

Abstract
Key points
Myotonic dystrophy type 1 (DM1), the second most common muscular dystrophy and most prevalent adult form of muscular dystrophy, is characterized by muscle weakness, wasting and myotonia.
A microsatellite repeat expansion mutation results in RNA toxicity and dysregulation of mRNA processing, which are the primary downstream causes of the disorder.
Recent studies with DM1 participants demonstrate that exercise is safe, enjoyable and elicits benefits in muscle strength and function; however, the molecular mechanisms of exercise adaptation in DM1 are undefined.
Our results demonstrate that 7 weeks of volitional running wheel exercise in a pre‐clinical DM1 mouse model resulted in significantly improved motor performance, muscle strength and endurance, as well as reduced myotonia.
At the cellular level, chronic physical activity attenuated RNA toxicity, liberated Muscleblind‐like 1 protein from myonuclear foci and improved mRNA alternative splicing.
Abstract
Myotonic dystrophy type 1 (DM1) is a trinucleotide repeat expansion neuromuscular disorder that is most prominently characterized by skeletal muscle weakness, wasting and myotonia. Chronic physical activity is safe and satisfying, and can elicit functional benefits such as improved strength and endurance in DM1 patients, but the underlying cellular basis of exercise adaptation is undefined. Our purpose was to examine the mechanisms of exercise biology in DM1. Healthy, sedentary wild‐type (SED‐WT) mice, as well as sedentary human skeletal actin‐long repeat animals, a murine model of DM1 myopathy (SED‐DM1), and DM1 mice with volitional access to a running wheel for 7 weeks (EX‐DM1), were utilized. Chronic exercise augmented strength and endurance in vivo and in situ in DM1 mice. These alterations coincided with normalized measures of myopathy, as well as increased mitochondrial content. Electromyography revealed a 70–85% decrease in the duration of myotonic discharges in muscles from EX‐DM1 compared to SED‐DM1 animals. The exercise‐induced enhancements in muscle function corresponded at the molecular level with mitigated spliceopathy, specifically the processing of bridging integrator 1 and muscle‐specific chloride channel (CLC‐1) transcripts. CLC‐1 protein content and sarcolemmal expression were lower in SED‐DM1 versus SED‐WT animals, but they were similar between SED‐WT and EX‐DM1 groups. Chronic exercise also attenuated RNA toxicity, as indicated by reduced (CUG)n foci‐positive myonuclei and sequestered Muscleblind‐like 1 (MBNL1). Our data indicate that chronic exercise‐induced physiological improvements in DM1 occur in concert with mitigated primary downstream disease mechanisms, including RNA toxicity, MBNL1 loss‐of‐function, and alternative mRNA splicing.
Keywords: Myotonic dystrophy type 1, Exercise physiology, RNA toxicity, RNA processing, Muscleblind‐like 1, Mitochondria
Key points
Myotonic dystrophy type 1 (DM1), the second most common muscular dystrophy and most prevalent adult form of muscular dystrophy, is characterized by muscle weakness, wasting and myotonia.
A microsatellite repeat expansion mutation results in RNA toxicity and dysregulation of mRNA processing, which are the primary downstream causes of the disorder.
Recent studies with DM1 participants demonstrate that exercise is safe, enjoyable and elicits benefits in muscle strength and function; however, the molecular mechanisms of exercise adaptation in DM1 are undefined.
Our results demonstrate that 7 weeks of volitional running wheel exercise in a pre‐clinical DM1 mouse model resulted in significantly improved motor performance, muscle strength and endurance, as well as reduced myotonia.
At the cellular level, chronic physical activity attenuated RNA toxicity, liberated Muscleblind‐like 1 protein from myonuclear foci and improved mRNA alternative splicing.
Introduction
Myotonic dystrophy type 1 (DM1) affects ∼1/8000 individuals, making it the most common form of myotonic dystrophy (Chau & Kalsotra, 2015). It is also the most prevalent adult form of muscular dystrophy, as well as the second most common type of muscular dystrophy after Duchenne muscular dystrophy (DMD; Chau & Kalsotra, 2015). DM1 is an autosomal dominant trinucleotide repeat neuromuscular disorder with multisystem involvement, which is most prominently characterized by skeletal muscle weakness, wasting, myotonia and insulin resistance. DM1 is caused by a CTG microsatellite repeat expansion mutation in the 3ʹ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene (Cho & Tapscott, 2007). Healthy individuals have between 5–37 repetitions of the CTG trinucleotide sequence, whereas pre‐mutation symptoms will manifest with 38–49 repeats and individuals with 50 repeats or more are diagnosed with DM1 (Cho & Tapscott, 2007). Generally, the greater the number of repeats portends an earlier age of symptom onset, as well as a more severe phenotype (Brook et al. 1992). Furthermore, the appearance and severity of the disorder is also related to the extent and pattern of epigenetic methylation of CpG islands upstream of the repeats (Barbé et al. 2017).
The DM1 mutation results in expanded DMPK transcripts, which form stable, double‐stranded hairpin structures that aggregate as foci within nuclei. Via a toxic gain‐of‐function mechanism, these CUG expansions result in the dysregulation of several important RNA‐binding proteins (RNABPs), namely Muscleblind‐like 1 (MBNL1; Chau & Kalsotra, 2015). MBNL1 becomes sequestered by the expanded CUG nuclear aggregates, which leads to MBNL1 loss‐of‐function (Kalsotra et al. 2014; Chau & Kalsotra, 2015). MBNL1 plays critical roles in many steps of RNA metabolism, including primarily pre‐mRNA processing, as well as in the stability and transport of newly synthesized transcripts. Defects in these events due to MBNL1 loss‐of‐function contribute significantly to the clinical presentation of the disorder. Thus, the downstream functional consequences of the DM1 mutation are ultimately due to the toxic gain‐of‐function of DMPK mRNA within nuclei that alters RNABP function. For example, the myotonia of DM1 is largely attributed to the fetal isoform splicing pattern of the muscle‐specific chloride channel (CLC‐1; Chau & Kalsotra, 2015). The MBNL1 loss‐of‐function contributes to the aberrant splicing pattern of CLC‐1 mRNA, which includes exon 7a. CLC‐1 exon 7a, which is normally excluded from the mature transcript in adult skeletal muscle, contains a premature stop codon resulting in nonsense‐mediated decay of the mRNA (Wheeler et al. 2007). An adult‐to‐fetal switch in pre‐mRNA splicing patterns characterizes the misregulated mRNA processing events associated with the DM1 myopathy. These immature isoforms are unable to meet the requirements of adult skeletal muscle, which then manifest into DM1 symptoms.
There is no cure for DM1, which signifies a critically unmet clinical need. Exercise is a safe, effective, accessible and therefore practical lifestyle intervention that reduces all‐cause mortality and improves quality of life, in part by evoking favourable systemic adaptations via the stimulation of myriad gene expression programmes (Egan & Zierath, 2013). Studies examining the efficacy of exercise training in DM1 participants clearly demonstrate that physical activity can elicit modest, but significant physiological benefits, such as improved strength, endurance, function and quality of life metrics (Lindeman et al. 1995; Tollbäck et al. 1999; Aldehag et al. 2005, 2013; Ørngreen et al. 2005; Kierkegaard & Tollbäck, 2007; Brady et al. 2014; Ng et al. 2018; Dial et al. 2018a). Most recently, work in a pre‐clinical murine model of DM1 demonstrated that chronic exercise improved the spliceopathy of select genes germane to the disorder, such as CLC‐1, sarco/endoplasmic reticulum Ca2+‐ATPase 1 (SERCA) and ryanodine receptor 1 (RYR1; Ravel‐Chapuis et al. 2018). Other than these data, the molecular mechanisms of exercise adaptation in DM1 are unknown. Understanding the cellular processes that drive exercise‐induced remodelling in DM1 is important because it (1) will increase our knowledge of the basic biological mechanisms of the disorder, and (2) may assist in the discovery of more effective lifestyle and/or pharmacological interventions to mitigate the disease. Therefore, the purpose of this study was to examine the cellular and molecular mechanisms of exercise‐induced neuromuscular plasticity in a pre‐clinical model of DM1. We hypothesized that chronic exercise would ameliorate the disease phenotype at the physiological, cellular and molecular levels.
Methods
Ethical approval
All experiments performed in the current study are listed in the investigators’ Animal Utilization Protocol No. 18‐05‐25, which was approved by the Animal Research Ethics Board of McMaster University operating under the auspices of the Canadian Council for Animal Care (CCAC). Furthermore, this study complies with the animal ethics checklist as outlined in ‘Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology’.
Animals
Three‐ to six‐month‐old human skeletal actin‐long repeat (HSALR; DM1) mice and wild‐type (WT) FVB/N animals (The Jackson Laboratory, Bar Harbor, ME, USA) were utilized in this study. DM1 mice within this age range recapitulate the same degree of pathogenic muscle impairment and severity of DM1 symptoms (Jones et al. 2012; Brockhoff et al. 2017; Ravel‐Chapuis et al. 2017, 2018). DM1 mice were a kind gift from Dr Charles Thornton at the University of Rochester (Mankodi et al. 2000). All animals were reared and transported under conditions laid out by the CCAC. Three groups (n = 12–18/group) were defined: (1) sedentary WT mice (SED‐WT; 6 males, 6 females), (2) SED‐DM1 (9 males, 9 females, and (3) DM1 mice with access to a home cage running wheel for 7 weeks (EX‐DM1; 8 males, 8 females). Food and water were provided ad libitum.
Chronic exercise
EX‐DM1 mice were individually housed with volitional access to a home cage running wheel (Columbus Instruments, Columbus, OH, USA). The number of revolutions was recorded every 10 min daily for 7 weeks. The effects of chronic, volitional exercise in WT mice were not examined in the current study as there is an abundance of literature investigating the impact of exercise training on healthy, non‐dystrophic animals (Egan & Zierath, 2013), and our focus here is the effects of habitual physical activity in dystrophic context.
Performance testing
In order to assess functional, physiological adaptations to volitional exercise in DM1 mice, a battery of tests and measures were applied to examine muscle strength and balance. One day prior to tissue collection, pen and grip tests were performed as previously described to assess muscular endurance and balance, as well as strength, respectively (Willmann et al. 2011). Briefly, for the pen test, mice were placed on a pen suspended ∼50 cm above a foam mat and the duration that the animal remained on the pen before falling was recorded. The average of three attempts for each mouse was applied. For grip strength, mice grasped a wire mesh with either all limbs, or forelimbs only, and the animal was slowly pulled away horizontally by the base of the tail. A transducer recorded the force exerted against the mesh (Columbus Instruments), and the average of three trials was utilized.
