

Abstract
There is currently no blood-based marker in routine use for endometrial cancer patients. Such a marker could potentially be used for early detection, but it could also help to track tumor recurrence following hysterectomy. This is important, as extra-vaginal recurrence of endometrial endometrioid adenocarcinoma is usually incurable. This proof-of-principle study was designed to determine if tumor-associated mutations could be detected in cell-free DNA from the peripheral blood of early and late stage endometrial endometrioid carcinoma patients. Approximately 90% of endometrioid carcinomas have at least one mutation in the genes CTNNB1, KRAS, PTEN, or PIK3CA. Using a custom panel targeting 30 hot spot amplicons in these 4 genes, next-generation sequencing was performed on cell-free DNA extracted from plasma obtained from a peripheral blood draw at the time of hysterectomy and the matching tumor DNA from 48 patients with endometrioid endometrial carcinomas. At least one mutation in the tumor was detected in 45/48 (94%) of patients. Fifteen of 45 patients (33%) had a mutation in the plasma that matched a mutation in the tumor. These same mutations were not detected in the matched negative control buffy coat. Presence of a plasma mutation was significantly associated with advanced stage at hysterectomy, deep myometrial invasion, lymphatic/vascular invasion and primary tumor size. Detecting a plasma-based mutation was independent of the amount of cell-free DNA isolated from the plasma. Overall, 18% of early stage patients had a mutation detected in the plasma. These results demonstrate that mutations in genes relevant to endometrial cancer can be identified in the peripheral blood of patients at the time of surgery. Future studies can help to determine the post-operative time course of mutation clearance from the peripheral blood and if mutation re-emergence is predictive of recurrence.
INTRODUCTION
Blood-based biomarkers, such as CA 125 (ovarian cancer), CEA (colorectal cancer), and PSA (prostate cancer), have established clinical utility as tools for early diagnosis and inexpensive, non-invasive methods of monitoring tumor recurrence or efficacy of therapy. While none of these biomarkers are 100% specific or sensitive, they are tremendously helpful in individual patients and have contributed to the more optimal care of patients with these cancer types. This is reflected in the fact that the mortalities from these 3 cancer types have been significantly decreasing (1). Endometrial cancer is the most common gynecological malignancy (2). The vast majority of endometrial cancers are endometrioid carcinomas diagnosed at an early stage (3). In contrast to colorectal cancer, prostate cancer, and ovarian cancer, the annual incidence and annual deaths from endometrial cancer are increasing (1,2). The reasons for this increase in endometrial cancer-associated deaths are multi-factorial. One likely contributing factor is the lack of blood-based biomarkers in routine clinical use for these patients.
Analysis of circulating cell-free DNA via the “liquid biopsy” is emerging as a useful method to characterize tumor mutations. Plasma cell-free DNA is released into the peripheral blood by tumor cells and cells in the tumor microenvironment that undergo apoptosis or necrosis. While a number of recent publications have documented the utility and feasibility of analysis of mutations in cell-free DNA in pancreatic cancer (4,5), colorectal cancer (6), and lung cancer (7), this approach has not yet been examined in endometrial cancer. Cicchillitti et al. (8) demonstrated that it is feasible to measure cell-free DNA from the plasma of endometrial cancer patients, but they did not attempt to identify plasma-based mutations. From previous studies of endometrial cancer patients treated at our institution, we have identified that alterations in PTEN, PIK3CA, KRAS, and CTTNB1 are four of the most common molecular events in endometrioid-type endometrial carcinoma (9, 10, 11). These 4 genes have good sequencing coverage in a 50 gene hotspot next-generation sequencing panel employed in our clinical laboratory (12). Thus, we hypothesized that a small sequencing panel that covered these 4 genes could detect endometrioid-type endometrial carcinoma-associated mutations in the plasma of endometrial cancer patients.
METHODS AND MATERIALS
Study cohort and patient data collection
Forty-eight matched tumor-plasma-buffy coat samples from women diagnosed with endometrioid-type endometrial adenocarcinoma from 2011–2016 at the University of Texas MD Anderson Cancer Center were included in this proof-of-principle study. Patients included in the study were chosen based on the availability of sufficient plasma, white blood cell buffy coat, and sufficient formalin-fixed, paraffin-embedded endometrial carcinoma for amplicon-based next-generation sequencing. Endometrial cancer tissue and peripheral blood were both obtained at the time of hysterectomy. Peripheral blood was drawn into EDTA tubes, and the plasma and buffy coat fractions were separated within 12 hours of collection and stored at −80ºC. None of the 48 patients had chemotherapy or radiation therapy prior to hysterectomy. Representative H&E stained slides from each hysterectomy were reviewed by a gynecological pathologist (RRB) to confirm endometrioid histology and grade and determine estimated tumor cellularity. The demographic and clinical information were obtained by review of the pathology reports and the electronic medical record for each patient. Stages I and II (tumor confined to the uterus) were designated as early stage disease, while stages III and IV (tumor detected outside the uterus) were designated as late stage or advanced disease. The study was approved by the University of Texas MD Anderson Cancer Center’s institutional review board (Protocol LAB01–718).
