

Abstract
A variety of methods have been employed to study the impact of posttranslational modifications on Tau protein function. Here, a semisynthesis strategy is described that enables selective modification within the central repeat domain of Tau4 (residues 291‐321), comprising a major interaction motive with tubulin as well as one of the key hexapeptides involved in Tau aggregation. This strategy has led to the preparation of four semisynthetic Tau variants with phosphoserine residues in different positions and one with a so far largely ignored carboxymethyllysine modification that results from a non‐enzymatic posttranslational modification (nPTM). The latter modification inhibits tubulin polymerization but exhibits an aggregation behavior very similar to unmodified Tau. In contrast, phosphorylated Tau variants exhibit similar binding to tubulin as unmodified Tau4 but show lower tendencies to aggregate.
Keywords: neurodegeneration, protein aggregation, protein modifications, protein semisynthesis, Tau protein
Tau is a central player in different tauopathies1 and is intimately involved in Alzheimer's disease (AD) by forming neurofibrillary tangles (NFTs), mainly consisting of hyperphosphorylated Tau.2 The latter results from kinase/phosphatase dysregulation3 and this process is thought to induce the release of abnormally phosphorylated Tau from microtubules and initiates Tau dimerization. This predisposes Tau to form larger oligomers, paired helical filaments (PHFs), and then NFTs. As an intrinsically disordered protein (IDP), with six isoforms found in humans,4 biological functions of Tau are regulated by interactions with other biomolecules and are linked to four regions of the protein (Scheme 1).5 Upon binding to microtubules and tubulin polymerization the microtubule‐binding region (MTBR) and the proline‐rich region (PRR) of Tau become more structured, but the N‐terminal region (NTR) and C‐terminal region (CTR) remain flexible.6 Many mutations of Tau have been linked to the development of neurodegenerative diseases, as have a large set of posttranslational modifications (PTMs) ranging from truncations to phosphorylation, glycosylation, glycation, and others.7 These PTMs occur throughout the protein, but those located within the MTBR are most relevant for tubulin binding and stabilization of microtubules.8
Scheme 1.
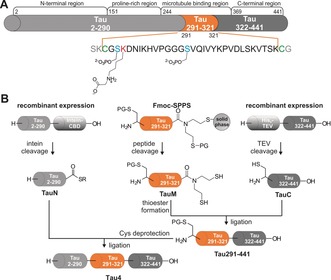
Semisynthetic scheme for introducing modifications in the MTBR between aa 291 and 321 in Tau4. A) Blue and red letters indicate modification sites and green letters show native cysteine residues for NCL. Three modifications were separately incorporated into TauM. B) Two ligation steps lead to semisynthetic Tau4 variants with different (n)PTMs. The SEA linker on TauM can be switched off to allow assembly from N‐ to C‐ as well as from C‐ to N‐terminus.
Furthermore, non‐enzymatic PTMs (nPTMs) have been identified on pathological Tau (NFTs), such as carboxymethylation of lysine side chains.9 However, no clear pattern of the effect of single phosphorylations or glycations within the MTBR has been identified yet, mainly due to the lack of homogeneously, site‐selectively modified Tau. Various strategies to generate Tau proteins with PTMs, or mimics thereof, have been pursued, including simple mutations to glutamic acid, enzymatic phosphorylations that have led to inhomogeneous Tau variants, as well as more complex semisynthesis strategies.10 Semisynthesis approaches have so far mostly targeted modifications within the C‐terminal domain of Tau.11 A recent, more elaborate semisynthesis of the longest Tau variant (Tau4, 441 aa) by the Lashuel group covers amino acids 246 to 441 by linking 4 synthetic peptide segments through convergent native chemical ligation (NCL) steps and provides access to four PTMs (AcK280, pY310, pS396, and pS404).11c Here, we establish a strategy that allows the site‐selective incorporation of modifications into the MTBR (aa 291–321) of Tau4 by taking advantage of the two native cysteines in the longest Tau variant. This strategy facilitates a 3‐segment semisynthesis, without any sequence manipulations (Scheme 1).
