

Abstract
This study is to investigate the genomic methylation in cervical adenocarcinoma in Xinjiang, China, using the DNA methylation analysis chips.
Methylation of 5 cases of cervical adenocarcinoma tissues and 5 cases of normal cervical tissues were analyzed by the Illumina 850K methylation chip. The genes with abnormal methylation modification were screened out and analyzed by the gene ontology (GO) functional annotation analysis. Enrichment analysis of kyoto encyclopedia of genes and genomes (KEGG) signal transduction pathways was also performed.
Totally 4056 sites showed differential expression patterns in cervical adenocarcinoma tissues compared to normal cervical tissues, of which 3738 were hypermethylated, and 318 were hypomethylated. The distribution of these sites covered from the 1st to 22nd chromosomes. GO functional annotation analysis showed that the differentially expressed genes in cervical adenocarcinoma tissues were mainly involved in the processes of tumor growth, development, metabolism, ion transport, transcriptional regulation, cell division, cell cycle regulation, and signal transduction. KEGG signaling pathway analysis showed that the most significantly different signaling pathway was the neuroactive ligand–receptor interaction. Gene-net-work analysis suggested that CCND1, CTNNB1, MAPK10, and PRKCA were involved.
Methylated genes are specifically expressed in cervical adenocarcinoma tissues in Xinjiang, China. Four of these genes (CCND1, CTNNB1, MAPK10, and PRKCA) with differential expression patterns may play important regulatory roles in cervical adenocarcinoma development through affecting the neuroactive ligand–receptor interaction.
Keywords: cervical adenocarcinoma, chip, gene, methylation, signal pathway
1. Introduction
With the wide application of cervical thin-layer cytology in cervical cancer screening, the incidence of cervical squamous cell cancer is declining, but the incidence of cervical adenocarcinoma is rising.[1–4] It has been shown that the detection rate of cervical cancer increased from 5% to 10%–20% and showed a trend to occur in younger people in the past 40 years.[5] Cervical adenocarcinoma originates from the cervical canal.[6] The early diagnosis of cervical adenocarcinoma is difficult due to its endogenous growth.[7] The disease is more common in young women.[8] The main clinical manifestations are the vaginal bleeding and vaginal discharge, most of which are nonspecific cases.[9] Therefore, the study about the pathogenesis of cervical adenocarcinoma is particularly urgent.
Most scholars believe that cervical adenocarcinoma is related with the human papilloma virus (HPV) infection and estrogen.[10,11] De Sanjose has summarized the data from 10,575 cases of cervical cancer HPV infection in 38 countries, including the North America, Central America, Europe, Asia, Africa, and Oceania.[12] The HPV positive rate in 951 cases of cervical adenocarcinoma is 65.7%.[12] Thus, the occurrence and development of cervical adenocarcinoma still need further study.
DNA methylation is an important part of epigenetics and regulation of genomic function.[13] Some papers have studied the mechanism and methylation of cervical adenocarcinoma,[14,15] but they only focused on one or a few genes.[16]
In this study, the genomic methylation of cervical adenocarcinoma was analyzed using the Illumina 850K methylation chip. The specific genes and their potential targets were analyzed by the bioinformatic analysis. Our findings may provide the basis for the further study of the molecular mechanism of cervical adenocarcinoma.
2. Materials and methods
2.1. Clinical data
Patients with cervical adenocarcinoma (n = 5) hospitalized at Affiliated Tumor Hospital of Xinjiang Medical University from January 2016 to June 2017 were enrolled in this study. Their age ranged from 42 to 60 years old, with the median age of 44 years old. They were treated with laparoscopic surgery or laparotomy, and adenocarcinoma tissues were collected during surgery. Inclusion criteria: patients with cervical adenocarcinoma confirmed by pathology; patients not receiving chemotherapy, radiotherapy, or hormone treatment, prior to surgery; and patients without other malignancies or severe medical conditions. Exclusion criteria: patients pathologically diagnosed as nonprimary cervical adenocarcinoma; the specimens failing to meet the experimental requirements; and carcinoma merged with other tumors. For the control, normal cervical tissues were collected from 5 patients who were hospitalized during the same period and received hysterectomy due to noncervical lesions of other gynecologic benign diseases. The control patients matched with the cervical adenocarcinoma patients in terms of geography, age, educational level, economic income, and body mass index. All patients and their families signed the informed consent form. The study was approved by the ethics review board of the Affiliated Tumor Hospital of Xinjiang Medical University.