DNA extraction
For each of the 48 cases, DNA was isolated from plasma, formalin-fixed, paraffin-embedded tumor, and buffy coat white blood cells. Circulating cell-free DNA was extracted from 3 ml of frozen plasma using the QIAamp Circulating Nucleic Acid Kit (QIAGEN, Valencia, CA). Using a mapped H&E stained slide as a guide, unstained slides of formalin-fixed, paraffin-embedded endometrial carcinoma were macro-dissected and then DNA extracted from the scraped tissue using the Arcturus PicoPure DNA Extraction Kit (Applied Biosystems, Thermo Fisher Scientific). Starting tumor percentage (per cent tumor cells in the mapped H&E stained slide) ranged from 50–90%. DNA from white blood cells was extracted from 300µl of thawed buffy coat using the Maxwell 16 LEV Blood DNA kit (Promega, Madison, WI). Quantification of DNA from all samples was performed on a Qubit Flourometer (Thermo Fisher Scientific) using the QUBIT DNA High-sensitivity (tumor DNA and cell-free DNA) and Broad Range (white blood cell DNA) assay kits (Life Technologies, Illkrich, France).
Next-generation sequencing
A custom-designed Accel-Amplicon NGS panel 2.6 kb in size (Swift Biosciences, Ann Arbor, MI) covering hotspots in PTEN (exons 1,3,5,6,7,8), CTNNB1 (exon 3), KRAS (exons 2,3,4), and PIK3CA (exons 5,7,8,10,14,19,21) for Illumina platforms was used to profile tumor DNA, cell-free DNA, and white blood cell DNA. The average amplicon size for the 30 covered amplicons was 142 bp (range 119–187 bp). For these 4 genes, amplicon regions covered was similar to that for a 46 gene hotspot next-generation sequencing platform that we have previously described (12). Sequencing libraries were prepared and quantified as previously described (13). Quantified libraries were pooled, and the 20 pmol/l library was loaded on MiSeq Micro flow cell for paired-end sequencing using the MiSeq 300 Cycles version 2 kit (Illumina). Data analysis was performed using a custom bioinformatics pipeline (13). In cases with gene mutations detected at lower allelic fraction (less than 1%) in plasma cell-free DNA, libraries were re-sequenced in a standard flow cell at a higher depth to confirm the results obtained in the first analysis. For variant calling in the cell-free DNA, the filtering parameters were set as a minimum 250× sequencing depth and 25× variant coverage if the gene mutation allelic frequency was below 0.5%. Mutations called had to be present in the matching tumor DNA at ≥5% allelic frequency. All mutation calls were verified by visual inspection of the sequencing results using IGV.
Assessment of DNA mismatch repair
Mismatch repair protein and microsatellite instability testing were performed according to methodology previously described (14). Immunohistochemistry of mismatch repair proteins was performed using standard techniques for MLH1 (G168–15 1:25; BD Biosciences Pharmingen), MSH2 (FE11, 1:100; Calbiochem), MSH6 (44, 1:300; BD Biosciences Pharmingen), and PMS2 (Alb-4, 1:125; BD Biosciences Pharmingen). Immunohistochemistry was scored as mismatch repair protein intact or deficient using light microscopic examination. Complete absence of mismatch repair protein expression was required in order for a case to be designated as mismatch repair deficient. Stromal cells served as an internal positive control. PCR-based microsatellite instability analysis was performed using 7 microsatellites (TGFBR2 and the 6 National Cancer Institute recommended microsatellites, BAT25, BAT26, BAT40, D2S123, D5S346, and D17S250). A tumor was considered microsatellite instability-high if three or more of the seven markers demonstrated allelic shift. Tumors without allelic shift were designated as microsatellite stable.
Statistical analyses
Comparisons of cell-free DNA yield in relation to clinical and pathological characteristics were performed using ANOVA when there was homogeneity of variances. When the assumption of homogeneity was violated, the Kruskal Wallis test was applied. The Pearson correlation test was performed to determine the relationship between cell-free DNA yield and tumor size and tumor cellularity. Associations between tumor stage, tumor grade, myometrial invasion, and lymphatic/vascular space invasion with the detection of mutation in the plasma were performed using the Chi square test. When any of the assumptions of the Chi square test were violated, Fisher’s exact test was performed. The association between mutation status, tumor size, and the initial cell-free DNA yield was determined by ANOVA with intra-class correlation measurements. For all analyses, p < 0.05 was considered significant.
RESULTS
Clinical and pathological characteristics for the 48 endometrial cancer patients are summarized in Table 1. The mean age of the cohort was 58 years (range, 26–86), and most of the patients had early stage (Stage I or II), grade 2 tumors with less than 50% myometrial invasion at hysterectomy. The mean tumor size was 5.3 cm. Myometrial lymphatic/vascular space invasion was present in nearly half the tumors and was much more common in advanced stage cases. Note that no patients with grade 1 endometrioid tumors were included, as we did not have collected plasma for patients in this grade category. Mean patient age and mean tumor size were comparable between early stage and late stage patients. Overall, sixty-three per cent of the patients had mismatch repair intact tumors, while 37% were mismatch repair deficient.
Table 1.
