Kawashima J, Kato S. Ghrelin and gastric acid secretion. World J Gastroenterol. 2008;14(41):6334–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saqui-Salces M, Dowdle WE, Reiter JF, Merchant JL. A high-fat diet regulates gastrin and acid secretion through primary cilia. FASEB J. 2012;26(8):3127–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doherty GM. Stomach & Duodenum In: Doherty GM, ed. CURRENT Diagnosis & Treatment: Surgery, 14e. New York, NY: McGraw-Hill Education; 2015. [Google Scholar]
- 77.Swami Sunil Y. AAB, Narwade Sandhya B, and Valand Arvind G. Icing Sugar Spleen/Perisplenitis Cartilaginea: A Case Report. Journal of Cytology & Histology. 2018;9(1). [Google Scholar]
- 78.Cystitis. [Website]. 2018; https://www.betterhealth.vic.gov.au/health/conditionsandtreatments/cystitis. Accessed December 13, 2018, 2018.
- 79.The Truth About Online Gout Remedies. 2018; https://www.everydayhealth.com/gout-pictures/truth-online-gout-remedies.aspx#02. Accessed December 14, 2018.