2.2. Detection of methylation by chip
Genome-wide methylation was detected by the Illumina Human Methylation 850K Beadchip (Zhuoli Tech, Shanghai, China). Briefly, the genomic DNA was extracted using QIAamp DNA MiniKit (Qiagen, Hilden, Germany) and treated with bisulfite to convert cytosine to uracil. MSA4 was produced by alkali denaturation and genomic amplification. Probes were designed for the converted sequence and hybridized with the chip. The methylation status was determined by calculating the ratio of fluorescent signals.
2.3. Bioinformatic analysis
The differentially expressed genes were annotated with gene ontology (GO) and analyzed through kyoto encyclopedia of genes and genomes (KEGG) database. The GO annotation analysis included the biologic process, cellular component, and molecular function, which was performed with the DAVID database. P ≤ .05, which was given directly in the database, was used as the significance threshold to screen the results and to verify the P-value.
2.4. Statistical analysis
Data were processed using the SPSS 19.0 statistical software. Independent sample t test was used for comparison. The results of Illumina methylation chip were analyzed by the GenomeStudio software (Illumina, San Diego, CA). The methylation level of each site was calculated. Difference scores >13 or <−13, with Δβ > 0.2 or <−0.2, were used as cut-off points to screen the differential methylation sites. The similarity between the samples was analyzed by the principal component analysis (PCA) after beta value standardization through the dimensionality reduction method. The major signaling pathways by KEGG were selected according to the criteria of P < .05 and false discovery rate (FDR) < 0.05.
3. Results
3.1. Genome-wide detection of methylation in the cervical adenocarcinoma tissue by the methylation chip
The data from cervical adenocarcinoma and normal cervical tissue samples were preprocessed and normalized. Two-dimensional PCA analysis results were obtained and shown in Figure 1. Different colors indicate different samples. The closer the distance between samples was, the closer the expression pattern of sample gene was. Thus, the gene methylation profile of cervical adenocarcinoma samples was significantly different from that of the control samples.
Figure 1.
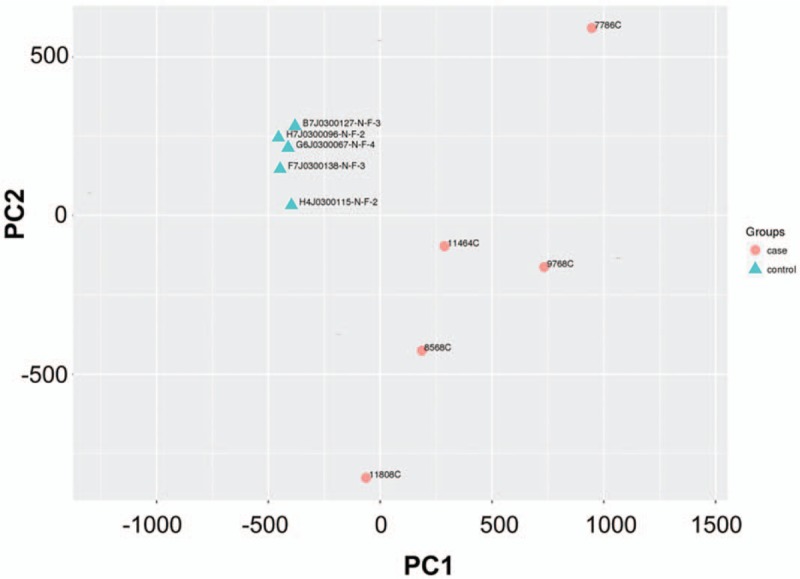
Principal component analysis (PCA) analysis of beta values after standardization of 2 groups of samples. PC1 = principal component 1, PC2 = principal component 2.
The 850K methylation sites in the cervical and normal adenocarcinoma tissues were analyzed using the Illumina 850K methylation chip. We found that 4056 sites showed differential expression in cervical adenocarcinoma tissues compared to normal cervical tissues, of which 3738 were hypermethylated and 318 were hypomethylated (Fig. 2). The different methylation sites in cervical adenocarcinoma were widely distributed, from chromosome 1 to the chromosome 22 (Fig. 3). Among them, chromosome 1 and chromosome 2 contained the largest number of methylation sites (418 and 271, respectively), while chromosome 21 and chromosome 18 contained the least number of methylation sites (21 and 42, respectively).
Figure 2.