Clinical and pathological characteristics of the study cohort
| Characteristic | All patients (n=48) | Early stage (n = 35) |
Late stage (n=13) |
|---|---|---|---|
| Age in years, mean (s.d.) | 58.0 (12.0) | 58.3 (11.7) | 58.1 (13.9) |
| Endometrioid grade, n (%) | |||
| 1 | 0 (0%) | 0 (0%) | 0 (0%) |
| 2 | 30 (62%) | 24 (69%) | 6 (46%) |
| 3 | 18 (38%) | 11 (31%) | 7 (54%) |
| aMyometrial invasion, n (%) | |||
| <50% | 30 (64%) | 27 (77%) | 3 (25%) |
| ≥ 50% | 17 (36%) | 8 (23%) | 9 (75%) |
| bLVSI, N (%) | |||
| No | 25 (53%) | 24 (69%) | 1 (8%) |
| Yes | 22 (47%) | 11 (31%) | 11(92%) |
| cTumor size in cm, mean (s.d.) | 5.3 (3.4) | 5.1 (3.7) | 5.8 (2.7) |
| DNA mismatch repair | |||
| Intact | 30 (63%) | 24 (69%) | 6 (46%) |
| Deficient | 18 (37%) | 11 (31%) | 7 (54%) |
Data missing for one patient.
LVSI, lymphatic/vascular space invasion; data missing for one patient.
Data missing for two patients
The mean cell-free DNA yield extracted from the plasma samples was 15.6 ng/ml (range, 4.5–30 ng/ml). The presence of deep myometrial invasion was associated with significantly higher mean plasma cell-free DNA yield (Figure 1). Pathological stage, tumor grade, and lymphatic/vascular space invasion, however, had no significant impact on mean plasma cell-free DNA (Figure 1). Increased tumor size trended towards being associated with cell-free DNA yield (p=0.06). No correlation was found between the cell-free DNA yield and the starting tumor cellularity before tumor DNA extraction (p=0.416). Increasing patient age had no significant effect on the cell-free DNA yields (p= 0.212; data not shown).
Figure 1.
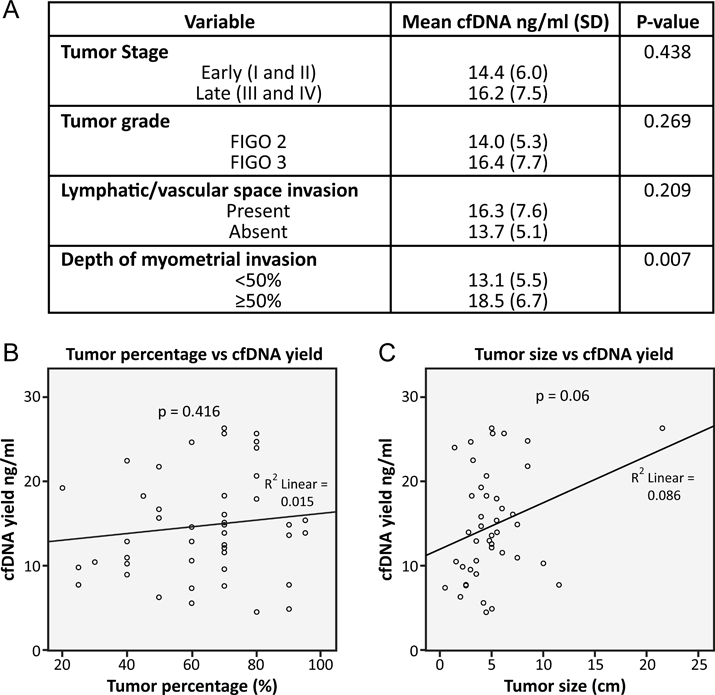
Plasma cell-free DNA (cfDNA) yield according to various tumor variables. Higher levels of plasma cell-free DNA were associated with deeper myometrial invasion (A), but not starting primary tumor per cent cellularity (B) or primary tumor size (C).
The molecular findings for both tumor and plasma for the entire cohort are presented in Figure 2. At least one mutation was detected in 45/48 (94%) of the tumors analyzed. Thirty patients (30/45, 66.7%) had at least one mutation in the tumor, but no matching mutations in the plasma. Fifteen patients (15/45, 33.3%) had at least one mutation in the tumor and the same matching mutation in the plasma. For 2 patients (9 and 18), a gene mutation was not detected in either tumor or plasma. For patient 21, KRAS and PIK3CA mutations were detected, but only in the plasma. Patients 1 and 32 had mutations in the tumor but different mutations in the plasma. Patient 1 had PTEN and PIK3CA mutations in the tumor that were also detected in the plasma. The plasma had a KRAS mutation and a second PTEN mutation; these were also in the tumor, but at allelic frequencies below 1%, well below our established 5% cut-off for calling mutations in the tumor. Patient 32 had only a PTEN mutation in the tumor, but the plasma had a KRAS mutation. This KRAS mutation was present in the tumor, but at 0.1% allelic frequency and therefore below the 5% cut-off for calling tumor mutations. Presence of mismatch repair deficiency in the tumor does not appear to be related to detetion of a mutation in the plasma. From Figure 2, 31 patients did not have a mutation present in plasma. Ten of 31 (32%) of these had mismatch repair deficient tumors, while 21/31 (68%) had mismatch repair intact tumors. These percentages are nearly the same as the percentage of mismatch repair intact and deficient in the entire study cohort.
Figure 2.