Electromyography (EMG)
To determine the effect of exercise on a cardinal symptom of DM1 biology, EMG was utilized to quantify the severity of myotonia in DM1 mice. EMG recordings were obtained using two tungsten intramuscular electrodes (6–8 MΩ, FHC, Bowdoinham, ME, USA) with a fixed interelectrode distance of 2 mm. Insulation was removed from the tip of each electrode to expose the bare tungsten as a recording surface. This pair of electrodes was inserted into the muscle belly to rest just beneath the fascia in anaesthetized (150 mg/kg ketamine/10 mg/kg xylazine (K/X) via intraperotineal (i.p.) injection) mice. A ground electrode (a third tungsten electrode with 1 cm of insulation removed) was positioned under the retracted skin in the inguinal region. The intramuscular EMG signal was preamplified at × 35 (EQ Inc., Chalfont, PA, USA) and passed through a second‐stage, variable‐gain amplifier (×20, Model D423A, York University Electronics Shop, Toronto, Ontario, Canada). The EMG signal was digitized at 1000 Hz (Micro3 1401, Cambridge Electronics Design, Cambridge, UK), and high‐pass filtered offline at 25 Hz (Spike2 version 7, Cambridge Electronics Design). To elicit a myotonic response, a 50 Hz train of twenty 200 μs square‐wave pulses (Digitimer constant‐current stimulator, model DS7A, Hertfordshire, UK) were applied using a small custom‐built probe positioned proximal to the recording electrodes on the surface of the muscle. Five separate trains were applied to the medial gastrocnemius (GAST), tibialis anterior (TA), and triceps brachii (TRI). Muscle activity before and after each train of stimuli was assessed offline (Spike2 version 7). The response to tetanic stimulation was characterized by both burst amplitude and burst duration. Root mean square (RMS) was used as a measure of EMG amplitude for a 1 s period after each train of 50 Hz stimulation (Spike2 version 7). Baseline EMG, recorded over a 1 s period prior to stimulation, was subtracted from the poststimulus EMG. The burst duration value provides a measure of the time it took for EMG to return to baseline when stimulation led to a burst of EMG activity. Anaesthetized mice were killed by cervical dislocation at the end of the experiments.
In situ skeletal muscle fatigue assessment
In situ force assessment of the triceps surae complex was performed to investigate muscle‐specific performance adaptations in DM1 animals. Separate cohorts of the three experimental groups were anaesthetised (i.p K/X injection) and their triceps surae complex was distally attached to a force transducer (Grass Instruments, West Warwick, RI, USA) and a fatigue protocol was employed as described earlier (Krause et al. 2008). The protocol consisted of eliciting maximal tetanic contractions at 30 Hz for 333 ms in each 1 s for a total duration of 5 min as previously described (Shortreed et al. 2009). Force output was expressed relative to the tension produced during the initial contraction. Anaesthetized mice were killed by cervical dislocation at the end of the experiments.
Tissue collection
Skeletal muscles from all experimental groups were collected 24 h after access to the wheel was removed in order to perform a battery of cellular and molecular analyses. Animals were killed via cervical dislocation and the TA, extensor digitorum longus (EDL), soleus (SOL), quadriceps and GAST muscles were harvested and immediately snap frozen in liquid nitrogen. Contralateral EDL and SOL muscles were immersed in Optimal Cutting Temperature compound (OCT; VWR, Mississaugua, Ontario, Canada) and frozen in melting isopentane pre‐cooled in liquid nitrogen. Muscles examined in the EMG and in situ force production experiments were not used for further cellular and molecular analyses. GAST, EDL, QUAD and TA were used for cellular and molecular analyses because they share a similar fibre‐type composition (Bloemberg & Quadrilatero, 2012), facilitating complementary analyses and allowing for a more thorough investigation into the exercise‐induced adaptations in DM1 biology. Indeed, by using muscles of reasonably similar function and metabolic profile, conclusions reached for each experiment, regardless of the specific muscle used, can therefore be linked for a more comprehensive understanding of the effects of volitional exercise. In addition, multiple studies have shown that these muscles are recruited in mice during running and adapt significantly to exercise (Allen et al. 2001; Call et al. 2010).
Protein extraction and Western blotting
Western blotting was performed to measure both the levels of proteins that are known to respond to exercise, as well as those germane to the DM1 biology. TA muscles were added to a solution of RIPA buffer (20 μL of RIPA per 1 mg muscle weight; Sigma‐Aldrich, Oakville, Ontario, Canada) with dissolved protease and phosphatase inhibitor cocktail tablets (Roche, Mississauga, Ontario, Canada). Samples were then homogenized using steel ball bearings and a motorized tissue lyser (Qiagen, Hilden, Germany). Following centrifugation at 14,000 g for 15 min and aspiration of the supernatant, protein concentration was determined via a bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA). Muscle protein content was analysed via standard Western blotting techniques, using SDS‐PAGE to separate proteins by size, and transferred onto a nitrocellulose membrane (Thermo Fisher Scientific). Commercially available Ponceau S solution (Sigma‐Aldrich) was used to assess equal loading between samples. Membranes were blocked for 1 h in 5% bovine serum albumin (BSA) in Tris‐buffered saline with 1% Tween‐20 (TBS‐T) solution. Membranes were then incubated with a diluted solution of primary antibodies in a 5% BSA in TBS‐T buffer. Antibodies and dilutions were as follows: mitochondrial oxidative phosphorylation (OXPHOS) complex cocktail (1:1000; Abcam, Toronto, Ontario, Canada), AMP‐activated protein kinase (AMPK; 1:1000; CST, Beverly, MA, USA), peroxisome proliferator‐activated receptor γ coactivator‐1α (PGC‐1α; 1:1000; EMD, Etobicoke, Ontario, Canada), pan Ca2+/calmodulin‐dependent protein kinase II (CAMKII: 1:10,000; CST), skeletal muscle‐specific chloride channel 1 (CLC‐1: 1:200; ADI, San Antonio, TX, USA), and MBNL1 (1:1000; Santa Cruz Biotechnology, Dallas, TX, USA). Following overnight incubation at 4˚C, membranes were washed, incubated with the appropriate horseradish peroxidase‐linked secondary antibody (Thermo Fisher Scientific), washed again and visualized via reaction with enhanced chemiluminescence solution (Bio‐Rad, Mississauga, Ontario, Canada) using a FluorChem SP Imaging System (Alpha Innotech Corporation, San Leandro, CA, USA). Densitometry was performed using ImageStudio Lite software (LI‐COR Biosciences, Lincoln, NE, USA) or Image Lab (Bio‐Rad). All blots were normalized to Ponceau S solution, in line with standard practice (Romero‐Calvo et al. 2010; Stouth et al. 2018; Vanlieshout et al. 2018; Dial et al. 2018b).
Immunofluorescence (IF) microscopy
Standard IF procedures were performed in order to assess any changes in the expression and localization of key proteins either affected by DM1 or that may be implicated in mediating exercise‐induced adaptations. EDL muscle samples embedded in OCT were sectioned into 10 μm cross‐sections at −20˚C using a cryostat (Thermo Fisher Scientific). Microscope slides were blocked in a 10% goat serum in 1% BSA in phosphate‐buffered saline with Tween‐20 solution (PBS‐T). Slides were then incubated with a CLC‐1 antibody (1:100 dilution in 1% BSA in PBS‐T; ADI) overnight. Following washing, slides were blocked once again and then incubated with a dystrophin antibody (1:1000 dilution in 1% BSA in PBS‐T; Abcam) overnight. Following washing, slides were incubated with an Alexa‐conjugated secondary antibody (1:500 in 1% BSA in PBS‐T; Thermo Fisher Scientific) and 4ʹ,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI; 1:20,000 in 1% BSA in PBS‐T; Thermo Fisher Scientific). After the slides were dried, fluorescent mounting media (Agilent Technologies, Mississauga, Ontario, Canada) was applied to mount the slide with a cover slip. Slides were imaged by confocal microscopy (60×, 1.4 n.a. oil emersion; Nikon Instruments, Mississauga, Ontario, Canada). Z‐Plane imaging was used to compress 13 images with 0.9 μm into a single plane (Nikon Instruments). Slides were imaged by widefield microscopy (20×, 1.4 n.a.; Nikon Instruments). For each sample, four regions of interest (ROI) were created, each representing approximately 10% of the total cross‐sectional area (CSA) of the muscle cross‐section. A threshold was applied to CLC‐1 protein where only the mean intensity within the dystrophin boundaries was calculated, which denotes the amount of CLC‐1 localized to the sarcolemma of each myofibre, as previously performed in our laboratory (Dial et al. 2018b). The mean intensity of CLC‐1 protein localized to this threshold within each ROI was added and compared between the three mice groups. Mean intensity was used to normalize for differences in CSA of individual myofibres within the ROIs.
For IF microscopy analyses of AMPK and PGC‐1α, samples were fixed in 4% paraformaldehyde (PFA), washed and blocked as described above and incubated overnight with the respective primary antibody (AMPK: 1:1000 in 5% BSA in 1× PBS‐T; CST; PGC‐1α: 1:1000 in 5% BSA in 1× PBS‐T; SCB). Following incubation with the secondary (1:500 in 1% BSA in PBS‐T; Thermo Fisher Scientific), slides were stained with a fluorophore‐conjugated wheat‐germ agglutinin antibody against laminin (1:500 in 1% BSA in 1× PBS‐T; Vector Laboratories, Burlington, Ontario, Canada) and DAPI (1:20,000 in 1% BSA in PBS‐T; Thermo Fisher Scientific) for 15 min. Samples were subsequently washed and dried. Slides were imaged by wide‐field microscopy (20×, 1.4 n.a.; Nikon Instruments). For analysis, four ROIs were created, each representing 10% of muscle CSA. A threshold was applied to laminin and DAPI in order to assess the cellular localization of AMPK and PGC‐1α as either cytosolic or myonuclear, as performed previously (Dial et al. 2018b).
Histochemical staining
EDL muscle cross‐sections were stained with Haematoxylin and Eosin, dehydrated with successive 70%, 95% and 100% ethanol exposures, further dried with xylene and mounted with Permount (Thermo Fisher Scientific) to analyse the DM1‐associated myopathy. Stains were imaged using light microscopy (Nikon) at 20× magnification. For each sample, 100 myofibres were analysed across three ROIs, by quantifying the corresponding CSA via tracing the perimeters of myofibres and counting the presence of centrally nucleated fibres. Centrally nucleated fibres were defined as a muscle fibre containing at least one myonuclei not on the periphery. We further defined central as at least one myonucleus diameter length away from the border of the muscle fibre.