Within the synthetic segment (aa 291–321, TauM), two serine phosphorylations were incorporated in position 293 (part of a KXGS motif crucial for binding to MTs) and in position 305 (frequently found in AD patient brains).1b, 7e
To study carboxymethylation on lysine, the most abundant advanced glycation end product (AGE) in vivo,9b and a modification that has been repeatedly found on Tau isolated from NFTs, but that can neither be generated by enzymes nor mimicked by a natural mutation,7d lysine residue 294 was selected, which has been recently reported as a major target for acetylation and is located in the R2 repeat of the MTBR.12 Carboxymethylation is quite similar in size to acetylation but generates a different electrostatic environment with a negatively charged carboxyl group in close proximity to a positively charged secondary amine and has been described to interfere with tubulin association of Tau.9, 12
To obtain modified semisynthetic Tau4, the C‐terminal Tau segment (TauC, aa 322–441, Scheme 1) was cloned into a pET28a vector containing an N‐terminal hexahistidine tag (His6‐tag) and a tobacco etch virus (TEV) protease cleavage site. TauC bearing an N‐terminal cysteine residue was obtained with a yield of 2 mg L−1 of culture after optimized expression in E. coli. LC‐MS analysis confirmed the presence and sufficient purity of TauC (Supporting Information, Figure S1). The N‐terminal Tau segment (TauN, aa 1–290) fused to an Mxe GyrA intein and a chitin‐binding domain (CBD) was also obtained by E. coli expression using a pTWIN1 vector. TauN‐α‐thioester was released by incubation with 200 mm mercaptoethanesulfonate (MESNa) at pH 7.5 for 48 hours and obtained in 1 mg L−1 yield (Supporting Information, Figure S2). To take full advantage of the flexibility of our semisynthesis strategy, synthetic TauM peptides were equipped with controllable N‐ and C‐terminal functional groups for ligation, allowing the attachment of TauN and TauC segments in any order.
A bis(2‐sulfanylethyl)amido (SEA) linker produced TauM peptides with a C‐terminal thioester precursor that can be inactivated by forming an intramolecular disulfide (SEAoff).13 N‐terminal protection was initially achieved with l‐thiazolidine‐4‐carboxylic acid (Thz) but subsequently switched to acetamidomethyl (Acm)‐protected cysteine for stability reasons (see below). Phosphorylated serine residues (pS) were incorporated into TauM as Fmoc‐Ser(PO(OBzl)OH)‐OH (Supporting Information, Figures S3–5). To generate the nPTM on lysine 294, alkylation with bromoacetic acid t‐butyl ester was used. This reaction gave a mixture of unmodified, mono‐, double‐, and triple‐alkylated lysine 294. The best results were obtained when treating resin‐bound TauM peptide with 0.8 equiv. of bromoacetic acid t‐butyl ester and 1.8 equiv. of DIEA at 4 °C overnight (Supporting Information, Figures S6 and S7). Separation from non‐ and over‐alkylated peptides was possible by HPLC but resulted in only 1 % overall yield, compared to 6–18 % for other TauM peptides. HPLC analysis of purified TauM peptides always showed two peaks due to the isomers generated by the SEA‐amide/thioester equilibrium on the C‐terminus (Scheme 1 B).14
Assembly of full length Tau4 was explored from both directions, but the reaction of unmodified TauM carrying a C‐terminal SEAoff with TauN‐thioester produced only trace amounts of product after 24 h. We found no evidence that the inactivation of the SEA moiety and the simultaneous Thz deprotection by treatment with I2 caused this low reaction yield. C‐to‐N assembly with TauC and N‐terminally protected TauM resulted in much better yields after 48 h (Figure 1 A).15 The N‐terminal protecting group was subsequently removed by treatment with N‐methylhydroxylamine (for the Thz group16) or with AgOAc in acetic acid/water (for the Acm group17). All four variants of deprotected Tau291–441 were obtained in high purity and in yields of isolated product of between 31 and 60 % (Supporting Information, Figure S8). The second ligation reaction of Tau291–441 with TauN gave 70 % conversion after 4 h at 37 °C (Figure 1 B). All Tau variants were isolated in yields of 11–28 % over both steps, corresponding to 1.5 to 2.8 mg of protein (Supporting Information, Figure S9). However, the final products generated with Thz‐protected TauM segments showed a low intensity band at slightly higher molecular weight in SDS‐PAGE (Supporting Information, Figure S10) that could be identified by MS analysis as Tau4 containing an additional TauM segment. Partial removal of the Thz‐protecting group during the long 48 h ligation reaction, due to the presence of bis(2‐sulfanylethyl)amine released from TauM with a C‐terminal SEA group, is most likely the reason for this double incorporation of TauM. The SEA moiety can form a thiazolidine and compete with the N‐terminal protection of TauM.13b As this side product could not be separated from Tau4, a switch in the N‐terminal cysteine protecting group from Thz to Acm was necessary. With Acm‐protection, no undesired side‐products were observed in any of the four Tau4 variants generated here: Tau4, Tau4 [pS293], Tau4 [p305], and Tau [CML294] (Figure 2 A, S11). With these variants in hand, the effect of (n)PTMs on aggregation and tubulin binding was analyzed. As Tau is an IDP, no folding steps were required.