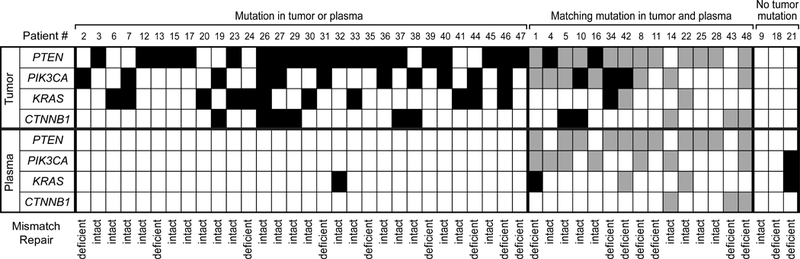
Summary of tumor and plasma mutations identified in the study cohort. White box, no mutation present; black box, mutation identified only in tumor or plasma; gray box, same mutation identified in both tumor and the matching plasma
Table 2 summarizes the comparisons of gene mutational frequencies between tumor and plasma. The most common mutations in the tumor were in PTEN and PIK3CA, with lower frequency of mutations in KRAS and CTNNB1, similar to our previous published work (11). In the plasma, PTEN and PIK3CA mutations were also the most common. For PTEN and KRAS, there was substantial drop-off in the detection of mutations in the plasma compared to tumor. For PTEN, 70.8% (34/48) of patients had at least one tumor mutation. Only 11/34 (32.4%) patients with a tumor PTEN mutation had the same PTEN mutation detected in plasma. For KRAS, 29.2% (14/48) of patients had a tumor mutation, but only 2/14 (14.3%) of these patients had the same KRAS mutation detected in plasma. The tumor-plasma mutational comparisons were more equitable for CTNNB1 and PIK3CA, although for both of these genes mutations were more often detected in the tumor than plasma.
Table 2.
Detection of gene mutation in tumor and plasma for 48 patients
| Tumor Tissue n (%) | ||||
|---|---|---|---|---|
| KRAS | Mutant | WT | Total | |
| Plasma cfDNA |
Mutant | 2 (14%)a | 3 | 5 (10%)b |
| WT | 12 (86%)a | 31 | 43 (90%)b | |
| Total | 14 (29%)b | 34 (71%)b | 48 | |
| PTENc | ||||
| Plasma cfDNA |
Mutant | 11 (32%)a | 0 | 11 (23%)b |
| WT | 23 (68%)a | 14 | 37 (77%)b | |
| Total | 34 (71%)b | 14 (29%)b | 48 | |
| PIK3CA | ||||
| Plasma cfDNA |
Mutant | 7 (33%)a | 1 | 8 (17%)b |
| WT | 14 (67%)a | 26 | 40 (83%)b | |
| Total | 21 (44%)b | 27 (56%)b | 48 | |
| CTNNB1 | ||||
| Plasma cfDNA |
Mutant | 3 (27%)a | 0 | 3 (6%)b |
| WT | 8 (73%)a | 37 | 45 (94%)b | |
| Total | 11 (23%)b | 37 (77%)b | 48 | |
The percentage of plasma gene mutation or wildtype detected was calculated of the total number of mutant cases in the tumor tissue column (n=14 for KRAS; n=34 for PTEN; n= 21 for PIK3CA; n=11 for CTNNB1).
The percentage was calculated of the total number of patients (n=48).
One patient (Patient 1 in Tables 3 and 4) had 2 PTEN mutations detected in the plasma, R130G and P213fs*8. R130G was present at high allelic fraction in the tumor, while P213fs*8 was not detected in the tumor. Only the R130G mutation is considered for this table, since it was present in both tumor and plasma.
Fifteen of 45 patients (33%) had the same mutation detected in both the tumor and plasma. A more detailed summary of these patients is presented in Table 3. Remarkably, 8/15 (53.3%) of the patients with mutations detected in the plasma at the time of surgery presented at early stage (stage I or II), when the tumor is confined to the uterus. Overall, there were 24 stage IA patients in this cohort. Four of these (17% of stage IA) had a mutation detected in the plasma. Patients 5,10, and 42 were stage IA patients with the same mutations detected in the tumor and plasma (Table 3). The fourth stage IA patient (patient 32) had a KRAS mutation in the plasma that was present at very low allelic fraction in the tumor (Table 4). None of these patients with early stage disease and presence of plasma gene mutations had recurrence following surgery, but follow-up for most patients was limited (Table 3; mean follow-up time for early stage patients 2.4 years, range 0–6 years). A wide spectrum of mutations was identified in the 4 genes, suggesting that presence of mutation in plasma is not restricted to specific mutations in specific genes. Six of the patients had more than one mutation detected in both tumor and plasma.
Table 3.