Fluorescence in situ hybridization (FISH)‐MBNL1 IF
Combination FISH with IF targeting MBNL1 was implemented as described by Mankodi et al. (2001) to assess the effect of exercise on the presence of myonuclear (CUG)n foci and sequestered MBNL1. Briefly, 10 μm cross‐sections of EDL muscles embedded in OCT were fixed in 3% PFA for 30 min, washed with PBS and fixed again in chilled 2% acetone. Slides were then incubated in a pre‐hybridization solution for 10 min before incubating in the hybridization solution at 45˚C for 2 h. The hybridization solution contained a modified DNA probe complementary to 10 CUG repeats with a 5′ end‐labelled Texas Red fluorescein (Integrated DNA Technologies, Coralville, IA, USA), allowing for detection using confocal microscopy. Samples were then washed in a post‐hybridization solution and a saline‐sodium citrate wash buffer. To probe for MBNL1, slides were blocked in a 1% goat serum in 1% BSA in PBS‐T and then incubated in the antibody solution (1:1000 in 1% BSA in PBS‐T; a generous gift from Dr Thornton) overnight. Following overnight incubation, slides were washed and subsequently incubated with an Alexa‐conjugated secondary antibody (1:500 in 1% BSA in PBS‐T; Thermo Fisher Scientific) and DAPI (1:20,000 in 1% BSA in PBS‐T; Thermo Fisher Scientific). After the slides were dried, fluorescent mounting media was applied, and a cover slip added. Slides were imaged by confocal microscopy (60× magnification, 1.4 n.a. oil emersion). Four 60× magnification Z‐plane images were taken per animal and used for analysis. Images were taken every 0.9 μm throughout the entire muscle cross‐section. The number of myonuclei containing at least one (CUG)n focus, and (CUG)n foci overlaying a MBNL1 puncta, were counted and expressed as a percentage of total myonuclei in the image. In total, 50–100 myonuclei were counted per stack for a total of approximately 1200 myonuclei analysed per group.
RNA purification and quantitative real‐time polymerase chain reaction (qPCR) and endpoint polymerase chain reaction (EPPCR)
To assess DM1‐associated alternative splicing in response to exercise training, 5–10 mg of GAST muscle was utilized to extract RNA as described previously (Stouth et al. 2018). All samples were homogenized using 1 ml of TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in Lysing D Matrix tubes (MP Biomedicals, Solon, OH, USA) at a speed of 6 m/s for 40 s. Samples were then shaken vigorously for 15 s with 200 μl of acetone, incubated at room temperature for 5 min and then centrifuged at 12,000 g for 10 min. The aqueous RNA phase was collected and purified using the Total RNA Omega Bio‐Tek kit (VWR). RNA concentration was determined using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Concentrations were normalized, and RNA was reverse transcribed using a high‐capacity cDNA reverse transcription kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
For qPCR, all samples were run in triplicate where each reaction contained GoTaq qPCR Master Mix (Promega, Madison, WI, USA). Data were analysed using the comparative CT method (Schmittgen & Livak, 2008) where glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and ribosomal protein S11 (RPS11) served as housekeeper genes. Data were analysed as the ΔΔCT score for the targeted exon relative to the ΔΔCT score for an intron spanning a sequence included in both splicing variants (pan), as previously described (Brockhoff et al. 2017). qPCR primer sequences (Sigma‐Aldrich) were as follows: SERCA (also known as ATP2A1) +ex22: forward (F) GCCCTGGACTTTACCCAGTG, reverse (R) ACGGTTCAAAGACATGGAGGA; ATP2A1 pan: F GCCCTGGACTTTACCCAGTG, R CCTCCAGATAGTTCCGAGCA; CAMKIIβ ‐ex13: F TTTCTCAGCAGCCAAGAGTTT, R TTCCTTAATCCCGTCCACTG; CAMKIIβ pan: F GCACGTCATTGGCGAGGAT, R ACGGGTCTCTTCGGACTGG; CLC‐1 +ex7a: F GGGCGTGGGATGCTGCTACTTTG, R AGGACACGGAACACAAAGGC; CLC‐1 pan: F CTGACATCCTGACAGTGGGC, R AGGACACGGAACACAAAGGC; GAPDH: F AACACTGAGCATCTCCCTCA, R GTGGGTGCAGCGAACTTTAT; RPS11: F CGTGACGAACATGAAGATGC, R GCACATTGAATCGCACAGTC; as previously reported (Wheeler et al. 2007; Nakamori et al. 2013; Brockhoff et al. 2017).
For EPPCR, cDNA was added to a reaction mixture (Thermo Fisher Scientific) containing Taq polymerase (Thermo Fisher Scientific) and primers. EPPCR products were resolved on a 2% agarose gel with electrophoresis at 110 mV for 80 min. The percentage of misspliced mRNA was determined using Image Lab (Biorad), where the intensity of the misspliced product was determined relative to the total band intensities of both the alternatively spliced and correctly spliced products. EPPCR primer sequences were as follows: CLC‐1: F GGAATACCTCACACTCAAGGCC, R CACGGAACACAAAGGCACTGAATGT; ATP2A1: F ATCTTCAAGCTCCGGGCCCT, R CAGCTTTGGCTGAAGATGCA; CAMKIIβ: F AAGTCGAGTTCCAGCGTGCA, R AGGTCCTCATCTTCTGTGGTGG; RYR1: F GACAATAAGAGCAAAATGGC, R CTTGGTGCGTTCCTGATCTG; dihydropyridine receptor (DHPR): F GAGATCCTTGGAATGTGTTTGACTTCCT, R GGTTCAGCAGCTTGACCAGTCTCAT; bridging integrator 1 (BIN1): F TCAATGATGTCCTGGTCAGC, R GCTCATGGTTCACTCTGATC; as previously published (Wheeler et al. 2007; Brockhoff et al. 2017; Jauvin et al. 2017; Ravel‐Chapuis et al. 2017).
Citrate synthase assay
Quadriceps muscle citrate synthase (CS) maximal enzyme activity was measured spectrophotometrically using a microplate reader at 37˚C and 412 nm, as previously described (Biotek Instruments, Winooski, VT, USA; Brooks et al. 2008). This technique is a well‐established marker for mitochondrial adaptions to exercise in skeletal muscle (Hood, 2009).
Statistical analyses
One‐way analysis of variance (ANOVA) and Student's t tests were employed to compare means between experimental groups, as appropriate. Statistical tests were performed on the raw data before any conversion to ‐fold differences that appear in the graphical summaries. To compare the force‐frequency and fatigue curves generated by the in situ skeletal muscle force production experiment, a regression analysis was performed to statistically compare the line of best fit for each group. Statistical analyses were performed with the GraphPad Prism software package (GraphPad Software, La Jolla, CA, USA). Significance was accepted at P < 0.05. Data are presented as means ± SEM as previously done (Knuth et al. 2018; Stouth et al. 2018; Yamada et al. 2018).
Results
DM1 mice run less than healthy animals
DM1 mice were given access to a voluntary running wheel for 7 weeks. Following a short period of steady increase in running volume, the daily distance ran plateaued at a consistent rate (Fig. 1 A). The mean running distance over the experimental time course was 5.3 ± 1.2 km/day. This is less than the distance healthy WT mice run, which averages over 10 km/day (Lerman et al. 2002; Steiner et al. 2013). The EX‐DM1 animals engaged in wheeling running primarily during their nocturnal phase (Fig. 1 B), similar to healthy animals (Landisch et al. 2008; Smythe & White, 2012). Females demonstrated modestly greater daily running distance (∼20%) and total distance ran (∼20%) versus males, but these differences were not statistically significant (Fig. 1 C and D).
Figure 1.
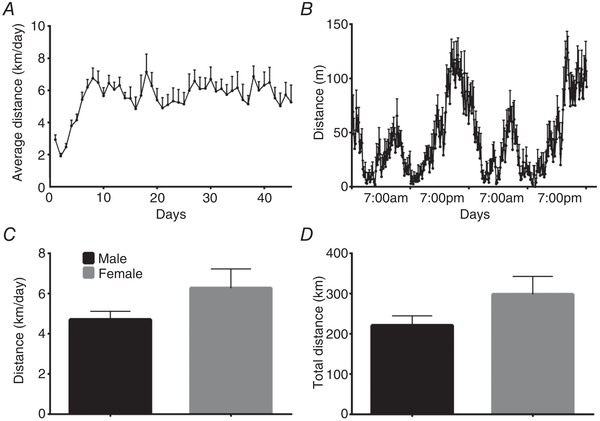
Running behaviour of mice with myotonic dystrophy type 1
A, average daily volitional running distance of human skeletal actin‐long repeat (HSALR) mice, a murine model of myotonic dystrophy type 1 (DM1), given access to a running wheel for 7 weeks. B, 48 h recording of volitional running. C and D, average running distance (km/day; C) and total run distance (km; D) of male and female DM1 animals. n = 16.
Chronic exercise improves strength and function in DM1 mice
We first assessed functional adaptations to chronic exercise by examining balance and muscular endurance, as well as strength via the pen and grip strength tests, respectively. As expected, mice in the SED‐WT group outperformed the SED‐DM1 animals in all three measures of functionality (Fig. 2 A–C). Both the SED‐DM1 and EX‐DM1 groups had significantly shorter latencies to fall from the pen as compared to SED‐WT mice (15.3 ± 1.6 s; P < 0.05; Fig. 2 A). However, EX‐DM1 animals increased time on the pen by 118% (from 4.7 ± 1.6 s to 10.3 ± 1 s; P < 0.05), compared to SED‐DM1 mice. Next, we assessed the maximum grip strength produced by the forelimbs, as well as by all limbs, using a force transducer. When measuring the resistance produced by all limbs, SED‐WT mice produced 13.1 ± 0.7 mN/mg of force, which was 28% and 24% greater (P < 0.05) relative to SED‐DM1 (9.2 ± 1 mN/mg) and EX‐DM1 (9.9 ± 0.4 mN/mg) animals, respectively (Fig. 2 B). There was no difference between the two DM1 groups. For the forelimb grip test, SED‐WT mice outperformed SED‐DM1 mice by 28% (4.9 ± 0.4 vs. 3.6 ± 0.2 mN/mg; P < 0.05) but produced a comparable force to the EX‐DM1 group, which was 4.5 ± 0.2 mN/mg (Fig. 2 C). Forelimb grip strength was significantly increased by 21% in the EX‐DM1 group compared to SED‐DM1.
Figure 2.
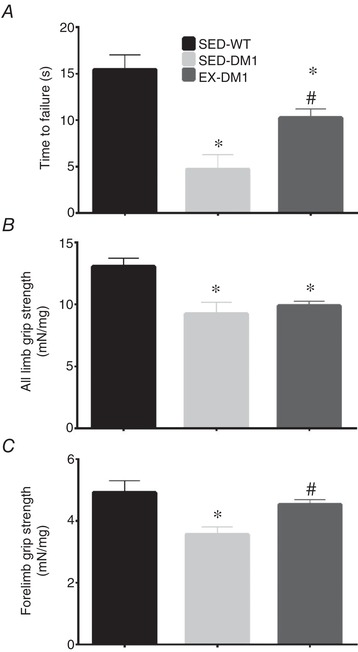
Assessment of muscle strength and endurance
A, latency to fall during pen test assay for sedentary wild‐type (SED‐WT), sedentary HSALR (SED‐DM1), and DM1 mice given access to an in‐cage running wheel for 7 weeks (EX‐DM1) animals. B, maximal grip strength exerted by all limbs relative to body weight in the three experimental groups. C, peak forelimb grip strength corrected for body weight in mice from all groups. n = 8–13; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1.