Figure 1.
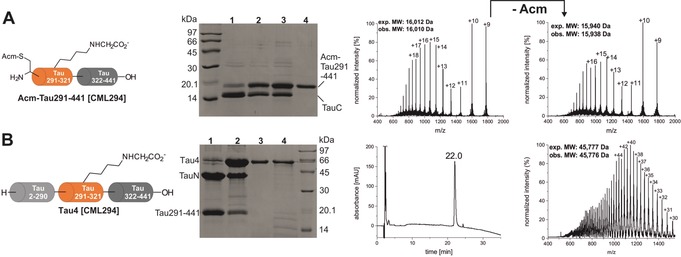
Semisynthesis of Tau4 [CML294]. A) First NCL with TauM [CML294] (4 mm) and TauC (2 mm) for 48 h at 37 °C, monitored by SDS‐PAGE. Lane 1: reaction at t=0; 2: reaction at 24 h; 3: reaction at 48 h; 4: purified Tau291‐441 [CML294]. Mass spectra show the ligation product before and after removal of the thiol‐protecting group Acm. B) Second ligation reaction between Tau291–441 [CML294] and TauN at 2 mm concentration each for 4 h at 37 °C, monitored by SDS‐PAGE. Lane 1: reaction at t=0; 2: reaction at 4 h; 3: purified Tau4 [CML294]; 4: recombinant Tau4. HPLC and mass spectrum of the purified ligation product Tau4 [CML294] are shown on the right. For analytical data of all four Tau4 variants, see Figures S8–11 in the Supporting Information.
Figure 2.
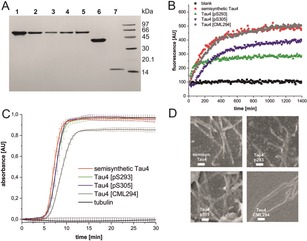
Analysis of full length Tau4 variants. A) SDS‐PAGE of Tau proteins: Lane 1: recombinantly produced Tau4; 2: Semisynthetic Tau4; 3: Tau4 [CML294]; 4: Tau4 [pS293]; 5: Tau4 [pS305]; 6: TauN; 7: TauC. B) Aggregation behavior of Tau4 variants in ThT assays (error bars omitted for clarity—the same figure with error bars is shown in Figure S13 in the Supporting Information). C) Tubulin‐binding of Tau4 variants measured by turbidity at 340 nm. Protein concentration and homogeneity was checked by SDS‐PAGE prior to tubulin polymerization (Supporting Information, Figure S15). D) SEM images of semisynthetic Tau4 variants after ThT aggregation assays (scale bar: 200 nm).