Summary of patients with same mutation detected in both tumor and matched plasma. AF, allelic frequency of mutation detected
| Case # | Stage | Endometrioid Grade |
Tumor Recurrence |
Gene Mutation | Tumor AF |
Plasma AF |
Buffy Coat AF |
|---|---|---|---|---|---|---|---|
| 5 | I | 2 | None | PIK3C H1047Y | 22 | 1.1 | 0 |
| PTEN H93R | 26 | 1 | 0 | ||||
| PTEN C136fs*11 | 26 | 1 | 0 | ||||
| 10 | I | 2 | None | PTEN R130G | 45 | 1 | 0 |
| 14 | I | 2 | None | PIK3CA H1047R | 22 | 1.2 | 0 |
| CTNNB1 D32Y | 20 | 2 | 0 | ||||
| 25 | I | 3 | None | PTEN I203fs*14 | 32 | 0.5 | 0 |
| 28 | I | 2 | None | PTEN L57fs*42 | 40 | 0.6 | 0 |
| 42 | I | 2 | None | PTEN T319* | 21 | 0.8 | 0 |
| 48 | I | 3 | None | PTEN Q219* | 27 | 3 | 0 |
| PTEN L146* | 60 | 13 | 0 | ||||
| CTNNB1 S37C | 39 | 3 | 0 | ||||
| 16 | II | 3 | None | PIK3CA E81K | 36 | 0.2 | 0 |
| 1 | III | 3 | Yes, vaginal cuff |
PIK3CA Q546H | 47 | 0.3 | 0 |
| PTEN R130G | 75 | 0.6 | 0 | ||||
| 4 | III | 3 | None | PIK3CA R88Q | 34 | 0.3 | 0 |
| 8 | III | 3 | Yes, pelvic | PIK3CA Y1021C | 28 | 3.5 | 0 |
| PTEN V290* | 80 | 3.6 | 0 | ||||
| 11 | III | 3 | None | PTEN L267 fs*9 | 34 | 0.5 | 0 |
| 22 | III | 2 | Yes, colon | PTEN T131N | 38 | 1.1 | 0 |
| KRAS G12D | 22 | 1.1 | 0 | ||||
| 34 | III | 3 | No follow- up |
PTEN R130G | 35 | 0.9 | 0 |
| 43 | III | 3 | Yes, colon | CTNNB1 S33F | 30 | 0.2 | 0 |
Table 4.
Mutations detected in the plasma and at very low allelic frequency (AF) in the tumor. LVSI, lymphatic/vascular invasion
| Case # | Stage | Grade | Tumor Size (cm) |
Invasion Depth (% Myometrial thickness) |
LVSI | Gene mutation | Tumor AF |
Plasma AF |
Buffy Coat AF |
|---|---|---|---|---|---|---|---|---|---|
| 1 | III | 3 | 6.2 | 85 | yes | KRAS G13D | 0.1 | 0.6 | 0 |
|
PTEN P213fs*8 |
0.6 | 0.8 | 0 | ||||||
| 21 | III | 2 | 6.5 | 100 | yes | PIK3CA R88Q | 0.2 | 0.6 | 0 |
| KRAS G12D | 0.1 | 10 | 0 | ||||||
| 32 | I | 2 | 1.6 | 7 | no | KRAS G13S | 0.1 | 0.6 | 0 |
Note from Table 3 that 10/23 (43.5%) of the detected plasma mutations are at lower allelic frequency (less than 1%). A number of lines of evidence indicate that these low level mutations are not sequencing artifacts. One, the same mutation in each instance was identified in the matching tumor. Second, we never identified the same mutation when the matched buffy coat germline control was sequenced. Third, sequencing was repeated for these cases at higher depth, and the identical mutations at approximately the same allelic frequencies were identified. Fourth, using the same 4 gene next-generation sequencing platform, limiting dilution studies using the DLD1 colon cancer cell line detected mutations in PIK3CA and KRAS at 1% allelic frequency. Fifth, although we did not have sufficient residual plasma DNA for the more sensitive sequencing technique, digital droplet PCR, our previously published studies demonstrated that plasma mutations identified by next-generation sequencing at lower allelic frequencies were all verified by digital droplet PCR (15). The presence of plasma mutations at higher allelic frequencies (1% or greater) was observed in 4/8 (50%) of early stage patients and 2/7 (28.6%) of advanced stage patients (Table 3). Therefore, disease outside the uterus was not associated with detection of greater gene mutation allelic frequencies in the plasma.
For 3 patients, mutations detected in the plasma were present at very low allelic frequencies, less than 1%, in the tumor (Table 4). In our clinical laboratory, tumor mutations detected at less than 5% allelic frequency by next-generation sequencing are typically not reported. Next-generation sequencing was repeated for these tumors using a different sequencing platform to confirm the absence of tumor mutations at 5% or greater allelic frequency. Note that the KRAS G12D mutation detected in the plasma of patient 21 was present at relatively high allelic frequency (10%), but the same mutation was not identified at greater than 5% allelic frequency in the tumor. Patient 1 had 2 PTEN mutations in the plasma; R130G was detected at higher allelic frequency in the tumor (Table 3), while P213fs*8 was detected at below 1% allelic frequency in the tumor (Table 4).
In Table 5, clinical/pathological characteristics of the 45 patients with at least one tumor mutation detected are stratified by mutation status of the plasma cell-free DNA. The plasma mutant group includes the 15 patients summarized in Table 3. Patients 9, 18, and 21 are excluded because no matching mutations were detected in their tumors at 5% or greater allelic frequency. Advanced tumor stage at the time of diagnosis, myometrial invasion ≥ 50% myometrial thickness, and presence of lymphatic/vascular invasion are associated with detection of a mutation in plasma. In addition, mean uterine primary tumor size in the plasma mutant group was significantly larger than tumor size in the plasma wildtype group. Importantly, pre-analytic factors such as total cell-free DNA yield, cell-free DNA input for sequencing library preparation, and primary tumor per cent cellularity had no significant impact on the ability to detect a gene mutation in plasma.
Table 5.