Physical activity ameliorates myotonia
We next examined muscle adaptations to exercise by assessing myotonia, a hallmark pathophysiological sign in DM1 patients and pre‐clinical murine models. Specifically, in situ needle EMG of the TA, GAST and TRI muscles was employed to reveal muscle electrical activity after a stimulation‐induced contraction in anaesthetized mice from the three experimental groups. In all muscles examined, action myotonia was virtually non‐existent in SED‐WT animals, but present in both DM1 groups (Fig. 3 A–G). However, GAST EMG burst duration was significantly reduced by 80% in the EX‐DM1 mice compared to SED‐DM1 animals (Fig. 3 B). In addition, exercise elicited a ∼70% decrease in EMG burst duration in the TRI muscle of EX‐DM1 versus SED‐DM1 mice (Fig. 3 D). TA muscle action myotonia was 74% lower in the EX‐DM1 group relative to SED‐DM1 mice, but this did not reach statistical significance (P = 0.07; Fig. 3 F). Examining the RMS amplitude of a 1 s sample of electrical activity following stimulation‐induced contraction showed no difference between SED‐DM1 and EX‐DM1 in any of the muscles (Fig. 3 C, E and G).
Figure 3.
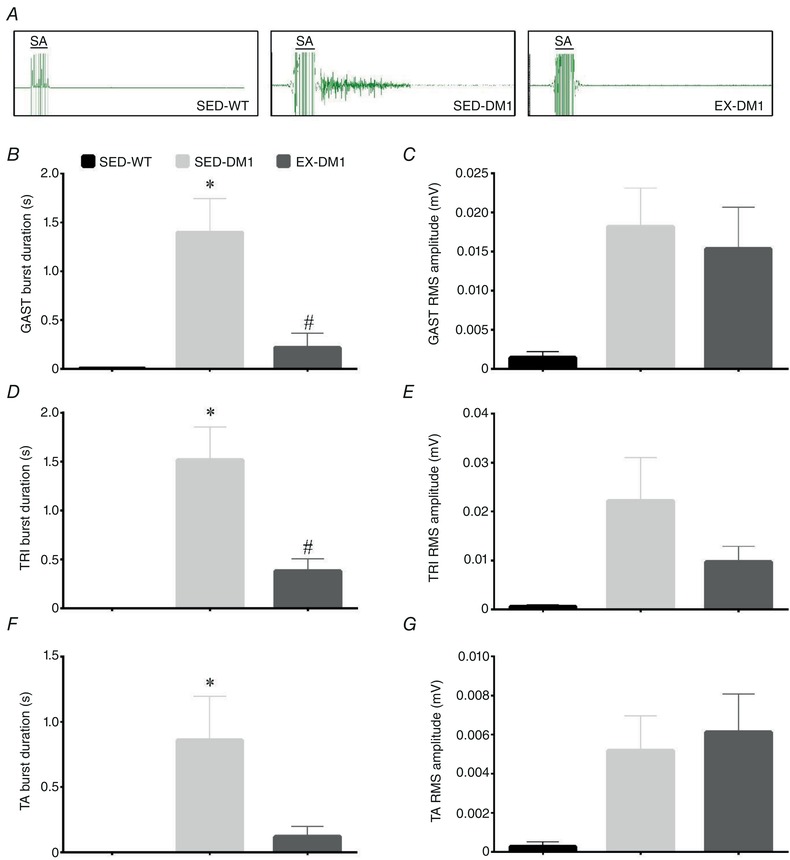
Effects of chronic exercise on myotonia
A, representative intramuscular electromyogram (EMG) tracings of gastrocnemius (GAST) muscles in SED‐WT, SED‐DM1 and EX‐DM1 mice. Note the stimulation artifact (SA) for 50 Hz stimulation present at left of each EMG. B–G, graphical summaries of the burst duration and root mean squared (RMS) amplitude of a 1 s stimulation in the GAST (B and C), triceps (TRI; D and E), and tibialis anterior (TA) muscles (F and G). n = 6–11; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1. [Color figure can be viewed at wileyonlinelibrary.com]
Voluntary running attenuates the DM1‐associated myopathy
Here, we first assessed the effects of chronic exercise on in situ muscle fatigue kinetics of the triceps surae complex. EX‐DM1 mice demonstrated a greater (P < 0.05) resistance to fatigue during repetitive stimulation‐induced contractions, as evidenced by a 65% reduction in force production versus a 72% and 75% reduction in SED‐WT and SED‐DM1 animals, respectively (Fig. 4 A). We continued to examine the effects of chronic exercise on characteristic features of the DM1 myopathy by examining myofibre CSA, as well as the presence of centrally nucleated fibres in EDL muscles. Volitional exercise in DM1 mice decreased the prevalence of myofibres with a CSA between 500–1,000 μm2 compared to SED‐DM1 mice (P < 0.05; Fig. 4 B). Otherwise, the overall distribution of myofibre CSA range was unremarkable between all three groups. As such, the average CSA was similar between groups (Fig. 4 C and E). Finally, habitual exercise did not alter the presence of centrally nucleated fibres, as both DM1 groups had ∼3 centrally nucleated fibres per 10 myofibres analysed (P < 0.05 vs. SED‐WT; Fig. 4 D).
Figure 4.
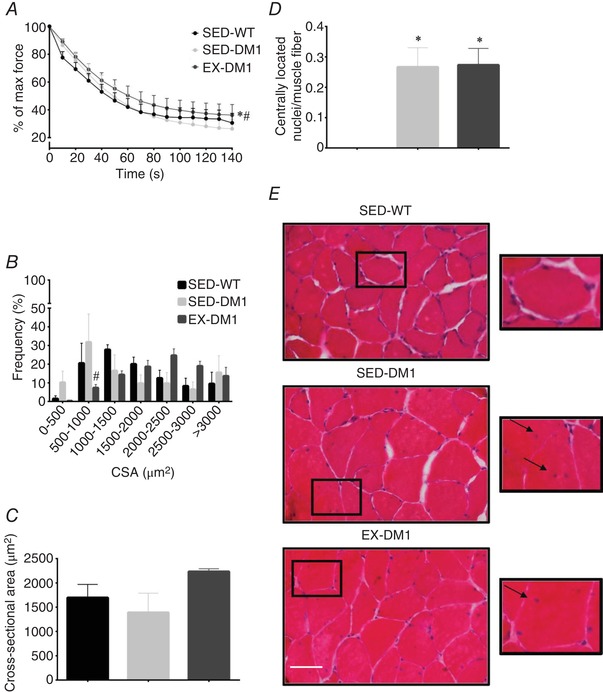
Effects of chronic exercise on skeletal muscle fatigability and histological metrics of the DM1 myopathy
A, fatigue kinetics in triceps surae complex during unilateral in situ stimulation of SED‐WT, SED‐DM1 and EX‐DM1 mice, displayed as a percentage of the force produced during the initial contraction. B, frequency distribution of extensor digitorum longus (EDL) myofibre cross‐sectional area (CSA) based on the Haematoxylin and Eosin (H&E) staining technique. C, graphical summary of average CSA, based on data in panel B. D, number of myofibres with centrally located nuclei per total number of muscle fibres analysed. E, representative H&E images of EDL muscle transverse sections from SED‐WT, SED‐DM1, and EX‐DM1 mice. Inset, arrows indicate centrally located myonuclei. Scale bar = 50 μm. n = 4–6; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1. [Color figure can be viewed at wileyonlinelibrary.com]
Exercise‐induced increase in mitochondrial content and activity in DM1 muscle
Skeletal muscle mitochondrial adaptions are examples of favourable molecular alterations that are evoked by chronic exercise in healthy animals, including humans (Hood, 2001; Egan & Zierath, 2013). We observed that the protein content of representative subunits of mitochondrial OXPHOS complexes I–V in the TA muscles were similar between SED‐WT, SED‐DM1, and EX‐DM1 animals (Fig. 5 A and B). However, we also noted a trend (P = 0.06) towards the normalization of complex IV expression with exercise. Thus, when the data for complexes I, II, III and V were pooled, since their expression levels appeared to be affected by exercise in the same positive direction, they revealed that EX‐DM1 mice had significantly higher OXPHOS expression than SED‐WT (+21%) and SED‐DM1 (+30%) animals. Another biomarker associated with mitochondrial density is mitochondrial CS enzyme activity (Larsen et al. 2012). Quadriceps muscle CS activity was 63% and 88% greater (P < 0.05) in the EX‐DM1 group compared to the SED‐DM1 and SED‐WT mice, respectively (Fig. 5 C).
Figure 5.
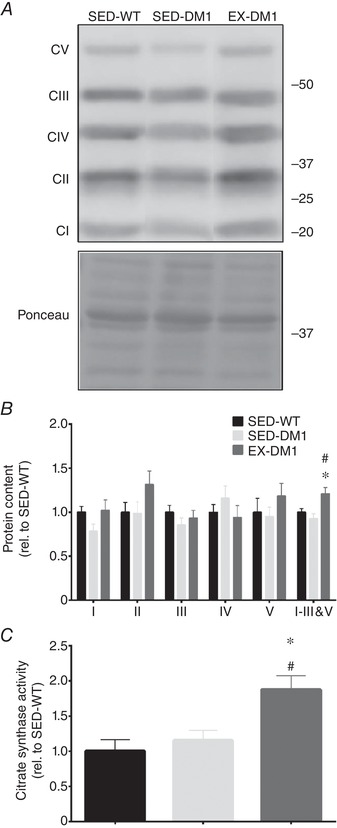
Exercise‐induced mitochondrial adaptations in DM1 mice
A, representative Western blot of mitochondrial oxidative phosphorylation (OXPHOS) complexes I–V (CI‐CV) in TA muscles from SED‐WT, SED‐DM1 and EX‐DM1 mice. A typical Ponceau stain is shown below to indicate equal loading between samples. Molecular weight (kDa) markers appear on the right of blots. B and C, graphical summaries of CI‐CV levels, including pooled CI, II, III and V data (B) and citrate synthase enzyme activity (C). Data are expressed relative to the SED‐WT group. n = 8; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐WT.
Physical activity selectively normalizes alternative splicing events associated with DM1
Using complementary EPPCR and qPCR analyses to examine mRNA splicing (Wheeler et al. 2007; Brockhoff et al. 2017; Jauvin et al. 2017; Ravel‐Chapuis et al. 2017), we found that volitional exercise had no effect on the missplicing of CAMKIIβ, ATP2A1, RYR1 and DHPR pre‐mRNAs, as the amount of misspliced variant did not differ between the two DM1 groups (Fig. 6 A–C). However, compared to SED‐DM1 mice, the EX‐DM1 group demonstrated a significant decrease in the abundance of CLC‐1 transcripts containing exon 7a, as revealed by both EPPCR (−35%) and qPCR (−21.3%) assays (Fig. 6 A–C). Additionally, chronic exercise normalized the inclusion of exon 11 in the BIN1 pre‐mRNA, as the percentage exon 11 inclusion was significantly different between SED‐DM1 (77%) and EX‐DM1 (90%) groups.
Figure 6.
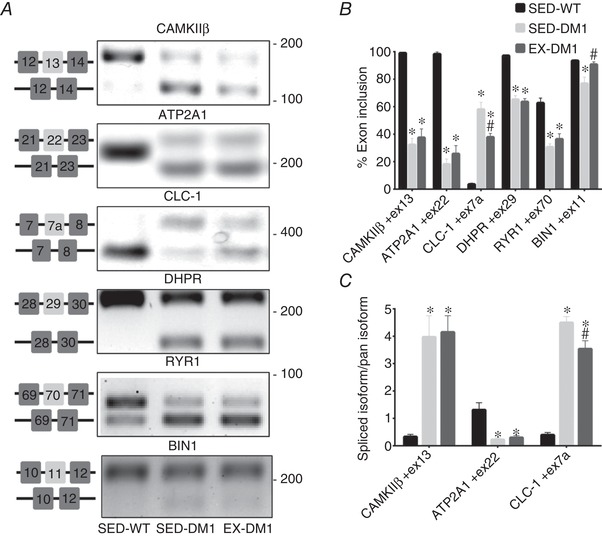
Effects of daily, volitional physical activity on mRNA splicing
A, representative endpoint polymerase chain reaction (EPPCR) gel electrophoresis UV images of mRNA splicing assays for Ca2+‐calmodulin‐dependent protein kinase IIβ (CaMKIIβ), sarco/endoplasmic reticulum Ca2+‐ATPase (ATP2A1), muscle‐specific voltage‐gated chloride channel (CLC‐1), dihydropyridine receptor (DHPR), ryanodine receptor 1 (RYR1), and Bridging integrator 1 (BIN1) in the GAST muscles from the three experimental groups. Specific exon alternative splicing diagrams are displayed to the right of each corresponding image. Molecular weight (bp) markers appear on the right of gels. B, graphical summaries of the percentage exon inclusion of EPPCR data in panel A. C, graphical summaries of quantitative, real‐time PCR analysis of CaMKIIβ, ATP2A1 and CLC‐1 spliced isoform abundance as indicated relative to the amount of the full‐length (pan) sequence. n = 8; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1.
Effect of exercise on proteins indicative of the DM1 pathology
We next sought to investigate the whole cell expression levels of various proteins that are specifically perturbed in DM1. Aligning with the exercise‐induced rescuing in its pre‐mRNA alternative splicing observed in Fig. 6, CLC‐1 protein content in TA muscles significantly increased (+55%) in the EX‐DM1 mice versus their SED‐DM1 littermates, reaching levels that were comparable to SED‐WT animals (Fig. 7 A and B). In contrast, CaMKIIα, ‐β, and pan CaMKII, as well as total MBNL1 content, were similar across all groups (Fig. 7 A, C and D). To complement the CLC‐1 Western blot analysis, quantitative immunofluorescence was employed to determine the localization of CLC‐1 at the sarcolemma (Fig. 8 A and B). SED‐DM1 mice displayed significantly less CLC‐1 protein in the sarcolemmal rim compared to both SED‐WT and EX‐DM1 mice. Chronic physical activity augmented sarcolemmal CLC‐1 content by 88% (P < 0.05) in DM1 animals. As a result, there was no statistical difference (P = 0.07) in CLC‐1 expression at the sarcolemma between SED‐WT and EX‐DM1 groups. Expression of the sarcolemmal marker dystrophin was similar between the three experimental cohorts (Fig. 8 C). Myofibre CSA, as determined using the dystrophin stain, was similar between groups (data not shown), in agreement with earlier findings (Fig. 4 C).
Figure 7.
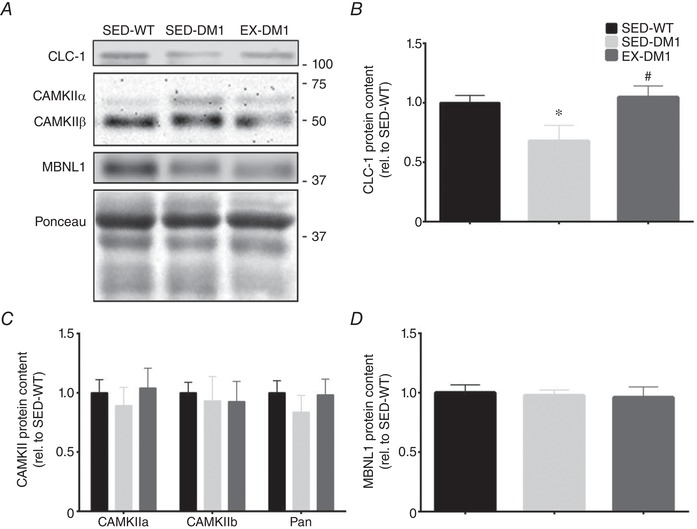
Effects of exercise on the DM1 molecular signature
A, typical Western blots of CLC‐1, CaMKIIα and ‐β isoforms, as well as Muscleblind‐like 1 (MBNL1) protein expression in TA muscles of SED‐WT, SED‐DM1 and EX‐DM1 animals. A representative Ponceau stain is displayed below. Molecular weight (kDa) markers appear on the right of blots. B–D, graphical summaries of CLC‐1 (B), CaMKIIα, ‐β, and pan‐CaMKII (C), as well as MBNL1 (D) protein levels. Data are relative to SED‐WT. n = 8; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1.
Figure 8.
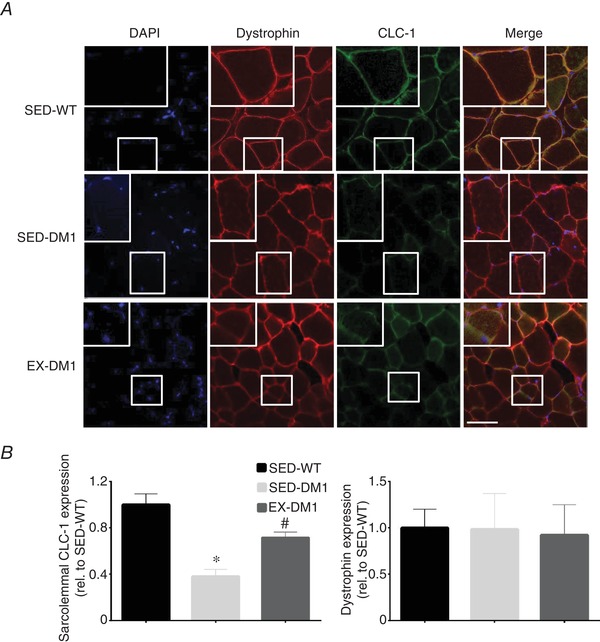
Effects of chronic exercise on sarcolemmal CLC‐1 expression
A, representative immunofluorescence (IF) images of EDL muscle cross‐sections from SED‐WT, SED‐DM1 and EX‐DM1 mice stained for 4',6‐diamidino‐2‐phenylindole, dihydrochloride (DAPI) to identify myonuclei, dystrophin as a marker of the sarcolemma, CLC‐1 protein, and merged images. Higher magnifications are inset. Scale bar = 50 μm. B, graphical summary of the mean fluorescence intensity of CLC‐1 protein localized specifically at the sarcolemma as determined by dystrophin overlay. C, average fluorescence intensity of dystrophin in the three experimental groups. Data are relative to SED‐WT. n = 3–4; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1. [Color figure can be viewed at wileyonlinelibrary.com]
Chronic physical activity attenuates (CUG)n foci and MBNL1 myonuclear sequestration
In an attempt to elucidate a potential mechanism by which chronic volitional exercise normalized CLC‐1 and BIN1 alternative splicing, we employed combination FISH/IF to examine the presence of (CUG)n‐dense myonuclei and MBNL1 myonuclear sequestration (Fig. 9 A and B), as done previously (Mankodi et al. 2000; Lin et al. 2006; Wheeler et al. 2009; Nakamori et al. 2011; Sobczak et al. 2013). Confocal microscopy analyses of the FISH/IF approach revealed that the prevalence of (CUG)n‐positive myonuclei in EDL muscles was significantly reduced (−25%) in the EX‐DM1 animals, compared to their SED‐DM1 littermates (Fig. 9 C). Additionally, 7 weeks of volitional running by the DM1 mice reduced sequestered MBNL1 by 45% (P < 0.05) versus their sedentary counterparts (Fig. 9 D).
Figure 9.
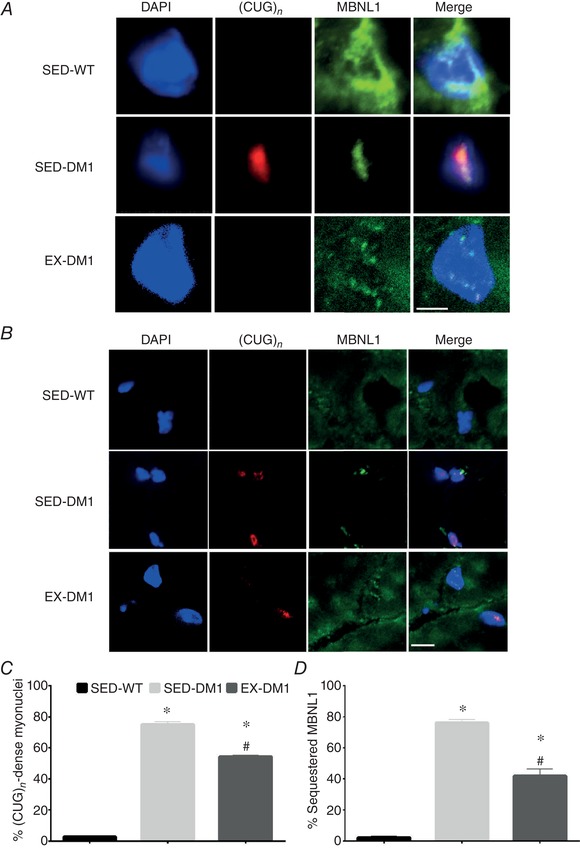
Exercise‐induced reduction in (CUG)n‐dense myonuclei and liberation of MBNL1 sequestration
A and B, representative high (A) and lower magnification (B) images of combination fluorescence in situ hybridization probing for CUG repeats ((CUG)n) with IF targeting MBNL1 in EDL muscle sections from SED‐WT, SED‐DM1 and EX‐DM1 animals. DAPI (denoting myonuclei) and merged images are also shown. Scale bar in A = 5 μm; scale bar in B = 50 μm. C and D, graphical summary of the percentage of myonuclei with (CUG)n foci (C) and myonuclei with sequestered MBNL1 (D). n = 4; * P < 0.05 vs. SED‐WT; # P < 0.05 vs. SED‐DM1. [Color figure can be viewed at wileyonlinelibrary.com]
Effect of chronic exercise on PGC‐1α and AMPK expression and subcellular localization
There is evidence to suggest that PGC‐1α and AMPK, which are activated with exercise, either directly or indirectly regulate mRNA splicing and MBNL1 myonuclear sequestration (Monsalve et al. 2000; Kim et al. 2005, 2014; Dial et al. 2018a). To further explore this, we employed IF analyses to examine potential exercise‐mediated subcellular translocation of these proteins. The cytosolic and nuclear abundance of PGC‐1α in EDL muscles was similar between the three experimental groups (Fig. 10 A and C). The subcellular distribution of AMPK was also similar among SED‐WT, SED‐DM1, and EX‐DM1 animals (Fig. 10 B and D). Western blot analyses revealed that total cellular PGC‐1α and AMPK content was not different between groups (Fig. 10 E).
Figure 10. Subcellular localization of peroxisome proliferator‐activated receptor γ coactivator‐1α (PGC‐1α) and AMP‐activated protein kinase (AMPK) in DM1 skeletal muscle.
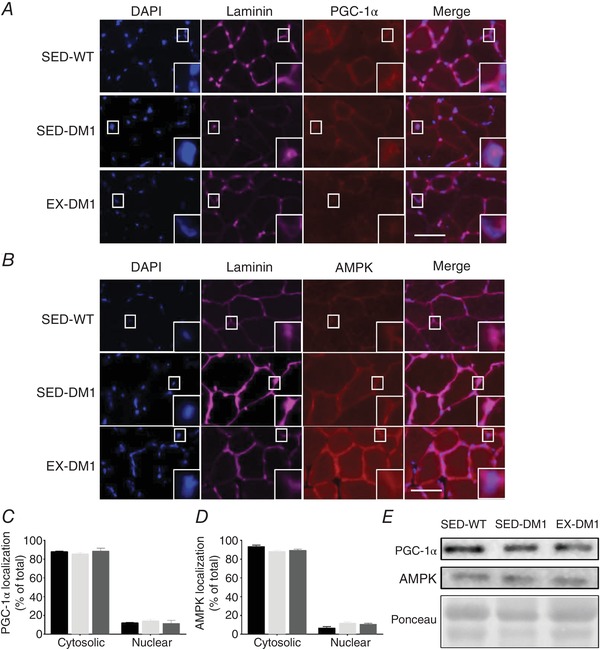
A and B, representative IF images of DAPI, laminin (denoting the sarcolemma), merged overlay, along with PGC‐1α (A) or AMPK (B) in EDL muscle sections from SED‐WT, SED‐DM1 and EX‐DM1 animals. Higher magnifications are inset. C and D, graphical summaries of PGC‐1α (C) and AMPK (D) subcellular localization in cytosolic and nuclear compartments of the EDL muscles from the three experimental groups. E, representative Western blots of whole cell PGC‐1α and AMPK content, as well as a typical Ponceau stain, in TA muscle homogenates. n = 4. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
This study is the first to demonstrate that following 7 weeks of daily volitional physical activity, DM1 mice exhibit significant beneficial adaptations to their skeletal muscle at the physiological, cellular and molecular levels. Specifically, chronic exercise led to an improvement in muscle strength and endurance, as well as an attenuation of myotonia, all hallmark signs of the DM1 pathology. At the cellular level, exercise augmented mitochondrial‐specific enzyme activity, increased the sarcolemmal localization of CLC‐1 protein, and significantly reduced the prevalence of (CUG)n‐dense myonuclear foci with sequestered MBNL1. Exercise‐induced molecular adaptations were exemplified by improved alternative splicing profiles of CLC‐1 and BIN1, which support the enhanced muscle physiology. Thus, these data indicate that chronic physical activity mitigates RNA toxicity and its resultant spliceopathy, fundamental disease mechanisms of DM1, which together form the cellular foundations of physiological improvements.
Ng and colleagues recently provided the first review of DM1 exercise biology (Ng et al. 2018). Although this body of literature is limited in number of reports, a consensus emerges that exercise is a safe and modestly effective, lifestyle‐based intervention to improve muscle strength, function and quality of life in DM1 patients. A notable caveat, however, is that the disparate training regimes employed in these studies, the small cohort sizes, as well as the varying experimental designs utilized, indicate that caution is indeed required when discussing in broad terms the impact of exercise in DM1. Nevertheless, in addition to observing physiological adaptations such as enhanced strength and , these DM1 participant studies have revealed some valuable clues as to the cellular alterations to exercise training, for example an increase in myofibre CSA and unaltered serum creatine kinase levels, a marker for muscle damage (Ørngreen et al. 2005). Very recently it was reported that 8 weeks of habitual, voluntary wheel running partially rescued a number of mRNA alternative splicing events in DM1 mouse skeletal muscle (Ravel‐Chapuis et al. 2018). This suggests that exercise positively affects the DM1 spliceopathy; however, the underlying mechanism(s) linking exercise to the attenuation of missplicing remains unknown. Moreover, the influence of chronic physical activity on other critical disease characteristics, such as myotonia and CLC‐1 expression, has not been examined. It is critical to thoroughly investigate the physiology of exercise adaptation DM1, as well as the cellular and molecular mechanisms that drive physical activity‐induced remodelling of DM1 skeletal muscle to increase our knowledge of the basic biology of the disorder and assist in the discovery of more effective lifestyle and/or pharmacological interventions to mitigate its severity.
Our data demonstrate that despite the presence of typical indicators of the DM1 myopathy such as muscle atrophy, weakness and reduced functional capacity (Jones et al. 2012; Timchenko, 2013), DM1 animals voluntarily ran consistently for 7 weeks, averaging 5.3 ± 1.2 km/day. This distance is less than than that achieved by healthy FVB/n mice (i.e. > 10 km/day; Lerman et al. 2002; Steiner et al. 2013; FVB/n mice are the background strain for the transgenic HSALR line). Nonetheless, these results are consistent with findings from participant studies that clearly indicate that those DM1 patients that are able to engage in a structured physical activity regimen will do so (i.e. high participant adherence rates) and find it enjoyable (e.g. high satisfaction scores; Wright et al. 1996; Ørngreen et al. 2005; Aldehag et al. 2013; Brady et al. 2014). Importantly, our data also show that the volume of activity completed by the DM1 animals, although modest relative to healthy mice, was indeed sufficient to elicit significant physiological improvements in strength and endurance as indicated by a battery of complementary in vivo and in situ phenotyping experiments. Physical activity‐induced increases in muscle strength and fatigue resistance in the healthy condition are correlated with muscle hypertrophy and mitochondrial biogenesis, respectively (Hawley et al. 2014). In DM1 mice, voluntary running led to a significant increase in skeletal muscle CS activity and OXPHOS protein levels, which exhibit stronger associations with mitochondrial volume than does PGC‐1α content (Larsen et al. 2012), which was unaffected here by chronic exercise. Additionally, volitional exercise did not exacerbate the prevalence of centrally nucleated fibres. It is reasonable to suspect, based on similar exercise‐induced alterations in healthy animals (Allen et al. 2001; Irrcher et al. 2003; Waters et al. 2004; Brooks et al. 2008; Hawley et al. 2014) that these adaptations contributed to the improved muscle strength and endurance observed in the EX‐DM1 group. Furthermore, as missplicing of BIN1 mRNA is highly correlated to muscle weakness in DM1 patients (Fugier et al. 2011; Nakamori et al. 2013), the complete correction of this alternative splicing event by chronic physical activity may also explain the increase in skeletal muscle performance. It is important to note that the observed recovery in forelimb strength in the exercise group has significant clinical relevance as grip weakness in DM1 patients is among the most common, debilitating symptoms affecting quality of life (Ansved, 2003).
Previously, the only metrics of myotonia in the DM1 condition in response to physical activity were self‐reported severity ratings before and after hand‐training interventions (Aldehag et al. 2005, 2013). Here, we provide quantitative data that demonstrate that chronic exercise ameliorates myotonia in vivo in DM1 mice. Studies employing pharmacological, cell‐based technologies such as antisense oligonucleotides or recombinant adeno‐associated viral vectors have shown that these interventions are able to significantly reduce myotonia in DM1 animals (Wheeler et al. 2012; Bisset et al. 2015). These strategies are remarkable for their efficacy in pre‐clinical contexts, but the results have yet to translate into the human condition. To our knowledge, we are the first to demonstrate that a physiological approach such as chronic exercise, a modality that is safe, low‐cost and accessible to patients, is also able to significantly reduce myotonia, a cardinal DM1 characteristic. These improvements in myotonia were associated with a rescuing of the depressed expression of mature CLC‐1 mRNA and protein in the EX‐DM1 cohort. Specifically, the data show that exercise significantly increased exon 7a exclusion, as well as total CLC‐1 protein content and its sarcolemmal localization. Healthy CLC‐1 levels and subcellular location are largely responsible for repolarizing and relaxing myocytes after contractions (Orengo et al. 2008). As such, its alternative pre‐mRNA splicing to include exon 7a and subsequent nonsense‐mediated mRNA degradation and sparse CLC‐1 protein expression accounts for the DM1‐associated myotonia (Chau & Kalsotra, 2015). Indeed, pharmacological induction of CLC‐1 in skeletal muscle significantly reduces myotonia in DM1 mice (Wheeler et al. 2007). Thus, the evidence strongly supports the hypothesis that the exercise‐evoked enhancement of skeletal muscle CLC‐1 expression provides the molecular basis for the attenuation of myotonia in DM1 animals. The induction of CLC‐1 protein in TA muscles along with the trend for improved TA myotonia in EX‐DM1 animals carries important translational relevance since this muscle is preferentially affected in DM1 patients, which presents as footdrop and detrimentally affects mobility and quality of life (Tang et al. 2012). Interestingly, we observed a mosaic expression pattern of sarcolemmal CLC‐1 after exercise, which indicates that CLC‐1 induction was more prevalent in some myofibres and less so in others.