To ensure that unmodified, semisynthetic Tau4 behaves similar to recombinantly produced Tau4, the in vitro aggregation properties of both proteins were compared using a Thioflavin T (ThT)‐based fluorescence assay at 37 °C.18 Six independent series of experiments at physiological Tau concentrations (2 μm) with all samples measured in triplicate were combined.19 No differences in the aggregation kinetics and maximum fluorescence of recombinant and semisynthetic Tau4 were found (Supporting Information, Figure S12). Phosphorylation at serine 293 or 305 induced different effects. Tau4 [pS293] aggregated slightly faster than unmodified Tau4 and reached maximum fluorescence after 3 h, whereas unmodified Tau4 reached the plateau after only 12 h (Figure 2 B). The overall fluorescence intensity for Tau4 [pS293] was reduced to approximately 40 % when compared to unmodified Tau4. HPLC analysis of Tau4 variants remaining in the supernatant of aggregation assays indicated that for Tau4 [pS293] approximately 30 % of the protein remained soluble (Supporting Information, Figure S16). Interestingly, Tau4 [pS305] exhibited a significantly slower onset of aggregation with a noticeable lag phase of 1.5 h. The reduction in maximum ThT fluorescence observed here could not be linked to remaining soluble Tau4 [pS305] by HPLC analysis (Figure 2 B). Therefore, this behavior might be induced by different binding of ThT to varying fibril structures and/or due the direct influence of PTMs on ThT. All aggregated samples were analyzed by electron microscopy, which confirmed that fibrillar structures were formed (Figure 2 D).
This impact of a single phosphorylation on the aggregation kinetics of a 441 aa protein can be explained by the proximity of pS305 to the aggregation‐prone hexapeptide motif 306VQIVYK311 implicated in the initial steps of Tau oligomerization.20 The effect on overall aggregation is in line with previous reports that single phosphorylations at residues Ser214, 235, 262, or Thr231 can decrease the tendency of Tau to form aggregates. In contrast, CML294 exhibited similar aggregation kinetics as unmodified Tau4 (Figure 2 B). Taken together, these results suggest that converting a positively charged lysine side chain into a zwitterionic, carboxymethylated moiety within the putative β‐strand‐forming region of Tau4 (290KCGSKDNIKHVPGGGS305) less significantly affects Tau aggregation than introducing a single phosphorylation site at the highly conserved Ser293 and in close proximity to the crucial β‐strand region 306VQIVYK311. Given the role of Tau interactions with microtubules in neuronal cytoskeleton stability and neurodegeneration,4b, 6a the effect of these (n)PTMs on Tau‐induced polymerization of tubulin was assessed in vitro. Again recombinant and semisynthetic Tau4 behaved identically as both induce the assembly of tubulin into microtubules (MTs; Supporting Information, Figure S14). Whereas pS293 or pS305 did not influence Tau‐mediated tubulin polymerization, CML294 considerably inhibited MT assembly (Figure 2 C). These findings suggest that single phosphorylations within the MTBR are not sufficient to disrupt Tau interactions with tubulin, which is consistent with previous studies that such a disruption is linked to hyperphosphorylation,21 but a single CML can have a disruptive effect.
This finding is corroborated by a recent cryo‐electron microscopy structure of the Tau–MT interaction that specifically concentrates on the R1 and R2 repeats, in which K294 interacts with the acidic C‐terminal tail of tubulin.22 Such an interaction would be severely impaired by a zwitterion such as CML294.
Overall, a new semisynthesis targeting the central repeat domains of the longest human Tau variant was established. Two recombinantly produced segments embrace a synthetic peptide that can carry different modifications. Three Tau4 variants with defined (n)PTMs were generated that are of special interest since the MTBR plays a critical role in regulating Tau aggregation and MT binding. Aggregation assays indicated that phosphorylation of Tau4 [pS293] and Tau [pS305] attenuates Tau fibril formation, as described for other phosphorylations.23 At the same time, these Tau variants exhibited similar tubulin‐polymerization properties as unmodified Tau4. Finally, we show that carboxymethylation results in significant inhibition of MT formation but does not alter the aggregation properties of Tau4, suggesting that this modification, alone or in concert with other PTMs, may contribute to regulating the dynamics of Tau–MT interactions and potentially its physiological functions or dysfunction in tauopathies. This finding illustrates that so far underinvestigated nPTMs can induce significant physiological effects, as recently reported for the nPTM argpyrimidine,24 and need to be studied in more detail to understand their implications for diseases, such as AD and other tauopathies.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge support from Manuel Felkl with peptide synthesis and from Christian Görner with cloning of the TauN and TauC segments. We thank Meder Kamalov for help with collecting and processing SEM pictures.