Pathology characteristics of patients with endometrioid endometrial cancer, stratified by presence or absence of a gene mutation in the plasma at time of hysterectomya. cfDNA, cell-free DNA
| Characteristic | Plasma mutant | Plasma wildtype | P-value |
|---|---|---|---|
| Tumor stage, n (%) | 0.009 | ||
| Early | 8 (18%) | 27 (60%) | |
| Late | 7 (16%) | 3 (7%) | |
| Tumor grade, n (%) | 0.077 | ||
| 1 | 0 (0%) | 0 (0%) | |
| 2 | 7 (16%) | 22 (49%) | |
| 3 | 8 (18%) | 8 (18%) | |
|
bMyometrial invasion, n (%) |
0.018 | ||
| <50% | 6 (14%) | 24 (55%) | |
| ≥ 50% | 8 (18%) | 6 (14%) | |
|
bLymphatic/vascular invasion, n (%) |
0.012 | ||
| yes | 10 (23%) | 9 (20%) | |
| no | 4 (9%) | 21 (48%) | |
| Tumor size in cm, mean (s.d.) |
7.1 (5.0) | 4.2 (2.0) | 0.019 |
| Patient age in years, mean (s.d) |
61 (14) | 58 (11) | 0.519 |
| Tumor percentage, mean (s.d.) |
70% (20) | 60% (19) | 0.119 |
| cfDNA yield, mean (s.d.) |
16.1 ng/ml (7.7) | 14.2 ng/ml (5.5) | 0.463 |
| cfDNA input for library preparation, mean (s.d.) |
0.8 ng/ul (0.2) | 0.8 ng/ul (0.2) | 0.818 |
The 45 patients for which at least one tumor mutation was identified are included in this analysis. Patients 9,18, and 21 are excluded.
Data missing for one patient.
DISCUSSION
To our knowledge, this is the first publication in the peer-reviewed English literature which has identified endometrial cancer-associated mutations in the plasma of patients. Remarkably, a very limited panel of only 4 genes identified tumor mutations in over 90% of the cases studied. It is possible that by adding a few more genes more commonly mutated in endometrioid-type endometrial carcinomas, such as ARID1A and PIK3R1 (11), an even higher percentage of endometrioid tumors could be captured by this panel. The ability to detect plasma-based mutations was dependent upon primary tumor characteristics, such as increased tumor size, advanced stage, deeper myometrial invasion, and the presence of lymphatic/vascular space invasion. The presence of mismatch repair deficiency in the tumor was not associated with increased likelihood of detecting mutations in the plasma. While the majority of the mutation-positive plasma cases were from patients with advanced stage disease (stages III and IV; endometrial cancer has spread beyond the uterus), we were surprised to find that nearly 20% of the early stage (stages I and II; endometrial cancer confined to the uterus) patients also had mutations detected in the plasma. We do not have follow-up blood samples from these patients. A more formal prospective study with multiple blood draws would be necessary to determine how soon after hysterectomy the mutations detected in plasma at the time of hysterectomy are cleared. Prospective studies could also help to determine if early stage patients with plasma-based mutations detected at the time of surgery are at increased risk for recurrence. These studies could also specifically investigate the incidence of plasma mutations in patients with grade 1 endometrioid tumors, which were not represented in this study. It would be expected that such patients would have smaller tumors and a lower incidence of myometrial lymphatic/vascular space invasion, which would likely impact detection of tumor-associated mutations in plasma in early stage patients.
For patients 1, 21, and 32, mutations detected in the plasma were not identified at 5% or greater allelic frequency in the tumor. The clinical significance of these mutations is unknown. As is the norm for molecular diagnostics testing, we examined mutations from one representative paraffin block of tumor. It is possible that if other paraffin blocks were available, one might harbor the mutations that were detected in the plasma. Such intra-tumoral heterogeneity has been identified in numerous systems, but how this heterogeneity should be accounted for in the clinical laboratory is currently uncertain. In one study, 3/69 (4.3%) of the colon cancers examined were wildtype for KRAS codon 12,13 mutations in the first block examined, but a mutation was detected in at least one additional block examined (16). Nearly 3% of the colon cancers examined had discordant BRAF V600E mutation status when assessing multiple paraffin blocks (15). Use of a 20 gene next-generation sequencing panel and testing 4 different areas of 87 colorectal adenocarcinomas demonstrated that 16% of mutation-positive cases had discordant mutations among the 4 areas sequenced (17). The most frequent genes demonstrating intra-tumoral heterogeneity were PIK3CA and KRAS (17). Sample pooling of DNA from multiple paraffin blocks (16,17) has been proposed to mitigate such intra-tumoral heterogeneity. In theory, the use of liquid biopsy analysis of peripheral blood as a more comprehensive way to assess a tumor’s molecular profile could substitute for the molecular testing of multiple paraffin blocks of tumor if the initial analyses demonstrated no mutation. This idea, however, has not been rigorously examined in a clinical setting. An alternative approach for the clinical laboratory would be to incorporate molecular barcodes, also known as unique identifiers, during library preparation for next-generation sequencing, as this would allow for more confidence in the assessment of low level mutations (18,19,20). The use of such molecular barcodes and digital droplet PCR (15) would be especially interesting to apply to patients with grade 1 endometrioid carcinomas, as these tumors tend to be smaller and with lower incidence of lymphatic/vascular space invasion.
Importantly, pre-analytical variables such as starting tumor per cent cellularity, cell-free DNA yield from plasma extraction, and mean cell-free DNA input for library preparation, were not significantly different between patients who had mutations detected in the plasma versus those who did not. In the current study, plasma was isolated from peripheral blood within 12 hours of the blood being collected into EDTA tubes, as per our standard tumor banking protocols. It is known that cell lysis after venipuncture when using standard EDTA tubes may result in contamination of tumor DNA with normal cell DNA (higher cell-free DNA) and decreased chances of detecting low level mutations (21,22). Previously, it was shown that when plasma is separated from peripheral blood after sitting in EDTA tubes for more than 16 hours, the amount of isolated cell free DNA is significantly higher compared to plasma isolated from blood collected in Streck tubes (13). It is possible that some of the cases in which no mutations were detected in the plasma may be false negatives due to our use of EDTA collection tubes. Thus, another benefit of prospective studies is that the more optimal Streck collection tube can be used.
Although our work represents the first attempt to identify tumor-associated mutations in the plasma of endometrial cancer patients, there have been several prior studies that employed different approaches to molecularly characterize a tumor without studying the tumor itself. Two prior studies examined cervico-vaginal secretions collected on tampons for methylated gene sequences known to occur in endometrial cancer (23,24). All 15 endometrial cancer patients examined had methylation of 3 or more genes detected in the cervico-vaginal secretions (23). In 38 patients with endometrial cancer, cervico-vaginal fluid gene methylation was significantly higher in 9/12 genes compared to individuals with benign endometrium (24). Analysis of the uterine cavity lavage fluid from 6 patients known to have endometrial cancer revealed mutations in PTEN, PIK3CA, CTNNB1 or APC in all 6 cases (25). Uterine lavage fluid from 7 endometrial cancer patients yielded mutations in PTEN, PIK3CA, KRAS, TP53, or CTNNB1 for each of the patients (26). For the tampon and uterine lavage fluid studies, presumably the source of gene mutations is from the primary tumor itself. It is unknown if mutations from the peripheral blood from a metastatic site outside the uterus could be detected in the tampons or uterine lavage fluid, although this possibility seems unlikely. Thus, while detecting mutations in tampons or uterine lavage fluid may have some utility as a means of early diagnosis of a primary endometrial cancer, these techniques would not be useful longitudinally for monitoring therapy of advanced disease that has spread outside the uterus. Detection of circulating tumor cells represents an alternative method to assessing mutations in plasma cell-free DNA. Previously, circulating tumor cells were identified from 6/40 (15%) endometrial cancer patients at the time of hysterectomy (27). Tumor stage, depth of myometrial invasion, and presence of lymphatic/vascular invasion, all factors influencing whether mutations were detected in plasma cell-free DNA in the current study, had no significant impact on detection of circulating tumor cells (27). For the 6 patients, only 1–2 circulating tumor cells were identified in each patient (27), which highlights a limitation to the widespread clinical applicability of this technique.
No matter the specific technique employed, the liquid biopsy approach has long been touted as a means of early cancer diagnosis or a way to perform cancer screening in at-risk individuals in a relatively non-invasive fashion. The use of liquid biopsy for this clinical application has pitfalls, however. It is known that somatic mutations in important driver genes are not restricted to malignancies. For example, it was recently demonstrated that 79% (19/24) of women with endometriosis had mutations in genes such as ARID1A, KRAS, and PIK3CA in the epithelial component of the endometriotic lesion tested (28). Mutations in each of these genes are relatively common in endometrioid endometrial carcinoma (11). Assuming the endometriosis-associated mutations could also be detected in peripheral blood, the finding of a plasma-based KRAS, ARID1A, or PIK3CA mutation in a woman during a screening test could not be assumed to mean that she had endometrial cancer. PIK3CA mutations can also occur in benign/premalignant skin lesions such as seborrheic keratosis (16%) and epidermal nevi (27%), but it is not known if the mutations from these skin lesions can be detected in a liquid biopsy (29). TP53 mutations are common in non-endometrioid and grade 3 endometrioid endometrial carcinomas, but they can also be detected in approximately 9% of grade 1 or 2, early stage endometrioid carcinomas (11). However, TP53 mutations can be found in the synovium of patients with rheumatoid arthritis (30,31). KRAS mutations are also identified in colorectal adenomas (32). Because of the prevalence of relevant gene mutations in benign and premalignant conditions, caution should be exercised when considering the use of liquid biopsy as a cancer screening tool (33). A more conservative, but still useful, clinical application for liquid biopsy in endometrial cancer could be as a non-invasive method of monitoring a patient for recurrence following hysterectomy. In this scenario, it would be necessary to first characterize the molecular changes in the primary tumor, as any mutations identified in the tumor could then be tracked longitudinally in blood samples. From the current study, using a next-generation sequencing panel of only 4 genes, more than 90% of patients with endometrioid-type endometrial carcinoma have at least one mutation identified and could be potentially followed with serial liquid biopsies. Prospective studies are necessary to determine if plasma-based mutations in these genes could be detected years after the hysterectomy and whether detection of such plasma mutations is an early indicator of tumor recurrence.
Acknowledgments
Financial Support: NIH SPORE in Uterine Cancer (RRB and WZ) NIH P50 CA09825; The Red and Charline McCombs Institute Center for Global Cancer Early Detection (RRB and WZ)
Footnotes
Disclosures: None
Portions of this manuscript were previously presented as a platform presentation at the United States and Canadian Academy of Pathology Annual Meeting, March 18–23, 2018, Vancouver, British Columbia, Canada.
REFERENCES
- 1.Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst 2017; 109: djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin 2018; 68: 7–30. [DOI] [PubMed] [Google Scholar]
- 3.Amant F, Moerman P, Neven P, et al. Endometrial cancer. Lancet 2005; 366: 491–505. [DOI] [PubMed] [Google Scholar]
- 4.Pietrasz D, Pecuchet N, Garlan F, et al. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res 2017; 23: 116–123. [DOI] [PubMed] [Google Scholar]
- 5.Sausen M, Phallen J, Adleff V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 2015; 6: 7686 10.1038/ncomms8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strickler JH, Loree JM, Ahronian LG, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Disc 2018; 8: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rolfo C, Mack PC, Scagliotti GV, et al. Liquid biopsy for advanced non-small cell lung cancer (NSLC): a statement paper from the IASLC. J Thorac Oncol 2018; June 6 pii: S1556-0864(18)30680-4. [DOI] [PubMed]
- 8.Cicchillitti L, Corrado G, De Angeli M, et al. Circulating cell-free DNA content as blood based biomarker in endometrial cancer. Oncotarget 2017; 8: 115230–115243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Djordjevic B, Hennessy BT, Li J, et al. Clinical assessment of PTEN loss in endometrial carcinoma: immunohistochemistry outperforms gene sequencing. Mod Pathol 2012: 25: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung LW, Hennessy BT, Li J, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism of regulation of PTEN protein stability. Cancer Discov 2011; 1: 170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurnit KC, Kim GN, Fellman BM, et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod Pathol 2017; 30:1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh RR, Patel KP, Routbort MJ, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn 2013; 15: 607–622. [DOI] [PubMed] [Google Scholar]
- 13.Mehrotra M, Singh RR, Chen W, et al. Study of preanalytic and analytic variables for clinical next-generation sequencing of circulating cell-free nucleic acid. J Mol Diagn 2017; 19: 514–524. [DOI] [PubMed] [Google Scholar]
- 14.Bartley AN, Luthra R, Saraiya D, et al. Identification of cancer patients with Lynch Syndrome: clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res 2012; 5: 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehrotra M, Singh RR, Loghavi S, et al. Detection of somatic mutations in cell-free DNA in plasma and correlation with overall survival in patients with solid tumors. Oncotarget 2018; 9: 10259–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richman SD, Chambers P, Seymour MT, et al. Intra-tumoral heterogeneity of KRAS and BRAF mutation status in patients with advanced colorectal cancer (aCRC) and cost-effectiveness of multiple sample testing. Anal Cell Pathol 2011; 34: 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson AC, Boone J, Cartwright D, et al. Optimal detection of clinically relevant mutations in colorectal carcinoma: sample pooling overcomes intra-tumoral heterogeneity. Mod Pathol 2018; 31: 343–349. [DOI] [PubMed] [Google Scholar]
- 18.Christensen E, Nordentoft I, Vang S, et al. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci Rep 2018; 8: 1917 10.1038/s41598-018-20282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vollbrecht C, Lehmann A, Lenze D, Hummel M. Validation and comparison of two NGS assays for the detection of EGFR T790M resistance mutation in liquid biopsies of NSCLC patients. Oncotarget 2018; 9: 18529–18539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016; 34: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toro PV, Erlanger B, Beaver JA, et al. Comparison of cell stabilizing blood collection tubes for circulating plasma tumor DNA. Clin Biochem 2015; 48: 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Messaoudi S, Rolet F, Mouliere F, et al. Circulating cell free DNA: preanalytical considerations. Clin Chim Acta 2013; 424: 222–230. [DOI] [PubMed] [Google Scholar]
- 23.Fiegl H, Gattringer C, Widschwendter A, et al. Methylated DNA collected by tampons – a new tool to detect endometrial cancer. Cancer Epidemiol Biomarkers Prev 2004; 13: 882–888. [PubMed] [Google Scholar]
- 24.Bakkum-Gomez JN, Wentzensen N, Maurer MJ, et al. Detection of endometrial cancer via molecular analysis of DNA collected with vaginal tampons. Gynecol Oncol 2015; 137: 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maritschnegg E, Wang Y, Pecha N, et al. Lavage of the uterine cavity for molecular detection of mullerian duct carcinomas: a proof-of-concept study. J Clin Oncol 2015; 33: 4293–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair N, Camacho-Vanegas O, Rykunov D, et al. Genomic analysis of uterine lavage fluid detects early endometrial cancers and reveals a prevalent landscape of driver mutations in women without histopathologic evidence of cancer: a prospective cross-sectional study. PLoS Med 2016; 13: e1002206 10.1371/journal.pmed.1002206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni T, Sun X, Shan B, et al. Detection of circulating tumour cells may add value in endometrial cancer management. Eur J Obstet Gynecol Reprod Biol 2016; 207: 1–4. [DOI] [PubMed] [Google Scholar]
- 28.Anglesio MS, Papdopoulos N, Ayhan A, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med 2017; 376: 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hafner C, van Oers JM, Hartmann A, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci USA 2007; 104: 13450–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reme T, Travaglio A, Gueydon E, et al. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol 1998; 111: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Firestein GS, Echeverri F, Yeo M, et al. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA 1997; 94: 10895–10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. [DOI] [PubMed] [Google Scholar]
- 33.Kato S, Lippman SM, Flaherty KT, Kurzrock R. The conundrum of genetic “drivers” in benign conditions. J Natl Cancer Inst 2016; 108: djw036. [DOI] [PMC free article] [PubMed] [Google Scholar]