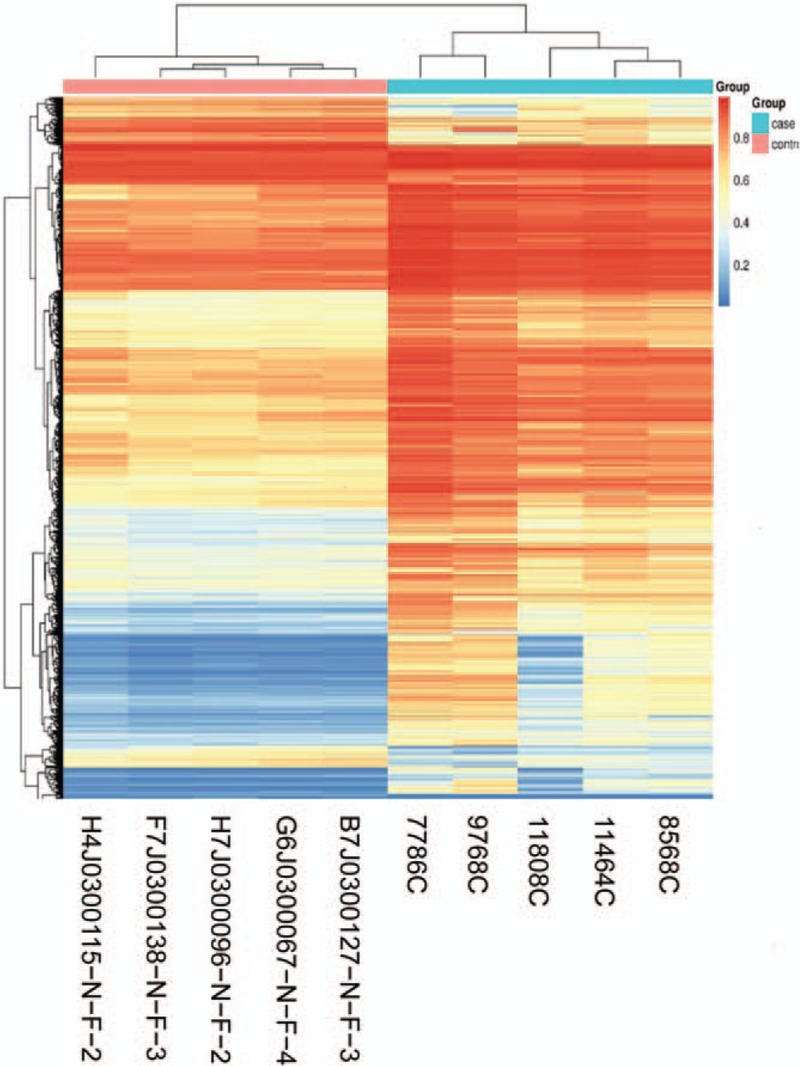
Heat map of 2 groups of differentially methylated sites and samples.
Figure 3.
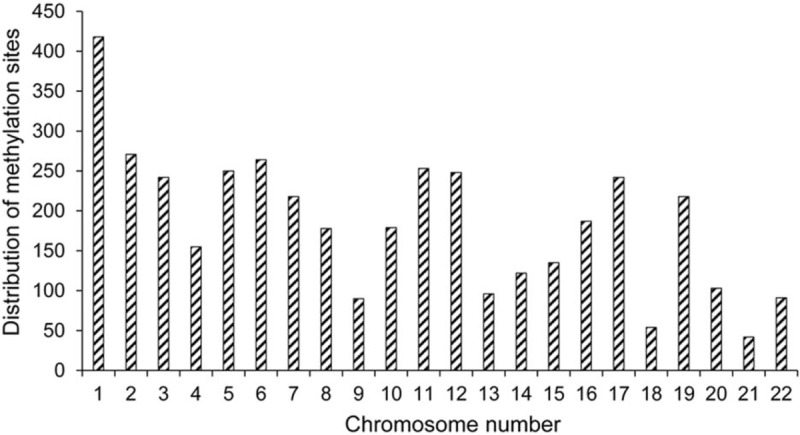
Distribution of methylation sites in the chromosomes.