D. Ellmer, M. Brehs, M. Haj-Yahya, H. A. Lashuel, C. F. W. Becker, Angew. Chem. Int. Ed. 2019, 58, 1616.
References
- 1.
- 1a. Lee V. M., Goedert M., Trojanowski J. Q., Annu. Rev. Neurosci. 2001, 24, 1121–1159; [DOI] [PubMed] [Google Scholar]
- 1b. Arendt T., Stieler J. T., Holzer M., Brain Res. Bull. 2016, 126, [DOI] [PubMed] [Google Scholar]; Part 3, 238–292. [Google Scholar]
- 2.
- 2a. Kosik K. S., Joachim C. L., Selkoe D. J., Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y.-C., Zaidi M. S., Wisniewski H. M., J. Biol. Chem. 1986, 261, 6084–6089. [PubMed] [Google Scholar]
- 3.
- 3a. Hardy J., Selkoe D. J., Science 2002, 297, 353–356; [DOI] [PubMed] [Google Scholar]
- 3b. Haass C., Selkoe D. J., Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R., Brain Res. Rev. 2000, 33, 95–130; [DOI] [PubMed] [Google Scholar]
- 4b. Goedert M., Jakes R., EMBO J. 1990, 9, 4225–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Cleveland D. W., Hwo S.-Y., Kirschner M. W., J. Mol. Biol. 1977, 116, 227–247; [DOI] [PubMed] [Google Scholar]
- 5b. Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W., Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Mandelkow E.-M., Mandelkow E., Cold Spring Harbor Perspect. Med. 2012, 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Kadavath H., Jaremko M., Jaremko Ł., Biernat J., Mandelkow E., Zweckstetter M., Angew. Chem. Int. Ed. 2015, 54, 10347–10351; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10488–10492; [Google Scholar]
- 6b. Li X.-H., Culver J. A., Rhoades E., J. Am. Chem. Soc. 2015, 137, 9218–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Šimić G., Leko M. B., Wray S., Harrington C., Delalle I., Jovanov-Milošević N., Bažadona D., Buée L., de Silva R., Di Giovanni G., Wischik C., Hof P. R., Biomolecules 2016, 6, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Min S.-W., Chen X., Tracy T. E., Li Y., Zhou Y., Wang C., Shirakawa K., Minami S. S., Defensor E., Mok S. A., Sohn P. D., Schilling B., Cong X., Ellerby L., Gibson B. W., Johnson J., Krogan N., Shamloo M., Gestwicki J., Masliah E., Verdin E., Gan L., Nat. Med. 2015, 21, 1154–1162; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Gong C.-X., Liu F., Iqbal K., Alzheimer′s Dementia 2016, 12, 1078–1089; [DOI] [PubMed] [Google Scholar]
- 7d. Gong C. X., Liu F., Grundke-Iqbal I., Iqbal K., J. Neural. Transm. 2005, 112, 813–838; [DOI] [PubMed] [Google Scholar]
- 7e. Martin L., Latypova X., Terro F., Neurochem. Int. 2011, 58, 458–471. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Cook C., Stankowski J., Carlomagno Y., Stetler C., Petrucelli L., Alzheimer's Res. Ther. 2014, 6, 29; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Carlomagno Y., Chung D.-e. C., Yue M., Castanedes-Casey M., Madden B. J., Dunmore J., Tong J., DeTure M., Dickson D. W., Petrucelli L., Cook C., J. Biol. Chem. 2017, 292, 15277–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Liu K., Liu Y., Li L., Qin P., Iqbal J., Deng Y., Qing H., Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 192–201; [DOI] [PubMed] [Google Scholar]
- 9b. Ikeda K., Higashi T., Sano H., Jinnouchi Y., Yoshida M., Araki T., Ueda S., Horiuchi S., Biochemistry 1996, 35, 8075–8083; [DOI] [PubMed] [Google Scholar]
- 9c. Ledesma M. D., Bonay P., Colaço C., J. Avila J. Biol. Chem. 1994, 269, 21614–21619. [PubMed] [Google Scholar]
- 10.