A main finding of the current study is that that voluntary running markedly diminished both the prevalence of myonuclei containing (CUG)n‐expanded RNA, as well as sequestered MBNL1. (CUG)n‐expansion‐mediated RNA toxicity is the prevailing primary downstream disease mechanism in DM1 (Chau & Kalsotra, 2015). Specifically, in DM1 patients, the microsatellite repeat expansion mutation within the 3ʹ‐UTR of the DMPK gene results in DMPK transcripts forming hairpin secondary structures that sequester MBNL1 proteins with high affinity, which causes MBNL1 loss‐of‐function (Chau & Kalsotra, 2015). MBNL1 is the most critical RNABP responsible for, among other functions, the proper splicing of pre‐mRNAs germane to DM1, for example CLC‐1 or SERCA proteins (Nakamori et al. 2013). Previous work has clearly demonstrated that the dissolution of toxic (CUG)n‐containing RNAs and dispersal of MBNL1 from myonuclear foci, either by pharmacological or transgenic means, attenuates the prevalence of alternatively spliced mRNAs and mitigates the DM1 myopathy (Thornton et al. 2017). For example, Coonrod et al. (2013) demonstrated the ability of pentamidine and its analogues to reduce (CUG)n RNAs, increase the concentration of free MBNL1, and selectively rescue certain MBNL1‐mediated splicing events, namely the partial rescue of SERCA exon 22 inclusion and full rescue of CLC‐1 exon 7a exclusion, in pre‐clinical cell and murine models of DM1. While speculative, we attribute the selective nature of the normalization of alternative splicing events elicited by chronic exercise to the amount of functional, liberated MBNL1 protein needed to regulate pre‐mRNA splicing, as well as to the sensitivity displayed by each misspliced pre‐mRNA towards the RNABP. This selectivity is wholly consistent with previous studies that have also observed a treatment‐induced rescue of assorted misspliced mRNAs in DM1 skeletal muscle (Kanadia et al. 2006; Coonrod et al. 2013; Brockhoff et al. 2017), while the notion that a critical threshold of free MBNL1 variably impacts on pre‐mRNA processing has been documented previously (Jog et al. 2012). Further investigation into this phenomenon of the selective correction of alternative splicing in DM1 is clearly warranted in order to optimize potential therapeutic strategies.
We postulate that AMPK and PGC‐1α are integral to the mechanisms responsible for the exercise‐induced attenuation of toxic RNAs, MBNL1 entrapment to myonuclear foci, and spliceopathy in DM1. In the healthy condition, exercise robustly stimulates the activity, expression and myonuclear translocation of these molecules with powerful, pleiotropic downstream effects (Kjøbsted et al. 2017; Dial et al. 2018a). For example, PGC‐1α determines, maintains and remodels neuromuscular phenotype by regulating transcriptional and posttranscriptional events, such as mRNA processing, including splicing (Monsalve et al. 2000; Martínez‐Redondo et al. 2016). A growing body of evidence strongly supports the concept that pharmacological AMPK activation has therapeutic potential in mitigating DM1 (Savkur et al. 2001; Laustriat et al. 2015; Brockhoff et al. 2017; Thornton et al. 2017; Ravel‐Chapuis et al. 2018; Dial et al. 2018a), in part via reduced (CUG)n RNA toxicity, but the mechanism for this is yet to be identified. AMPK directly interacts with a key nuclear (CUG)n RNA stabilizing protein, heterogeneous nuclear ribonucleoprotein H (hnRNP H), which reduces the stability of the expanded repeat tract, facilitating its export and thus liberation of MBNL1 from myonuclear aggregates (Kim et al. 2005, 2014). AMPK and PGC‐1α translocate to myonuclei in response to acute exercise, which in the DM1 context might promote the interaction between the kinase and ribonuclear protein, as well as PGC‐1α splicing activity. However, our data did not demonstrate a nuclear accumulation of AMPK or PGC‐1α, which we submit was probably due to the timing of the analysis (i.e. 24 s after the final exercise bout). This does not preclude the possibility that chronic exercise stimulates swift, but transient AMPK and PGC‐1α myonuclear localizations with each acute bout of activity, which may effectively reduce toxic RNAs and MBNL1 loss‐of‐function via an hnRNP H‐mediated mechanism, and/or a PGC‐1α‐induced missplicing correction. Indeed, exercise‐induced elevations in OXPHOS protein content and CS activity in the muscle of EX‐DM1 animals is reflective of chronic, enhanced activity of the AMPK‐PGC‐1α axis (Ljubicic et al. 2010). Consistent with this, we have observed in DM1 mice that a single bout of running augments AMPK activation status, PGC‐1α expression, and their myonuclear translocation during the immediate‐ to short‐term (≤ 3 h) post‐exercise recovery (A. Manta and V. Ljubicic, unpublished observations). Ongoing experiments in our laboratory pursue this line of inquiry further, as well as whether exercise: (1) evokes physical and functional interactions between AMPK and hnRNP H, and (2) elicits a PGC‐1α‐mediated splicing correction of DM1‐associated mRNAs.
In summary, chronic, volitional exercise results in favourable physiological, myocellular and molecular adaptations in the DM1 condition. In particular, the main findings demonstrate that in DM1 mice habitual physical activity elicited improved strength and endurance, and limited myotonia, which were associated with corrected splicing of the CLC‐1 and BIN1 mRNAs, as well as reduced (CUG)n RNA toxicity and the liberation of MBNL1 from myonuclear foci. Further exploration of the molecular mechanisms by which exercise causes these alterations is warranted, as is identifying the optimal exercise prescription (i.e. frequency, intensity, mode, duration) to obtain maximal benefits. Moreover, as exercise affords broad, systemic effects (Whitham & Febbraio, 2016; Vivar & van Praag, 2017), DM1 patients adopting a more physically active lifestyle have the potential to address other common, debilitating symptoms such as somnolence and cognitive impairment. Finally, the possibility that this safe, accessible and affordable physiological intervention provides additive or synergistic effects to current or experimental pharmacological or cell‐based therapeutic strategies is certainly worthy of future consideration, with the ultimate goal of improving the lives of individuals with DM1.
Additional information
Competing interests
None declared.
Author contributions
AM and VL conceived and designed experiments. AM and DWS collected samples. AM, DWS, DX, LC, IAR and JMK performed experiments. AM, DX, LC, IAR and JMK analyzed data. AM, IAR, JMK, TJH and VL interpreted results of experiments. AM prepared figures. AM, JMK, TJH and VL drafted the manuscript or revised it critically for important intellectual content. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Canadian Institutes of Health Research (CIHR) and Canada Research Chairs program. A.M. received a CIHR Canada Graduate Scholarship (CGS) during the course of this study. D.W.S. holds a Natural Science and Engineering Research Council of Canada (NSERC) CGS. L.C. was a NSERC Undergraduate Student Research Award recipient. I.A.R. received an Ontario Graduate Scholarship. V.L. is the Canada Research Chair (Tier 2) in Neuromuscular Plasticity in Health and Disease.
Biography
Alexander Manta completed his MSc in the Integrative Neuromuscular Biology Laboratory, part of the Exercise Metabolism Research Group in the Department of Kinesiology at McMaster University under the supervision of Dr Vladimir Ljubicic. His thesis focuses on the exercise biology of myotonic dystrophy type 1. He matriculated to medical school at the University of Ottawa. His main research interests include neuromuscular biology in health and disease, particularly neuromuscular disorders and ageing.

Edited by: Scott Powers & Troy Hornberger
References
- Aldehag AS, Jonsson H & Ansved T (2005). Effects of a hand training programme in five patients with myotonic dystrophy type 1. Occup Ther Int 12, 14–27. [DOI] [PubMed] [Google Scholar]
- Aldehag A, Jonsson H, Lindblad J, Kottorp A, Ansved T & Kierkegaard M (2013). Effects of hand‐training in persons with myotonic dystrophy type 1 a randomised controlled cross‐over pilot study. Disabil Rehabil 35, 1798–1807. [DOI] [PubMed] [Google Scholar]
- Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC & Leinwand LA (2001). Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J Appl Physiol 90, 1900–1908. [DOI] [PubMed] [Google Scholar]
- Ansved T (2003). Muscular dystrophies: influence of physical conditioning on the disease evolution. Curr Opin Clin Nutr Metab Care 6, 435–439. [DOI] [PubMed] [Google Scholar]
- Barbé L, Lanni S, López‐Castel A, Franck S, Spits C, Keymolen K, Seneca S, Tomé S, Miron I, Letourneau J, Liang M, Choufani S, Weksberg R, Wilson MD, Sedlacek Z, Gagnon C, Musova Z, Chitayat D, Shannon P, Mathieu J, Sermon K & Pearson CE (2017). CpG methylation, a parent‐of‐origin effect for maternal‐biased transmission of congenital myotonic dystrophy. Am J Hum Genet 100, 488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisset DR, Stepniak‐Konieczna EA, Zavaljevski M, Wei J, Carter GT, Weiss MD & Chamberlain JR (2015). Therapeutic impact of systemic AAV‐mediated RNA interference in a mouse model of myotonic dystrophy. Hum Mol Genet 24, 4971–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D & Quadrilatero J (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7, e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LI, MacNeil LG & Tarnopolsky MA (2014). Impact of habitual exercise on the strength of individuals with myotonic dystrophy type 1. Am J Phys Med Rehabil 93, 739–750. [DOI] [PubMed] [Google Scholar]
- Brockhoff M, Rion N, Chojnowska K, Wiktorowicz T, Eickhorst C, Erne B, Frank S, Angelini C, Furling D, Rüegg MA, Sinnreich M & Castets P (2017). Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J Clin Invest 127, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion J‐P, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ & Housman DE (1992). Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. [DOI] [PubMed] [Google Scholar]
- Brooks SV, Vasilaki A, Larkin LM, McArdle A & Jackson MJ (2008). Repeated bouts of aerobic exercise lead to reductions in skeletal muscle free radical generation and nuclear factor κB activation. J Physiol 586, 3979–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call JA, McKeehen JN, Novotny SA & Lowe DA (2010). Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 42, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau A & Kalsotra A (2015). Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Dev Dyn 244, 377–390. [DOI] [PubMed] [Google Scholar]
- Cho DH & Tapscott SJ (2007). Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim Biophys Acta 1772, 195–204. [DOI] [PubMed] [Google Scholar]
- Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, Siboni RB, Docter AG, Haley MM, Thornton CA & Berglund JA (2013). Reducing levels of toxic RNA with small molecules. ACS Chem Biol 8, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dial AG, Ng SY, Manta A & Ljubicic V (2018a). The role of AMPK in neuromuscular biology and disease. Trends Endocrinol Metab 29, 300–312. [DOI] [PubMed] [Google Scholar]
- Dial AG, Rooprai P, Lally JS, Bujak AL, Steinberg GR & Ljubicic V (2018b). The role of AMP‐activated protein kinase in the expression of the dystrophin‐associated protein complex in skeletal muscle. FASEB J 32, 2950–2965. [DOI] [PubMed] [Google Scholar]
- Egan B & Zierath JR (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184. [DOI] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade‐Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D & Charlet‐Berguerand N (2011). Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17, 720–725. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ & Zierath JR (2014). Integrative biology of exercise. Cell 159, 738–749. [DOI] [PubMed] [Google Scholar]
- Hood DA (2001). Invited Review: Contractile activity‐induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol 90, 1137–1157. [DOI] [PubMed] [Google Scholar]
- Hood DA (2009). Mechanisms of exercise‐induced mitochondrial biogenesis in skeletal muscle. Engineering 472, 465–472. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Adhihetty PJ, Joseph A‐M, Ljubicic V & Hood DA (2003). Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sport Med 33, 783–793. [DOI] [PubMed] [Google Scholar]
- Jauvin D, Chrétien J, Pandey SK, Martineau L, Revillod L, Bassez G, Lachon A, McLeod AR, Gourdon G, Wheeler TM, Thornton CA, Bennett CF & Puymirat J (2017). Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids 7, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jog SP, Paul S, Dansithong W, Tring S, Comai L & Reddy S (2012). RNA splicing is responsive to MBNL1 dose. PLoS One 7, e48825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K, Wei C, Iakova P, Bugiardini E, Schneider‐Gold C, Meola G, Woodgett J, Killian J, Timchenko NA & Timchenko LT (2012). GSK3β mediates muscle pathology in myotonic dystrophy. J Clin Invest 122, 4461–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ & Cooper TA (2014). The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep 6, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA & Swanson MS (2006). Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierkegaard M & Tollbäck A (2007). Reliability and feasibility of the six minute walk test in subjects with myotonic dystrophy. Neuromuscul Disord 17, 943–949. [DOI] [PubMed] [Google Scholar]
- Kim D‐H, Langlois M‐A, Lee K‐B, Riggs AD, Puymirat J & Rossi JJ (2005). HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic Acids Res 33, 3866–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Lee JO, Lee HJ, Lee SK, Moon JW, Kim SJ, Park SH & Kim HS (2014). AMPKα2 translocates into the nucleus and interacts with hnRNP H: Implications in metformin‐mediated glucose uptake. Cell Signal 26, 1800–1806. [DOI] [PubMed] [Google Scholar]
- Kjøbsted R, Hingst JR, Fentz J, Foretz M, Sanz M‐N, Pehmøller C, Shum M, Marette A, Mounier R, Treebak JT, Wojtaszewski JFP, Viollet B & Lantier L (2017). AMPK in skeletal muscle function and metabolism. FASEB J 32, 1741–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth CM, Peppler WT, Townsend LK, Miotto PM, Gudiksen A & Wright DC (2018). Prior exercise training improves cold tolerance independent of indices associated with non‐shivering thermogenesis. J Physiol 596, 4375–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause MP, Liu Y, Vu V, Chan L, Xu A, Riddell MC, Sweeney G & Hawke TJ (2008). Adiponectin is expressed by skeletal muscle fibers and influences muscle phenotype and function. Am J Physiol Cell Physiol 295, C203–C213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA & Lowe DA (2008). Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 38, 1290–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F & Hey‐Mogensen M (2012). Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590, 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laustriat D, Gide J, Barrault L, Chautard E, Benoit C, Auboeuf D, Boland A, Battail C, Artiguenave F, Deleuze JF, Bénit P, Rustin P, Franc S, Charpentier G, Furling D, Bassez G, Nissan X, Martinat C, Peschanski M & Baghdoyan S (2015). In vitro and in vivo modulation of alternative splicing by the biguanide metformin. Mol Ther Nucleic Acids 4, e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman I, Harrison BC, Freeman K, Hewett TE, Allen DL, Robbins J & Leinwand LA (2002). Genetic variability in forced and voluntary endurance exercise performance in seven inbred mouse strains. J Appl Physiol 92, 2245–2255. [DOI] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS & Thornton CA (2006). Failure of MBNL1‐dependent post‐natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Lindeman E, Leffers P, Spaans F, Drukker J, Reulen J, Kerckhoffs M & Köke A (1995). Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: A randomized clinical trial. Arch Phys Med Rehabil 76, 612–620. [DOI] [PubMed] [Google Scholar]
- Ljubicic V, Joseph A‐M, Saleem A, Uguccioni G, Collu‐Marchese M, Lai RYJ, Nguyen LM‐D & Hood DA (2010). Transcriptional and post‐transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochim Biophys Acta 1800, 223–234. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M & Thornton CA (2000). Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1772. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan Q‐P, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS & Thornton CA (2001). Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10, 2165–2170. [DOI] [PubMed] [Google Scholar]
- Martínez‐Redondo V, Jannig PR, Correia JC, Ferreira DMS, Cervenka I, Lindvall JM, Sinha I, Izadi M, Pettersson‐Klein AT, Agudelo LZ, Gimenez‐Cassina A, Brum PC, Dahlman‐Wright K & Ruas JL (2016). Peroxisome proliferator‐activated receptor γ coactivator‐1 α isoforms selectively regulate multiple splicing events on target genes. J Biol Chem 291, 15169–15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M & Spiegelman BM (2000). Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC‐1. Mol Cell 6, 307–316. [DOI] [PubMed] [Google Scholar]
- Nakamori M, Pearson CE & Thornton CA (2011). Bidirectional transcription stimulates expansion and contraction of expanded (CTG)•(CAG) repeats. Hum Mol Genet 20, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson MS, Moxley RT & Thornton CA (2013). Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Manta A & Ljubicic V (2018). Exercise biology of neuromuscular disorders. Appl Physiol Nutr Metab 43, 1194–1206. [DOI] [PubMed] [Google Scholar]
- Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ & Cooper TA (2008). Expanded CTG repeats within the DMPK 3ʹ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci U S A 105, 2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørngreen MC, Olsen DB & Vissing J (2005). Aerobic training in patients with myotonic dystrophy type 1. Ann Neurol 57, 754–757. [DOI] [PubMed] [Google Scholar]
- Ravel‐Chapuis A, Al‐Rewashdy A & Jasmin BJ (2018). Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscles. Hum Mol Genet 27, 3361–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel‐Chapuis A, Bélanger G, Côté J, Michel RN & Jasmin BJ (2017). Misregulation of calcium‐handling proteins promotes hyperactivation of calcineurin‐NFAT signaling in skeletal muscle of DM1 mice. Hum Mol Genet 26, 2192–2206. [DOI] [PubMed] [Google Scholar]
- Romero‐Calvo I, Ocón B, Martínez‐Moya P, Suárez MD, Zarzuelo A, Martínez‐Augustin O & de Medina FS (2010). Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal Biochem 401, 318–320. [DOI] [PubMed] [Google Scholar]
- Savkur RS, Philips AV & Cooper TA (2001). Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29, 40–47. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD & Livak KJ (2008). Analyzing real‐time PCR data by the comparative CT method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Shortreed KE, Krause MP, Huang JH, Dhanani D, Moradi J, Ceddia RB & Hawke TJ (2009). Muscle‐specific adaptations, impaired oxidative capacity and maintenance of contractile function characterize diet‐induced obese mouse skeletal muscle. PLoS One 4, e7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe GM & White JD (2012). Voluntary wheel running in dystrophin‐deficient (mdx) mice: Relationships between exercise parameters and exacerbation of the dystrophic phenotype. PLoS Curr 3, RRN1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak K, Wheeler TM, Wang W & Thornton CA (2013). RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther 21, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JL, Davis JM, McClellan JL, Enos RT & Murphy EA (2013). Effects of voluntary exercise on tumorigenesis in the C3(1)/SV40Tag transgenic mouse model of breast cancer. Int J Oncol 42, 1466–1472. [DOI] [PubMed] [Google Scholar]
- Stouth DW, Manta A & Ljubicic V (2018). Protein arginine methyltransferase expression, localization, and activity during disuse‐induced skeletal muscle plasticity. Am J Physiol Cell Physiol 314, C177–C190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT & Thornton CA (2012). Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Cav1.1 calcium channel. Hum Mol Genet 21, 1312–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA, Wang E & Carrell EM (2017). Myotonic dystrophy approach to therapy. Curr Opin Genet Dev 44, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko L (2013). Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol 45, 2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollbäck A, Eriksson S, Wredenberg A, Jenner G, Vargas R, Borg K & Ansved T (1999). Effects of high resistance training in patients with myotonic dystrophy. Scand J Rehabil Med 31, 9–16. [PubMed] [Google Scholar]
- Vanlieshout TL, Stouth DW, Tajik T & Ljubicic V (2018). Exercise‐induced protein arginine methyltransferase expression in skeletal muscle. Med Sci Sports Exerc 50, 447–457. [DOI] [PubMed] [Google Scholar]
- Vivar C & van Praag H (2017). Running changes the brain: the long and the short of It. Physiology 32, 410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters RE, Rotevatn S, Li P, Annex BH & Yan Z (2004). Voluntary running induces fiber type‐specific angiogenesis in mouse skeletal muscle. Am J Physiol Cell Physiol 287, C1342–C1348. [DOI] [PubMed] [Google Scholar]
- Wheeler TM, Leger AJ, Pandey SK, Mac Leod AR, Wheeler TM, Cheng SH, Wentworth BM, Bennett CF & Thornton CA (2012). Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Lueck JD, Swanson MS, Dirksen RT & Thornton CA (2007). Correction of ClC‐1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest 117, 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT & Thornton CA (2009). Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M & Febbraio MA (2016). The ever‐expanding myokinome: Discovery challenges and therapeutic implications. Nat Rev Drug Discov 15, 719–729. [DOI] [PubMed] [Google Scholar]
- Willmann R, Dubach J, Chen K & TREAT‐NMD Neuromuscular Network (2011). Developing standard procedures for pre‐clinical efficacy studies in mouse models of spinal muscular atrophy. Report of the expert workshop “Pre‐clinical testing for SMA”, Zürich, March 29–30th 2010. Neuromuscul Disord 21, 74–77. [DOI] [PubMed] [Google Scholar]
- Wright NC, Kilmer DD, McCrory MA, Aitkens SG, Holcomb BJ & Bernauer EM (1996). Aerobic walking in slowly progressive neuromuscular disease: Effect of a 12‐week program. Arch Phys Med Rehabil 77, 64–69. [DOI] [PubMed] [Google Scholar]
- Yamada R, Himori K, Tatebayashi D, Ashida Y, Ikezaki K, Miyata H, Kanzaki K, Wada M, Westerblad H & Yamada T (2018). Preconditioning contractions prevent the delayed onset of myofibrillar dysfunction after damaging eccentric contractions. J Physiol 596, 4427–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]