3.2. Gene ontology functional analysis
The GO functional annotation analysis of the differentially expressed methylation sites showed that these genes were mainly enriched in cellular processes, histomorphology, nervous system development, and biologic processes (Fig. 4). The results of cell component analysis showed that molecules distributed in the cytoplasm, cell protrusions, neurons, and intracellular components were significantly enriched. At the molecular level, functional annotation showed that the highly enriched genes were related with sequence-specific DNA binding, phosphotransferase, protein kinase, and anion transmembrane transport.
Figure 4.
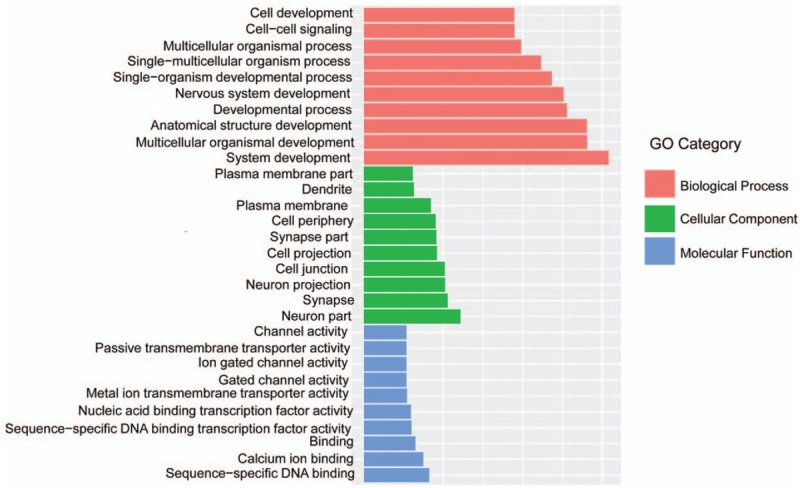
Significant enrichment of gene ontology histograms.
3.3. KEGG signaling pathway analysis
We further studied the signal pathways using the KEGG database. According to the criteria of P < .05 and FDR < 0.05, the most prominent 20 major signaling pathways were selected (Fig. 5). Furthermore, the 3000 methylation variable position genes with the most significant difference were further analyzed by KEGG. A total of 210 genes were screened from 10 pathways. The 210 genes were analyzed for inter-gene interaction, and 8 pathways were found to be matched best, including the neuroactive ligand–receptor interaction, calcium signaling pathway, focal adhesion, pathways in cancer, regulation of actin cytoskeleton, MAPK signaling pathway, Wnt signaling pathway, and tight junction. The most significant signal pathway was the neuroactive ligand–receptor interaction. Additionally, the Gene-net-work analysis showed that the genes of CCND1, CTNNB1, MAPK10, and PRKCA were involved in multiple pathways (Fig. 6), indicating their possible roles in cervical adenocarcinoma development.
Figure 5.
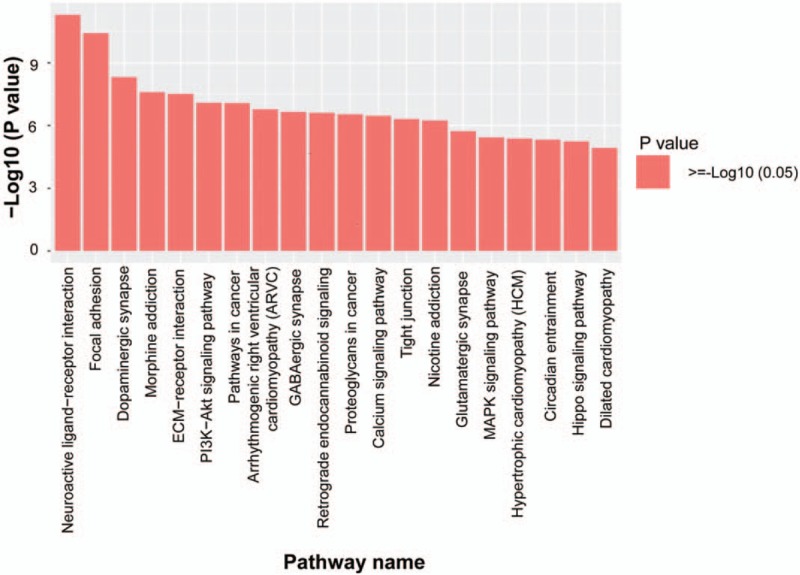
Significantly enriched KEGG pathway histogram. The abscissa represents the significantly enriched KEGG pathway name, and the ordinate represents −Log10 of the P-value. A larger ordinate indicates that the pathway is more enriched. KEGG = kyoto encyclopedia of genes and genomes.
Figure 6.
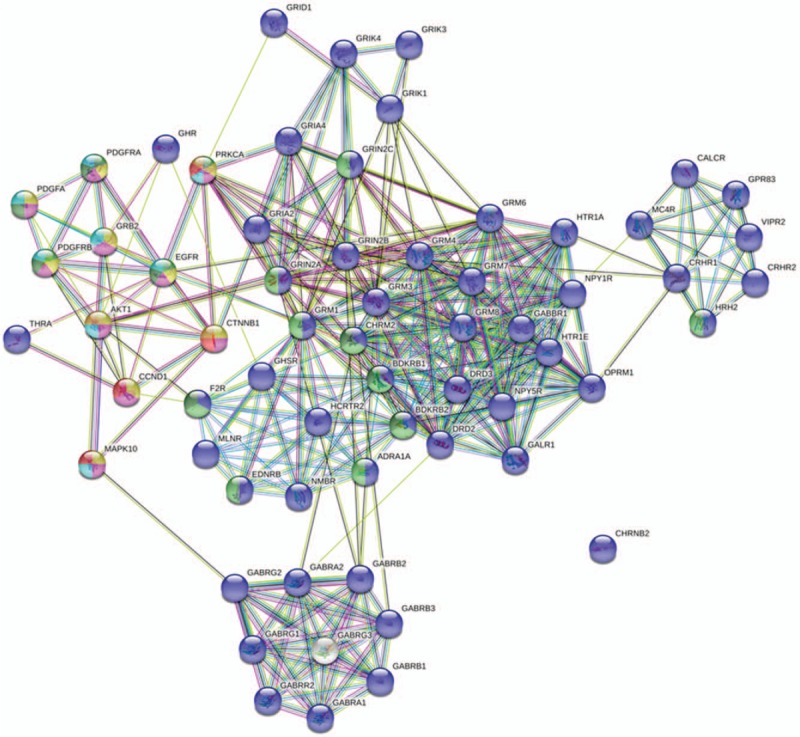
Analysis results of Gene-net-work.
4. Discussion
Genome-wide methylation is one of the most well-studied mechanisms in the field of epigenetics, which is closely related to the occurrence and development of diseases, including the cervical cancer. Jha et al found that the tumor suppressor genes p53 and p73 showed higher methylation degree in cervical cancer samples compared with normal samples.[17] Additionally, previous methylation analysis of 7 genes by QMSPCR showed that the detection sensitivities of SOX1, PAX1, ZNF582, PTPRR, AJAP1, HS3ST2, and POU4F3 were 100%, 86%, 71%, 86%, 86%, 57%, and 100%, respectively; and the specificities were 67%, 79%, 85%, 50%, 52%, 96%, and 52%, respectively.[18] Our study showed that 4056 sites showed differential expressions in cervical adenocarcinoma tissues compared to normal cervical tissues, of which 3738 were hypermethylated and 318 were hypomethylated. The different methylation sites in cervical adenocarcinoma were widely distributed, from chromosome 1 to chromosome 22. These results indicate that the methylation sites in cervical adenocarcinoma are widely distributed. The numbers and distribution of these sites in different chromosomes vary greatly.
We also analyzed these differentially expressed genes through the GO functional annotation. We found that differentially methylated genes in cervical adenocarcinoma were mainly involved in tumor growth, development, metabolism, ion transport, transcriptional regulation, cell division, cell cycle regulation, and signal transduction process. Many studies have shown that methylation is involved in the development of cervical cancer. For instance, several methylation genes like CDH1, CDKN2A, Rb1, and TP53 are involved in ion transport, cell cycle regulation, tumor growth, and metabolism in cervical cancer.[19] However, methylation of the p16 gene may play an important role in the carcinogenesis of cervical cancer in cooperation with HPV infection.[20]
It has been shown that the hypermethylation of genes can affect a variety of cellular pathways, such as DNA repair (mismatch repair gene and O6-methylguanine-DNA methyltransferase), apoptosis-related protein kinase (DAPK), apoptosis-activating factor, and cell cycle (p14 gene).[21–25] Pathway analysis in this study showed that differentially methylated genes were mainly involved in tumorigenesis, apoptosis, immune, and signal transduction. Among these pathways, the most significant one was the neural active ligand–receptor interaction pathway (neuroactive ligand–receptor interaction), which is a collection of all receptors and ligands on the plasma membrane associated with intracellular and extracellular signaling pathways.[26]
Further cluster analysis found that the differentially methylated genes covered a variety of different functional communities, indicating that there are many types of genes involved in regulation of the occurrence and development of cervical adenocarcinoma. Jha et al have found some driving genes highly relevant to cervical adenocarcinoma, using the next-generation sequencing technologies, including ND5, TG, AGXT, MYH7, FGA, APOC3, APOA1, C3, APCS, FBF1, SERPINA1, S100A9, and TXNIP GC.[27] More studies have shown that PAX1, SOX1, EPB41L3, ARHGAP6, HAND2, Lhx9 Hey2, NKX2-2, PCDH10, PITX2, Prox1, Tbx3, IKBKG, rab6c, and DAPK1 could be used as screening biomarkers for cervical cancer diagnosis.[28–30] Moreover, our Gene-net-work analysis showed that methylation-deficient genes such as CCND1, CTNNB1, MAPK10, and PRKCA were involved in multiple cervical adenocarcinoma-related pathways and may also serve as candidate molecular markers of cervical adenocarcinoma, which however needs more study.
In conclusion, we used the illumina Human Methylation 850K Beadchip methylation chip to detect the methylation sites in cervical adenocarcinoma and normal cervical tissues. Our findings show that the methylation modification in cervical adenocarcinoma cells is abnormal. The hypermethylation sites occur more frequently and are mainly enriched in functional categories such as the tumor growth, development, and metabolism. The most abundant signaling pathway is the signal pathway of neuroactive ligand–receptor interaction, indicating that the abnormal methylation may be involved in the development of cervical adenocarcinoma. Our work is only one of the beginning studies of genomic methylation in cervical adenocarcinoma. We need more comprehensive and more accurate results, and also need to expand the sample size for further analysis and verification. These could be very helpful for the early diagnosis, treatment, and prognosis of cervical adenocarcinoma, and for the development of targeted drugs.
Author contributions
Data curation: Lipa Mei.
Project administration: Guzhalinuer Abulizi.
Writing – original draft: Min Yuan, Jianlin Yuan.
Footnotes
Abbreviations: FDR = false discovery rate, GO = gene ontology, HPV = human papilloma virus, PCA = principal component analysis.
This study is supported by the Natural Science Foundation Project of Xinjiang Uygur Autonomous Region (2017D01C396, 2017.07.01-2020.06.30).
The authors have no conflicts of interest to disclose.
References
- [1].Gien LT, Beauchemin MC, Thomas G. Adenocarcinoma: a unique cervical cancer. Gynecol Oncol 2010;116:140–6. [DOI] [PubMed] [Google Scholar]
- [2].Pimenta JM, Galindo C, Jenkins D, et al. Estimate of the global burden of cervical adenocarcinoma and potential impact of prophylactic human papillomavirus vaccination. BMC Cancer 2013;13:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917. [DOI] [PubMed] [Google Scholar]
- [4].van der Horst J, Siebers AG, Bulten J, et al. Increasing incidence of invasive and in situ cervical adenocarcinoma in the Netherlands during 2004-2013. Cancer Med 2017;6:416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Howlett RI, Marrett LD, Innes MK, et al. Decreasing incidence of cervical adenocarcinoma in Ontario: is this related to improved endocervical Pap test sampling? Int J Cancer 2007;120:362–7. [DOI] [PubMed] [Google Scholar]
- [6].K K. Histopathological study of early cervical adenocarcinoma. Acta Med Kinki Univ 1983;8:37–46. [Google Scholar]
- [7].Obata N, Sasaki A, Takeuchi S, et al. Clinico-pathologic study on the early diagnosis of cervical adenocarcinoma [in Japanese]. Nihon Sanka Fujinka Gakkai Zasshi 1987;39:771–6. [PubMed] [Google Scholar]
- [8].McKeever A. Adenocarcinoma: yes cervical cancer can still happen in young women. J Obstet Gynecol Neonatal Nurs 2013;42:S92–3. [Google Scholar]
- [9].Weegar R, Kvist M, Sundstrom K, et al. Finding cervical cancer symptoms in Swedish clinical text using a machine learning approach and NegEx. AMIA Annu Symp Proc 2015;2015:1296–305. [PMC free article] [PubMed] [Google Scholar]
- [10].Q Wang MZ, Zeng S. Analysis of high-risk human papilloma virus infection in patients with cervical adenocarcinoma. China Med Herald 2013;10:57–9. [Google Scholar]
- [11].Kumar MM, Davuluri S, Poojar S, et al. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: a transcriptomic study. Tumour Biol 2016;37:4409–20. [DOI] [PubMed] [Google Scholar]
- [12].de Sanjose S, Quint WG, Alemany L, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010;11:1048–56. [DOI] [PubMed] [Google Scholar]
- [13].Laird PW, Jaenisch R. The role of DNA methylation in cancer genetic and epigenetics. Ann Rev Genet 1996;30:441–64. [DOI] [PubMed] [Google Scholar]
- [14].Lin YW, Chung MT, Lai HC, et al. Methylation analysis of SFRP genes family in cervical adenocarcinoma. J Cancer Res Clin Oncol 2009;135:1665–74. [DOI] [PubMed] [Google Scholar]
- [15].Ma D, Jiang C, Hu X, et al. Methylation patterns of the IFN-gamma gene in cervical cancer tissues. Sci Rep 2014;4:6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Banzai C, Nishino K, Quan J, et al. Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int J Clin Oncol 2014;19:127–32. [DOI] [PubMed] [Google Scholar]
- [17].Jha AK, Nikbakht M, Jain V, et al. Promoter hypermethylation of p73 and p53 genes in cervical cancer patients among north Indian population. Mol Biol Rep 2012;39:9145–57. [DOI] [PubMed] [Google Scholar]
- [18].Chang CC, Huang RL, Wang HC, et al. High methylation rate of LMX1A, NKX6-1, PAX1, PTPRR, SOX1, and ZNF582 genes in cervical adenocarcinoma. Int J Gynecol Cancer 2014;24:201–9. [DOI] [PubMed] [Google Scholar]
- [19].Cardoso MFS, Castelletti CHM, Lima-Filho JL, et al. Putative biomarkers for cervical cancer: SNVs, methylation and expression profiles. Mutat Res 2017;773:161–73. [DOI] [PubMed] [Google Scholar]
- [20].Ahmad A, Raish M, Shahid M, et al. The synergic effect of HPV infection and epigenetic anomaly of the p16 gene in the development of cervical cancer. Cancer Biomark 2017;19:375–81. [DOI] [PubMed] [Google Scholar]
- [21].Xiong HL, Liu XQ, Sun AH, et al. Aberrant DNA methylation of P16, MGMT, hMLH1 and hMSH2 genes in combination with the MTHFR C677T genetic polymorphism in gastric cancer. Asian Pac J Cancer Prev 2013;14:3139–42. [DOI] [PubMed] [Google Scholar]
- [22].Esposito A, Bardelli A, Criscitiello C, et al. Monitoring tumor-derived cell-free DNA in patients with solid tumors: clinical perspectives and research opportunities. Cancer Treat Rev 2014;40:648–55. [DOI] [PubMed] [Google Scholar]
- [23].Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol 2013;23:620–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hong L, Ahuja N. DNA methylation biomarkers of stool and blood for early detection of colon cancer. Genet Test Mol Biomarkers 2013;17:401–6. [DOI] [PubMed] [Google Scholar]
- [25].Chaar I, Amara S, Elamine OE, et al. Biological significance of promoter hypermethylation of p14/ARF gene: relationships to p53 mutational status in Tunisian population with colorectal carcinoma. Tumour Biol 2014;35:1439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lauss M, Kriegner A, Vierlinger K, et al. Characterization of the drugged human genome. Pharmacogenomics 2007;8:1063–73. [DOI] [PubMed] [Google Scholar]
- [27].Jha A, Khan Y, Mehdi M, et al. Towards precision medicine: discovering novel gynecological cancer biomarkers and pathways using linked data. J Biomed Semantics 2017;8:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kan YY, Liou YL, Wang HJ, et al. PAX1 methylation as a potential biomarker for cervical cancer screening. Int J Gynecol Cancer 2014;24:928–34. [DOI] [PubMed] [Google Scholar]
- [29].Clarke MA, Luhn P, Gage JC, et al. Discovery and validation of candidate host DNA methylation markers for detection of cervical precancer and cancer. Int J Cancer 2017;141:701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bhat S, Kabekkodu SP, Varghese VK, et al. Aberrant gene-specific DNA methylation signature analysis in cervical cancer. Tumour Biol 2017;39:1010428317694573. [DOI] [PubMed] [Google Scholar]