- 10a. Landrieu I., Lacosse L., Leroy A., Wieruszeski J.-M., Trivelli X., Sillen A., Sibille N., Schwalbe H., Saxena K., Langer T., Lippens G., J. Am. Chem. Soc. 2006, 128, 3575–3583; [DOI] [PubMed] [Google Scholar]
- 10b. Kamah A., Huvent I., Cantrelle F. X., Qi H., Lippens G., Landrieu I., Smet-Nocca C., Biochemistry 2014, 53, 3020–3032; [DOI] [PubMed] [Google Scholar]
- 10c. Jeganathan S., Hascher A., Chinnathambi S., Biernat J., Mandelkow E.-M., Mandelkow E., J. Biol. Chem. 2008, 283, 32066–32076. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Broncel M., Krause E., Schwarzer D., Hackenberger C. P. R., Chem. Eur. J. 2012, 18, 2488–2492; [DOI] [PubMed] [Google Scholar]
- 11b. Reimann O., Smet-Nocca C., Hackenberger C. P. R., Angew. Chem. Int. Ed. 2015, 54, 306–310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 311–315; [Google Scholar]
- 11c.A semisynthetic route to acetylated Tau variants targeting residues 246–389 was recently published—Haj-Yahya M., Lashuel H., J. Am. Chem. Soc. 2018, 140, 6611–6621. [DOI] [PubMed] [Google Scholar]
- 12. Kontaxi C., Piccardo P., Gill A. C., Front. Mol. Biosci. 2017, 4, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Ollivier N., Dheur J., Mhidia R., Blanpain A., Melnyk O., Org. Lett. 2010, 12, 5238–5241; [DOI] [PubMed] [Google Scholar]
- 13b. Dheur J., Ollivier N., Melnyk O., Org. Lett. 2011, 13, 1560–1563. [DOI] [PubMed] [Google Scholar]
- 14. Pira S. L., El Mahdi O., Raibaut L., Drobecq H., Dheur J., Boll E., Melnyk O., Org. Biomol. Chem. 2016, 14, 7211–7216. [DOI] [PubMed] [Google Scholar]
- 15. Ollivier N., Vicogne J., Vallin A., Drobecq H., Desmet R., El Mahdi O., Leclercq B., Goormachtigh G., Fafeur V., Melnyk O., Angew. Chem. Int. Ed. 2012, 51, 209–213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 213–217. [Google Scholar]
- 16. Seenaiah M., Jbara M., Mali S. M., Brik A., Angew. Chem. Int. Ed. 2015, 54, 12374–12378; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12551–12555. [Google Scholar]
- 17. Wang P., Dong S., Shieh J.-H., Peguero E., Hendrickson R., Moore M. A. S., Danishefsky S. J., Science 2013, 342, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smet-Nocca C. in Methods in Molecular Biology, 1st ed. (Ed.: S. S. B. M. N. York), Humana Press, Totowa, 2017. [Google Scholar]
- 19. Alonso A. d. C., Grundke-Iqbal I., Iqbal K., Nat. Med. 1996, 2, 783–787. [DOI] [PubMed] [Google Scholar]
- 20. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E., Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lathuilière A., Valdés P., Papin S., Cacquevel M., Maclachlan C., Knott G. W., Muhs A., Paganetti P., Schneider B. L., Sci. Rep. 2017, 10.1038/s41598-017-13786-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kellogg E. H., Hejab N. M. A., Poepsel S., Downing K. H., DiMaio F., Nogales E., Science 2018, 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Schneider A., Biernat J., von Bergen M., Mandelkow E., Mandelkow E. M., Biochemistry 1999, 38, 3549–3558; [DOI] [PubMed] [Google Scholar]
- 23b. Tepper K., Biernat J., Kumar S., Wegmann S., Timm T., Hübschmann S., Redecke L., Mandelkow E.-M., Müller D. J., Mandelkow E., J. Biol. Chem. 2014, 289, 34389–34407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matveenko M., Cichero E., Fossa P., Becker C. F., Angew. Chem. Int. Ed. 2016, 55, 11397–11402; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11569–11574. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary