

Abstract
Small single domain proteins that fold on the microsecond time scale have been the subject of intense interest as models for probing the complexity of folding energy landscapes. The villin headpiece subdomain (HP36) has been extensively studied because of its simple three helix structure, ultrafast folding lifetime of a few microseconds, and stable native fold. We have previously shown that folding as measured by a single 13C═18O isotopic label on residue A57 in helix 2 occurs at a different rate than that measured by global probes of folding, indicating noncooperative complexity in the folding of HP36. In order to determine whether this complexity reflects intermediates or parallel pathways over a small activation barrier, 13C═18O labels were individually incorporated at six different positions in HP36, including into all 3 helices. The equilibrium thermal unfolding transitions and the folding/unfolding dynamics were monitored using the unique IR signature of the 13C═18O label by temperature dependent FTIR and temperature jump IR spectroscopy, respectively. Equilibrium experiments reveal that the 13C═18O labels at different positions in HP36 show drastic differences in the midpoint of their transitions (Tm), ranging from 45 to 67 °C. Heterogeneity is also observed in the relaxation kinetics; there are differences in the microsecond phase when different labeled positions are probed. At a final temperature of 45 °C, the relaxation rate for 13C═18O A57 is 2.4e + 05 s−1 whereas for 13C═18O L69 HP36 the relaxation rate is 5.1e + 05 s−1, two times faster. The observation of sitedependent midpoints for the equilibrium unfolding transitions and differences in the relaxation rates of the labeled positions enables us to probe the progressive accumulation of the folded structure, providing insight into the microscopic details of the folding mechanism.
Graphical Abstract
■ INTRODUCTION
Small, single domain, ultrafast folding proteins often fold on a funneled energy landscape without populating intermediates in an equilibrium or kinetic sense.1,2 These so-called two-state folders have served as important models for testing protein folding theories. Their small size and fast folding times allow for extensive sampling of the folding dynamics in simulations extending to the millisecond time scale, providing atomic-level predictions of folding mechanism.3–7 It has been possible in many cases to test these predictions by connecting simulations with experiments, using a number of different experimental approaches.8–12 The transition state structure of two-state folders has been probed by measuring the effect of specific amino acid substitutions on the folding rate relative to their effect on protein stability to generate ø-values.13 Systematic application of this protein engineering approach yields an ensemble averaged degree of native structure acquired at a key point along the transition path, the maximum of the free energy barrier between unfolded and folded states, which can be compared with predictions from simulations.14 The simplest interpretation of ø-values, however, requires the assumption that mutants do not perturb the energetics of the unfolded ensemble.15 Folding on the downhill side of the transition state barrier has been studied using native-state H/D isotope exchange to measure what parts of a protein remain protected (folded) as the protein is progressively destabilized by denaturant.16 If one assumes that the time ordering of structure formation follows its relative free energy, with the most stable formed early and so on, this approach provides insight into the mechanism of structure formation in the late (post transition state) stages of folding.17 The most critical part of transition paths between unfolded and folded states, however, involves the initial search for the transition state because this is the rate limiting process. This part of the transition path is the most challenging to probe experimentally. Two approaches have been developed to study folding at every stage of the transition paths: single molecule methods and downhill protein folding.18–20 These methods offer the possibility of following the progressive accumulation of folded structure along the transition path, provided the spectroscopic probes have sufficient structural resolution. Here we report an alternative, nonperturbing approach, using site specific isotope labeling and temperature-jump (T-jump) infrared spectroscopy to probe the folding dynamics of the villin headpiece subdomain (HP36).
HP36 is an ultrafast folding protein that has been the subject of numerous studies due to its small size, simple α-helical topology, and fast folding rate.7,9,19,21–29 Despite this intense scrutiny, the folding mechanism of HP36 is not well established. Folding simulations of HP36 (or HP35, which lacks the N-terminal Met) have produced contradictory predictions of the folding mechanism, and there are cases where different force fields have produced different distributions of folding pathways.7,11,29–32. Experimental studies of HP36 folding by fluorescence and infrared (IR) T-jump21,33,34 and NMR line shape analysis35 observe two-state folding within a few microseconds and without population of intermediates. Both a balance of native and non-native residual structures and hydrophobic interactions in the unfolded state have been suggested to play important roles in HP36 folding.21,25,36–38 Different folding kinetics are observed using rapid dilution of denaturant to initiate folding rather than T-jump, interpreted as evidence for multiple folding pathways depending on the nature of unfolded conformational ensemble produced by temperature or denaturant.27 Other studies have suggested that formation of secondary structures, and the hydrophobic core is not the rate-limiting step but is likely preceded by the formation of a folding nucleus involving a turn between helices 2 and 3.26,39 Although these experimental studies have provided valuable insight into the folding process of HP36, the mechanism of folding is still not clear, and basic questions, such as the cooperativity of the folding process, have not been resolved.
Isotope-edited T-jump infrared spectroscopy provides a residue specific, nonperturbing tool to investigate protein folding dynamics.40,41 The amide I’ band of the polypeptide backbone is sensitive to both conformation and environment and has been extensively used for conformational analysis.42 While the amide I’ band is a sensitive indicator of secondary structure and backbone solvation,43 it is not a site-specific probe. Previously, we demonstrated that the site-specific introduction of a single 13C═18O isotope label in the amide backbone of a small protein enables residue specific resolution of the folding equilibrium and kinetics with high selectivity and sensitivity.41,44,45 The 13C═18O isotope label shifts the amide I’ signature of the labeled residue ~75 cm−1 to lower frequency, distinguishing it from the broad unlabeled amide I’ band. We demonstrated this approach using HP36, introducing a 13C═18O label at position A57 in the second helix.41 HP36 exhibits heterogeneous equilibrium unfolding behavior and folding kinetics when the labeled A57 position is compared with the unlabeled bulk, a surprising observation since global probes of HP36 folding (CD and fluorescence) suggest that it is a two-state folder that does not populate intermediate states.46 At elevated temperature HP36 folds over a very small barrier, which has been estimated as 1.6 kcal mol−1 for an unspecified reaction coordinate using Kramers theory to analyze the weakly temperature dependent T-jump kinetics data, and the 100 ns fast relaxation time to estimate the diffusion coefficient.24 This low barrier represents a nearly downhill folding scenario that offers the intriguing possibility of characterizing the multiplicity of parallel folding routes.24,47
In the present study, we have extended significantly our site-specific (13C═18O) labeling strategy to monitor equilibrium folding/unfolding and relaxation dynamics in other regions of HP36. We selectively labeled the carbonyl backbone in each of the helices (F47, A49, and F51 in helix 1; A57 in helix 2; and L63 and L69 in helix 3) to provide site-specific structural information within the context of the HP36 structure (Figure 1). We find that there are complex heterogeneities in the folding of HP36, with different apparent stabilities and kinetics observed depending on where the protein is probed.
Figure 1.
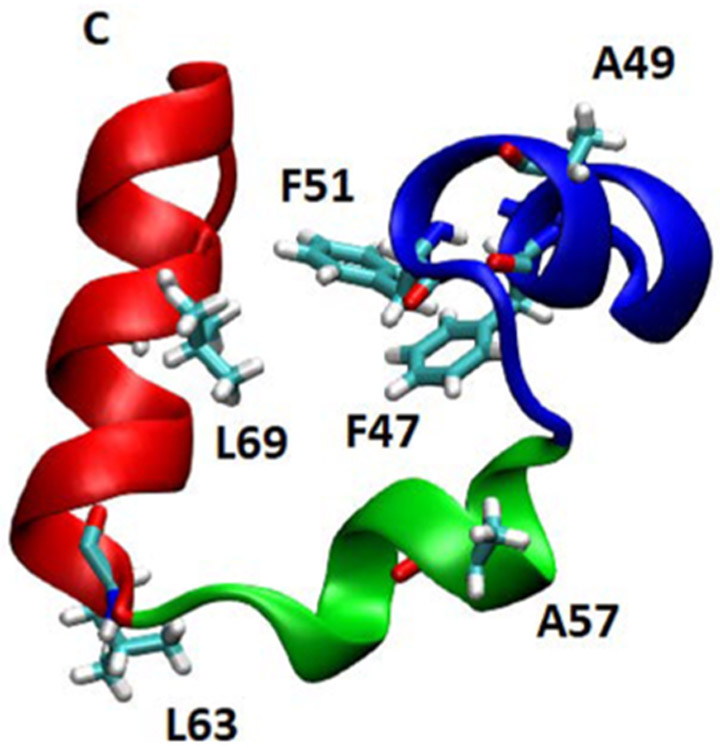
Ribbon diagram of HP36 from the X-ray structure (PDB ID 1YRF) showing the location of backbone amide 13C═18O labels on residues in stick format. The location of the C-terminus is indicated.
■ MATERIALS AND METHODS
Sample Preparation.
The villin headpiece subdomain (HP36 sequence MLSDEDFKAVFGMTRSA-FANLPLWKQQNLKKEKGLF) was prepared by solid-phase peptide synthesis using standard FMOC protocols and purified via reverse phase HPLC. The identity of the purified product was confirmed by mass spectrometry. The 13C═18O label was incorporated into the amide group of phenylalanine 47, alanine 49, phenylalanine 51, alanine 57, and leucine 63 and 69 according to a previously reported method48 with an isotope incorporation yield of 99% for 13C and 91% for 18O. The numbering system used here corresponds to that of the full-length villin headpiece. The first residue in the helical subdomain is Leu-42. Many studies of the headpiece subdomain have made use of recombinant protein that includes an additional Met at the N-terminus. This Met, which is the first residue in the construct used here, is designated as Met-41 for consistency with previously published studies. The protein was lyophilized and dissolved in D2O (Cambridge Isotope Laboratories) at least twice to allow complete deuterium—hydrogen exchange of the amide protons to occur. The protein was dissolved in a deuterated buffer containing 10 mM sodium phosphate and 150 mM sodium chloride at a pH* of 5.8 in D2O. pH* refers to the uncorrected (for D2O) pH-meter reading at 25 °C. This procedure is necessary to remove solvent interference in the amide I’ region (the prime indicates that D2O is the solvent). The protein solutions were filtered to remove any aggregates present and used without any further purification. The protein concentration for the IR experiments was 1.5 mM.
FTIR Spectroscopy.
Equilibrium FTIR temperature-dependent spectra were recorded on a Varian 3100 FTIR spectrometer equipped with a liquid nitrogen cooled mercury cadmium telluride (MCT) detector. The temperature-dependent FTIR spectra were the average of 256 scans recorded with 2 cm−1 spectral resolution. An IR cell composed of a 100 μm Teflon spacer sandwiched between two CaF2 windows held in a copper frame was utilized to record the temperature dependent IR spectra of each sample under identical conditions. The temperature of the IR cell was controlled by a water bath, and the sample temperature was measured by a thermocouple attached to the copper cell. The cell has two compartments, one containing the sample and the other containing a buffer reference; it is translated by a computer-controlled stage to allow acquisition of sample and reference spectra. The absorption spectra of the protein were determined from the negative logarithm of the ratio of the single beam spectra of the sample to the reference side of the IR split cell at each temperature. The equilibrium thermal unfolding of the protein was found to be reversible (absence of aggregation).
T-Jump Time-Resolved IR Kinetics Measurements.
The T-jump time-resolved IR apparatus used to measure the HP36 folding kinetics in this study has been described previously.49 This method was a pump-probe experiment in which a 1.91 μm, 10 ns Q-switched laser pulse constituted the pump beam that initiated a rapid T-jump in the sample, thereby perturbing the protein to a new equilibrium. A quantum cascade laser (Daylight Solutions Inc., Poway, CA) tunable in the 1535–1720 cm−1 region was used to probe structural changes in the sample, as the system relaxed in response to the T-jump to a new equilibrium at the final temperature. The changes in transmission of the IR probe beam were detected by a fast (200 MHz) photovoltaic (PV) MCT detector (Kolmar Technologies, Newburyport, MA). The 1.91 μm (10 ns pulse width, ~ 30 mJ/pulse) pump radiation was obtained from a H2 (g)-filled Raman shifter (1 Stokes shift) pumped by a 10 Hz repetition rate Q-switched DCR-4 Nd:YAG laser (Spectra Physics, Mountain View, CA) and was absorbed by weak combination bands in the D2O solution. This pump radiation was chosen due to its transmission properties (87% pump radiation transmitted through 100 μm path length sample cell) that allow for nearly uniform heating in the pump-probe overlap region.
The same CaF2 window cell used for the equilibrium FTIR experiments was used for the kinetic measurements with the reference D2O buffer compartment serving as an internal thermometer to determine the magnitude of the T-jump. The protein relaxation kinetic traces were extracted by subtracting the change in absorbance of the reference (1600 cm−1) from the sample (1632, 1572, 1585, 1588, and 1589 cm−1) in response to the T-jump. The kinetic traces were recorded from the nanosecond to tens of milliseconds time regime with the thermal energy diffusing from the pump-probe interaction volume in about 20 ms. The transients were fit to a single exponential function for the microsecond phase. Data analysis was performed using Igor Pro (Wavemetrics, Inc.).
■ RESULTS AND DISCUSSION
Site-Specific Labeling of HP36 and Characterization of Equilibrium Folding.
Isotope-edited FTIR difference spectra establish the local nature of the 13C═18O probes and their sensitivity to the specific structural environment of the labeled residue. We studied 6 different HP36 constructs with site-specific 13C═18O labels strategically located in each of the 3 helices at positions F47, A49, F51, A57, L63, and L69. We measured the temperature dependent FTIR spectra of each of these constructs from 10 to 90 °C in ~5 °C increments (shown for A57 in Figure 2A; others included in the SI, Figure S1). The FTIR difference spectrum (90–10 °C) of each construct is shown in Figure 2B; each has a negative peak between 1550–1590 cm−1, due to the 13C═18O-labeled site. Negative peaks in the difference spectra correspond to specific structures or interactions present in the folded state at low temperature that are lost as temperature is increased and the protein is unfolded. The position of this peak varies (summarized in Table 1), depending on the local structure of the labeled amino acid in the folded state. F47 and F51 HP36 each have a well-resolved peak due to the 13C═18O-labeled site at 1589 cm−1 (Figure 2B), shifted relative to the unlabeled 12C=16O band of buried helix (1650 cm−1). This result is consistent with the positions of F47 and F51 in the middle of the helix 1 and their burial in the hydrophobic core, where the amide groups are solvent-protected, and the side chains form key hydrophobic interactions with one another and with F58. The observed isotopic shift of ~61 cm−1 is less than the expected shift of 75 cm−1 for a local C═O oscillator.50 The geometry of a helix leads to extensive dipolar coupling among the aligned 12C═16O oscillators, causing the unlabeled helix band to shift to a lower frequency; in contrast a labeled site is decoupled because of its frequency mismatch and thus behaves more like a local oscillator.51 This decoupling of the labeled position results in a smaller than expected isotopic shift. A49 and A57 HP36 each have a 13C═18O labeled peak at 1572 cm−1, corresponding to a shift of 60 cm−1 from the unlabeled solvated helix component at 1632 cm−1, consistent with the solvent exposure of these backbone positions. There is an additional feature at 1560 cm−1 in the A49 spectrum that is likely due to a shift in the environment of a carboxylate (aspartate or glutamate) side chain, which has a strong absorption at 1558 cm−1, upon unfolding. The two labels in helix 3, L63 and L69, have a unique absorption centered at 1585 cm-1. These positions are solvent-protected in the folded structure, so this frequency represents a 65 cm−1 shift from the buried helix position. Since helix 3 is the longest helix, the dipolar coupling of C═O oscillators is likely the strongest in this case, which might explain the different shift compared to the helix 1 labels (F47 and F51).
Figure 2.

Isotope-edited FTIR of labeled HP36. (A) Temperature dependent difference FTIR spectra in the amide I’ spectral region of 13C═18O A57-HP36 as a function of temperature from 10 to 90 °C, in 5 °C increments. Difference spectra were generated by subtracting the lowesttemperature absorption spectrum from the spectra at the higher temperatures. (B) Temperature dependent difference FTIR spectra of 13C═18O F47, F51, A49, A57, L63, and L69 HP36, obtained by subtracting the 10 °C spectrum from the 90 °C one for each labeled protein. The positions of the labeled peaks are indicated by the vertical lines. (C) Thermal unfolding transitions monitored for each 13C═18O peak and compared with 1631 cm−1 as an internal standard.
Table 1.
Site-Specific (13C═18O) Stretching Frequency Assignments
| helix | 1 | 1 | 1 | 2 | 3 | 3 |
|---|---|---|---|---|---|---|
| residue | F47 | A49 | F51 | A57a | L63 | L69 |
| 13C═18O frequency (cm−1) | 1589 | 1572 | 1589 | 1572 | 1585 | 1585 |
| Tm (°C) | 63.6 | 57.2 | 64.5 | 45.5 | 66 | 67 |
The 1572 cm−1 assignment for A57 was made previously41
The unlabeled (12C═16O) region of the amide I’ difference spectra (1600–1700 cm−1) also shows variations depending on the location of the 13C═18O label (Figure 2B). These variations are due in part to the different chemical environments of the position substituted, for example a solvated helix at 1632 cm−1 versus a buried helix at 1650 cm−1, since labeling a specific site shifts part of the amide I’ intensity to a lower frequency, leaving a “hole” in the corresponding unlabeled spectral position. The 1632/1650 cm−1 intensity ratio (the ratio of unlabeled solvated/buried helix) shifts as expected for the position of the label: it decreases when a solvated helix position is labeled (A49, A57) and increases when a buried position is labeled (L63, L69). It is also possible that the dipolar coupling of C═O groups within each helix contributes to these variations, since this coupling depends on the length of the helix. For example, the position of the 13C═18O label in the middle of helix 3 (L69) should have a larger effect on the 12C═16O dipolar coupling along the helix since it divides the unlabeled helix length in half, whereas a position closer to the end (L63) does not disrupt the coupling along most of the length of the helix. In summary, it is clear from these results that the IR peaks due to the 13C═18O groups probe the unique structural environment of the specific amino acid that is labeled.
The site-specific thermal unfolding of HP36 was measured for each of the six 13C═18O-labeled constructs, and Figure 2C compares the thermal transitions of each one with the global unfolding transition measured at the solvated helix backbone position at 1631 cm−1. The local melt curves are different from one another and from the global transition, showing a heterogeneous distribution of Tm values ranging from 45 to 67 °C and varying sharpness of the local transitions. These results are consistent with our prior work, where we found that the global melt of HP36 (measured at 1648 cm−1, the 12C═16O frequency of buried helix) had a midpoint temperature (Tm) of 63.7 °C, whereas the midpoint of the transition of the A57 label at 1572 cm−1 was centered at 45.5 °C, or about 18 °C lower than the global Tm.41 A slightly broader transition with the same midpoint is observed for unlabeled HP36 when monitored at 1631 cm−1 (Figure 2C), which we attribute to temperature dependent changes in the hydrogen bonding between the solvent and the solvated components of the helices.52 The lower Tm for A57 indicates that the local structure involving this residue unfolds at a lower temperature than the global melting of the protein. F47 and F51 in helix 1 both have a midpoint temperature at 63 °C, similar to the global protein midpoint temperature, as measured at 1631 or 1648 cm−1. The Tm for A49 in helix 1, however, is 57 °C, which is 7 °C lower than the global Tm. The higher melting temperature observed for the F47 and F51 labels in helix 1 compared to A49 may be due to the residual structure comprising hydrophobic interactions of F47, F51, and F58 in the unfolded state of this protein.21 L63 and L69 located in helix 3 have local midpoint temperatures of 67 and 66 °C, respectively, which is slightly higher than the global Tm. It can also be seen in Figure 2C, that the local melting curve is steeper than the global melting curve for the helix 3 labels (L63 and L69) but less steep for the A47 (helix 1) label. This heterogenity in the unfolding thermodynamics is consistent with 2D 13C NMR studies on HP35, which observed a nonuniform population of the native secondary structure along the polypeptide chain by a rapid freeze method.53
A two-state folder should exhibit a melt curve that is a linear combination of the spectral signatures of the folded and unfolded conformations. The same melt curve should be observed, regardless of the nature of the spectral probe or the position of the label. In contrast, proteins that fold through a series of intermediates or that gradually populate an effective continuum of intermediates show distinct probe dependence of the folding thermodynamics. HP36 clearly fits the latter description. The deviation of site-specific thermal melt curves from the global transition indicates a non-two-state behavior characteristic of downhill folding or folding over a small barrier (incipient downhill folding), which allows the stability of local regions of the proteins structure to be probed independently. Analogous behavior has been reported for other downhill or nearly downhill folding proteins. For example, distinct, site-specific melt curves that depend on the position of the probe have been observed for BBL using NMR chemical shifts18 and a de novo helix-turn-helix motif using site-specific 13C isotopically edited IR.54,55 This interpretation is also consistent with the Ising-like, statistical mechanical model of the folding of HP36 developed by Kubelka et al.,46 which considers the ensemble of residue-specific microstates in terms of two possible conformations for each residue, native and nonnative. The success of this model in predicting the thermodynamics and kinetics of HP36 folding suggests that the behavior of a single-labeled site may closely approximate two-state behavior despite global non-two-state behavior.29 Therefore, the sitespecific thermal melt curves report on the relative stability of the local structure around the probe, indicating the most stable part of the HP36 structure is helix 3, followed by helix 1 and helix 2, respectively.
In summary, the results obtained from the local melt curves for each labeled position of HP36 clearly show the difference in the stability of labeled residues in each of the helices and the difference in melting between positions in the same helix. Some of the local unfolding curves are similar to the global transition, but at the global transition midpoint, there are clearly regions of the structure that are mostly unfolded, and others that are still native-like. The average of the local melt curves determined from site-specific labels is similar to the global melt determined from the unlabeled solvated helix band. Thus, the sum of the local unfolding processes monitored at positions in each of the helices corresponds well to the global process measured by the low resolution probe. These results are consistent with the statistical nature of protein folding, characterized by overlapping local events that span the full global transition instead of an all or nothing transition.
Residue Specific Resolution of Folding Dynamics.
Our previous T-jump IR measurements observed two kinetics phases in HP36, a 100 ns and a few μs relaxation phase.41 The microsecond (~5 μs) relaxation phase exhibits a small activation barrier and reports on the global folding and unfolding transition of the protein. The exact origin of the 100 ns phase is still uncertain. We have postulated that it involves the helix—coil transition in either the folded or unfolded free energy basin or both.21 Kubelka et al. favored an assignment to the fraying of helical structures in the folded free-energy basin.46 Changes in solvation around the solvated components of the helices might also contribute to this fast phase. Regardless of its exact origin, this 100 ns relaxation occurs on a much faster time scale than the global folding—unfolding transition and will not be considered further.
Figure 3 shows the T-jump IR relaxation transients monitored at the indicated 13C═18O label frequencies for F47, A49, F51, A57, L63, and L69 HP36. The decrease in the helix band absorption with time is consistent with a shift of the equilibrium toward the unfolded state with increasing temperature, as expected from the equilibrium melts. Although the net shift in equilibrium is toward the unfolded state, the relaxation response is dominated by the folding rate because the T-jump to 45 °C is well below the Tm of 63.7 °C for the global transition. For each site-specifically labeled construct, we also obtained the T-jump IR response at 1632 cm−1 (the unlabeled solvated-helix peak) as an internal reference (black trace in Figure 3). The 1632 cm−1 reference response is highly reproducible and is invariant within the noise of the measurement for all of the labeled constructs. It is immediately apparent that the transient response varies, depending on the position of the label. All of the transients are well fit with a single exponential function; only the fits at the extremes are shown in Figure 3 for clarity. The observed rates derived from these fits for T-jumps to final temperatures of 45 and 54 °C show that L69 (5.1e + 05 s−1; 7.14e + 05 s−1) in the middle of helix 3 exhibits the fastest relaxation rate, followed by F47 (3.59e+05 s−1; 4.82e+05 s−1), F51 (3.74e + 05 s−1; 4.73e + 05 s−1) in helix 1 and L63 (3.57e + 05 s−1; 4.39e + 05 s−1) at the end of the helix 3, followed by A49 (2.56e + 05 s−1; 3.39e + 05 s−1) in helix 1 and A57 (2.41e + 05 s−1; 3.12e + 05 s−1) in helix 2. The unlabeled reference that reports on the average helix backbone response shows a relaxation time that is intermediate between the two extremes.
Figure 3.
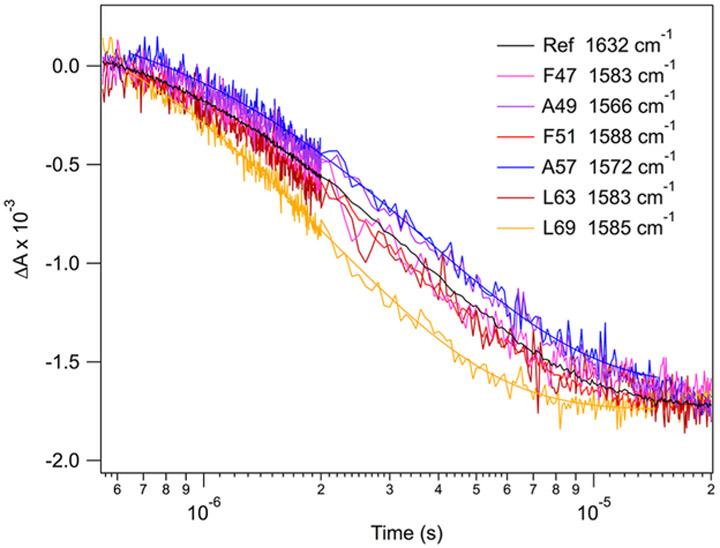
Infrared T-jump relaxation kinetics of 13C═18O F47, F51, A49, A57, L63, and L69 HP36, in response to a T-jump from 30 to 45 °C. The IR response is monitored at the indicated probe frequencies corresponding to the absorption of the labeled site and compared to the unlabeled reference response at 1632 cm−1. Each kinetic trace is the average of 2500 laser shots. Single exponential fits are shown for the fastest (L69) and slowest (A57) IR transients.
We also obtained relaxation transients for T-jumps to a final temperature near the midpoint (local Tm) of the thermal transition of each site-specific label for comparison (Figure S2). Table 2 reports the observed relaxation rates derived from single exponential fits of T-jumps to near the transition midpoint of each local probe. The observed rates for F47 and F51 are similar, but A49, which is in between these two residues, shows a slower rate. The faster rate for F47 and F51 may be due to the early formation of an ordered hydrophobic core consisting of the side chains of F47, F51, F58, and L69.53 Thus, we observed a heterogeneous distribution of relaxations rates that are different for each helix and even for different positions within a helix. Similar to the equilibrium behavior, the sum of the local transients monitored at positions in each of the helices corresponds well to the global transient monitored at the unlabeled backbone position. These results are consistent with the statistical nature of protein folding over a small barrier.
Table 2.
Relaxation Ratesa for a 15 °C T-Jump to Apparent Tm
| T (° C) | kr ref (×105 s−1) | kr F47 (×105 s−1 | kr A49 (×105 s−1 | kr F51 (×105 s−1 | kr A57 (×105 s−1) | kr L63 (×105 s−1 | kr L69 (×105 s−1) |
|---|---|---|---|---|---|---|---|
| 45 | 3.38 ± 0.05 | 2.41 ± 0.09 | |||||
| 54 | 4.24 ± 0.04 | ||||||
| 60 | 4.95 ± 0.04 | 4.0 ± 0.09 | |||||
| 63 | 5.78 ± 0.03 | 6.58 ± 0.1 | 6.21 ± 0.08 | ||||
| 66 | 6.10 ± 0.03 | 6.06 ± 0.09 | 8.55 ± 0.09 |
Determined from single exponential fits; errors are standard deviations from at least 3 separate trials.
The observed relaxation rates for the labeled positions are plotted as a function of the inverse temperature in Figure 4. Most of the relaxation rates are for temperatures below Tm, corresponding to the folding arm of the transition. The temperature dependence of the relaxation rates for the labeled positions are compared to the unlabeled solvated helix response at 1631 cm−1 as a reference. The solvated helix band gives a similar temperature dependence to what was reported previously for the buried helix band probed at 1650 cm−1.21 The temperature dependence is small in all cases, as expected for a process with a low activation barrier. There is some variation of the temperature dependence with the position of the label. A49 and A57 HP36 exhibit a steeper temperature dependence than the unlabeled reference, whereas it is shallower for L69. These differences could be due to position dependent differences in internal friction of the protein structure that contribute to the temperature dependence of the rate of the local response.
Figure 4.
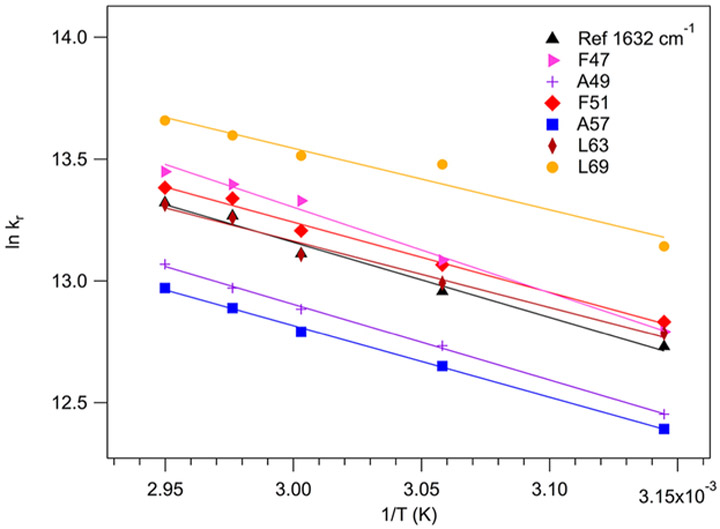
Arrhenius plot of the observed relaxation rates as a function of inverse temperature for (13C═18O) F47, A49, F51, A57, L63, and L69 HP36. The unlabeled solvated helix rates (1631 cm−1) are also shown for reference.
■ DISCUSSION
One of the experimental signatures of downhill or incipient downhill folding is heterogeneous, probe dependent transitions observed in equilibrium or kinetics measurements. T-jump infrared spectroscopy, in combination with site-specific, nonperturbing 13C═18O labels, serves as an excellent tool to probe differences in stability and folding kinetics with high structural resolution. Complex heterogeneity in the folding of HP36 is clearly observed by probing the thermal stabilities and folding kinetics of different regions of the protein. The different thermal melting profiles for the labeled positions (F47, A49, A57, L63, and L69) measured in this study suggest different stabilities for the three helices, or at least the parts of the helices that contain the isotope labels, with helix 3 being the most stable followed by helix 1 and then helix 2. The kinetics measured by T-jump IR follow this same trend, with folding rates correlated with helix stability.
Energy landscape theory predicts that there are many nearly equally likely transition paths to fold a small single domain protein on a well funneled landscape. In this view, HP36 folds through a large ensemble of pathways. Further support for this idea is provided by the small barrier separating the folded and unfolded basins, especially at the elevated temperature of the T-jump, such that the system can access many pathways. Ensemble kinetics experiments measure the average transition path, not the distributions, and thus are dominated by the pathways having the greatest flux. Therefore, the heterogeneity of the equilibrium and kinetics response of HP36 observed for the different 13C═18O probe positions is a consequence of the flux through these many possible pathways. The ensemble measurements reflect the underlying microscopic distributions.
Even for such a multipathway process, it is often possible to predict a general folding mechanism by using a coarse grain approach, for example, to analyze multiple trajectories from MD simulations. Such an approach has been used to address the question of whether there is a preferential ordering of the formation of the individual helices in the folding mechanism of HP36. If the assembly pathway of HP36 is defined in terms of the order in which the individual helices are formed, there are six possible pathways to fold the 3 helices of this protein. Henry et al. used both a simplified Ising-like model and all atom MD simulations to analyze the distribution of folding transition paths for HP36 in terms of the order of helix formation.29 They found significant population flux through all six pathways, although they observed the majority of flux through the 123, 213, and 231 transition paths. The distribution of transition paths observed in their analysis clearly favors the formation of helices 1 and 2 before helix 3, although there is significant overlap in the time scale of formation of all three. Other HP36 folding simulations found evidence of early formation of helix 1 or helix 2, but there are also conflicting reports that helix 3 forms first.31,56
The isotope-edited relaxation kinetics presented here are consistent with the microscopic model of Henry et al. in several respects. First, there is not a clear kinetic ordering of the individual helix formation. Rather, all three helices are formed with rates of the same order of magnitude, as might be expected from the model where all six pathways are accessible. Nevertheless, there are differences in the rates observed for probes placed in each of the helices, a consequence of the partitioning of the flux through multiple pathways having different orders of formation of the individual helices. These differences provide evidence for a kinetic bias for the early formation of different regions of the structure, particularly involving helices 3 and 1. There is even evidence of different folding rates for specific sites within the same helix (helix 1). Hence, it is clear from the kinetics that the helices cannot be considered as individual foldons that form in a well-ordered sequence. It is important to note that the microscopic model of Henry et al. defines an individual helix of n residues as folded when at least n – 2 contiguous residues are in their native conformations. The T-jump IR measurements are limited to a few local probes and therefore only provide a rough measure of the rate of helix formation. Nevertheless, the predictions of the microscopic model about the dominance of the 123, 213, and 231 transition paths are difficult to reconcile with our measurements. We observe the fastest relaxation kinetics for the labels in helix 3, evidence that the dominant pathways are ones for which this helix forms first.
The folding mechanism that emerges from our results is summarized in Figure 5. Formation and stabilization of helix 3 plays a crucial role in driving the folding of HP36. Folding of helix 3 appears to start from the middle of helix 3 (L69) and then propagate to its N-terminus (L63). This initiation point on helix 3 may also set up the interactions between the hydrophobic core residues comprised of L69, F58, F51, and F47, which in turn stabilize the formation of helix 1. Again, we emphasize that these are not sequential events, because their kinetics are strongly overlapped. The slower relaxation rates and the lower thermal stabilities for helices 1 and 2 suggest to us that these helices are probably stabilized later in the predominant folding pathways. The observation of faster folding rates for F47 and F51 when compared to A49 in helix 1 provides evidence that formation of the hydrophobic core precedes the complete formation of helix 1 and helix 2. The short helical segment helix 2 may dock with the preassembled structure to complete the folding of HP36. NMR studies from Tang et al.37,38 and Havlin and Tycko36 have indicated that there are residual helical structures within helices 1 and 2 in the unfolded state of this protein. It is possible that these residual helical structures play a role in the folding of this protein by reducing the conformational space that it has to sample early in the folding process. While early formation of these helices may occur, our data suggest that it is the formation and stabilization of helix 3 that dictates the fast folding of this protein.
Figure 5.

Ribbon cartoon representation of the HP36 folding mechanism: (A) thermal melt and (B) folding pathways.
In summary, the results presented here show that nonperturbing, site specific 13C═18O isotope labels can be utilized as infrared probes in combination with T-jump infrared spectroscopy to resolve equilibrium and kinetic heterogeneity in the folding of HP36. Different regions of the protein fold at different rates, suggesting preferred pathways through a funneled energy landscape, as shown in Figure 5B. It seems likely that other ultrafast folding proteins with low energy barriers and microsecond folding times may have multiple routes for the folding transition to the native state on a smooth energy landscape. This complexity should be easily resolvable using the methods developed in this study because they are completely general and easily applied to small, fast folding proteins.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a NIH Grant GM053640 (R.B.D.) and NSF Grant MCB-1330259 (D.P.R.).
ABBREVIATIONS
- T-jump
temperature-jump
- IR
infrared
- FTIR
Fourier transform infrared
- HP36
villin headpiece subdomain
- MD
molecular dynamics.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.jpcb.8b07683.
FTIR difference spectra of labeled HP36 and T-jump relaxation transients (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Jackson SE; Fersht AR Folding of chymotrypsin inhibitor 2.1. Evidence for a 2-state transition. Biochemistry 1991, 30, 10428–10435. [DOI] [PubMed] [Google Scholar]
- (2).Barrick D What have we learned from the studies of two-state folders, and what are the unanswered questions about two-state protein folding? Phys. Biol 2009, 6, 015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shaw DE; Maragakis P; Lindorff-Larsen K; Piana S; Dror RO; Eastwood MP; Bank JA; Jumper JM; Salmon JK; Shan Y; Wriggers W Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–6. [DOI] [PubMed] [Google Scholar]
- (4).Lindorff-Larsen K; Piana S; Dror RO; Shaw DE How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [DOI] [PubMed] [Google Scholar]
- (5).McKiernan KA; Husic BE; Pande VS Modeling the mechanism of CLN025 beta-hairpin formation. J. Chem. Phys 2017, 147, 104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pang YP How fast fast-folding proteins fold in silico. Biochem. Biophys. Res. Commun 2017, 492, 135–139. [DOI] [PubMed] [Google Scholar]
- (7).Mori T; Saito S Molecular Mechanism Behind the Fast Folding/Unfolding Transitions of Villin Headpiece Subdomain: Hierarchy and Heterogeneity. J. Phys. Chem. B 2016, 120, 11683–11691. [DOI] [PubMed] [Google Scholar]
- (8).Snow CD; Nguyen H; Pande VS; Gruebele M Absolute Comparison of Simulated and Experimental Protein-Folding Dynamics. Nature 2002, 420, 102–106. [DOI] [PubMed] [Google Scholar]
- (9).Beauchamp KA; Ensign DL; Das R; Pande VS Quantitative comparison of villin headpiece subdomain simulations and triplet-triplet energy transfer experiments. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12734–12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cellmer T; Buscaglia M; Henry ER; HofTichter J; Eaton WA Making connections between ultrafast protein folding kinetics and molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6103–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Piana S; Lindorff-Larsen K; Shaw DE Protein folding kinetics and thermodynamics from atomistic simulation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zanetti-Polzi L; Davis CM; Gruebele M; Dyer RB; Amadei A; Daidone I Parallel folding pathways of Fip35 WW domain explained by infrared spectra and their computer simulation. FEBS Lett 2017, 591, 3265–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Matouschek A; Kellis JT; Serrano L; Fersht AR Mapping the Transition-State and Pathway of Protein Folding by Protein Engineering. Nature 1989, 340, 122–126. [DOI] [PubMed] [Google Scholar]
- (14).Best RB; Hummer G Microscopic interpretation of folding varphi-values using the transition path ensemble. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cho JH; Meng WL; Sato S; Kim EY; Schindelin H; Raleigh DP Energetically significant networks of coupled interactions within an unfolded protein. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12079–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bai Y; Sosnick TR; Mayne L; Englander SW Protein folding intermediates: native-state hydrogen exchange. Science 1995, 269, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Englander SW; Mayne L The case for defined protein folding pathways. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8253–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sadqi M; Fushman D; Munoz V Atom-by-atom analysis of global downhill protein folding. Nature 2006, 442, 317–21. [DOI] [PubMed] [Google Scholar]
- (19).Zoldak G; Stigler J; Pelz B; Li H; Rief M Ultrafast folding kinetics and cooperativity of villin headpiece in single-molecule force spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chung HS; Eaton WA Protein folding transition path times from single molecule FRET. Curr. Opin. Struct. Biol 2018, 48, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Brewer SH; Vu DM; Tang YF; Li Y; Franzen S; Raleigh DP; Dyer RB Effect of modulating unfolded state structure on the folding kinetics of the villin headpiece subdomain. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 16662–16667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dyer RB Ultrafast and downhill protein folding. Curr. Opin. Struct. Biol. 2007, 17, 38–47. [DOI] [PubMed] [Google Scholar]
- (23).Cellmer T; Henry ER; Hofrichter J; Eaton WA Measuring internal friction of an ultrafast-folding protein. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18320–18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Godoy-Ruiz R; Henry ER; Kubelka J; Hofrichter J; Munoz V; Sanchez-Ruiz JM; Eaton WA Estimating free-energy barrier heights for an ultrafast folding protein from calorimetric and kinetic data. J. Phys. Chem. B 2008, 112, 5938–5949. [DOI] [PubMed] [Google Scholar]
- (25).Xiao SF; Bi Y; Shan B; Raleigh DP Analysis of Core Packing in a Cooperatively Folded Miniature Protein: The Ultrafast Folding Villin Headpiece Helical Subdomain. Biochemistry 2009, 48, 4607–4616. [DOI] [PubMed] [Google Scholar]
- (26).Bunagan MR; Gao JM; Kelly JW; Gai F Probing. the Folding Transition State Structure of the Villin Headpiece Subdomain via Side Chain and Backbone Mutagenesis. J. Am. Chem. Soc 2009, 131, 7470–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhu L; Ghosh K; King M; Cellmer T; Bakajin O; Lapidus LJ Evidence of Multiple Folding Pathways for the Villin Headpiece Subdomain. J. Phys. Chem. B 2011, 115, 12632–12637. [DOI] [PubMed] [Google Scholar]
- (28).Serrano AL; Bilsel O; Gai F Native state conformational heterogeneity of HP35 revealed by time-resolved FRET. J. Phys. Chem. B 2012, 116, 10631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Henry ER; Best RB; Eaton WA Comparing a simple theoretical model for protein folding with all-atom molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lei HX; Su Y; Jin LA; Duan Y Folding Network of Villin Headpiece Subdomain. Biophys. J. 2010, 99, 3374–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lee IH; Kim SY; Lee J Dynamic Folding Pathway Models of the Villin Headpiece Subdomain (HP-36) Structure. J. Comput. Chem 2010, 31, 57–65. [DOI] [PubMed] [Google Scholar]
- (32).Piana S; Lindorff-Larsen K; Shaw DE How robust are protein folding simulations with respect to force field parameterization? Biophys. J 2011, 100, L47–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kubelka J; Eaton WA; Hofrichter J Experimental tests of villin subdomain folding simulations. J. Mol. Biol 2003, 329, 625–630. [DOI] [PubMed] [Google Scholar]
- (34).Buscaglia M; Kubelka J; Eaton WA; Hofrichter J Determination of ultrafast protein folding rates from loop formation dynamics. J. Mol. Biol 2005, 347, 657–664. [DOI] [PubMed] [Google Scholar]
- (35).Wang MH; Tang YF; Sato SS; Vugmeyster L; McKnight CJ; Raleigh DP Dynamic NMR line-shape analysis demonstrates that the villin headpiece subdomain folds on the microsecond time scale. J. Am. Chem. Soc 2003, 125, 6032–6033. [DOI] [PubMed] [Google Scholar]
- (36).Havlin RH; Tycko R Probing site-specific conformational distributions in protein folding with solid-state NMR. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3284–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tang YF; Rigotti DJ; Fairman R; Raleigh DP Peptide Models Provide Evidence for Significant Structure in the Denatured State of A Rapidly Folding Protein: The Villin Headpiece Subdomain. Biochemistry 2004, 43, 3264. [DOI] [PubMed] [Google Scholar]
- (38).Tang YF; Grey MJ; McKnight J; Palmer AG; Raleigh DP Multistate folding of the villin headpiece domain. J. Mol. Biol 2006, 355, 1066–1077. [DOI] [PubMed] [Google Scholar]
- (39).Glasscock JM; Zhu YJ; Chowdhury P; Tang J; Gai F Using an amino acid fluorescence resonance energy transfer pair to probe protein unfolding: Application to the villin headpiece subdomain and the LysM domain. Biochemistry 2008, 47, 11070–11076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Williams S; Causgrove TP; Gilmanshin R; Fang KS; Callender RH; Woodruff WH; Dyer RB Fast events in protein folding: Helix melting and formation in a small peptide. Biochemistry 1996, 35, 691–697. [DOI] [PubMed] [Google Scholar]
- (41).Brewer SH; Song BB; Raleigh DP; Dyer RB Residue specific resolution of protein folding dynamics using isotope-edited infrared temperature jump spectroscopy. Biochemistry 2007, 46, 3279–3285. [DOI] [PubMed] [Google Scholar]
- (42).Dyer RB; Gai F; Woodruff WH; Gilmanshin R; Callender RH Infrared studies of fast events in protein folding. Acc. Chem. Res 1998, 31, 709–716. [Google Scholar]
- (43).Gilmanshin R; Callender RH; Dyer RB The core of apomyoglobin E-form folds at the diffusion limit. Nat. Struct. Biol 1998, 5, 363–365. [DOI] [PubMed] [Google Scholar]
- (44).Nagarajan S; Taskent-Sezgin H; Parul D; Carrico I; Raleigh DP; Dyer RB Differential Ordering of the Protein Backbone and Side Chains during Protein Folding Revealed by Site-Specific Recombinant Infrared Probes. J. Am. Chem. Soc 2011, 133, 20335–20340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Davis CM; Cooper AK; Dyer RB Fast Helix Formation in the B Domain of Protein A Revealed by Site-Specific Infrared Probes. Biochemistry 2015, 54, 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kubelka J; Henry ER; Cellmer T; Hofrichter J; Eaton WA Chemical, physical, and theoretical kinetics of an ultrafast folding protein. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18655–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Liu F; Du DG; Fuller AA; Davoren JE; Wipf P; Kelly JW; Gruebele M An experimental survey of the transition between two-state and downhill protein folding scenarios. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Torres J; Adams PD; Arkin IT Use of a new label C-13 = O-18 in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol 2000, 300, 677–685. [DOI] [PubMed] [Google Scholar]
- (49).Callender R; Dyer RB Probing protein dynamics using temperature jump relaxation spectroscopy. Curr. Opin. Struct. Biol 2002, 12, 628–633. [DOI] [PubMed] [Google Scholar]
- (50).Marecek J; Song B; Brewer S; Belyea J; Dyer RB; Raleigh DP A simple and economical method for the production of C-13, O-18-labeled Fmoc-amino acids with high levels of enrichment: Applications to isotope-edited IR studies of proteins. Org. Lett 2007, 9, 4935–4937. [DOI] [PubMed] [Google Scholar]
- (51).Bour P; Keiderling TA Vibrational spectral simulation for peptides of mixed secondary structure: Method comparisons with the TrpZip model hairpin. J. Phys. Chem. B 2005, 109, 23687–23697. [DOI] [PubMed] [Google Scholar]
- (52).Brewer SH; Tang YF; Vu DM; Gnanakaran S; Raleigh DP; Dyer RB Temperature Dependence of Water Interactions with the Amide Carbonyls of alpha-Helices. Biochemistry 2012, 51, 5293–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hu KN; Yau WM; Tycko R Detection of a Transient Intermediate in a Rapid Protein Folding Process by Solid-State Nuclear Magnetic Resonance. J. Am. Chem. Soc 2010, 132, 24–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kubelka GS; Kubelka J Site-Specific Thermodynamic Stability and Unfolding of a de Novo Designed Protein Structural Motif Mapped by C-13 Isotopically Edited IR Spectroscopy. J. Am. Chem. Soc 2014, 136, 6037–6048. [DOI] [PubMed] [Google Scholar]
- (55).Lai JK; Kubelka GS; Kubelka J Sequence, structure, and cooperativity in folding of elementary protein structural motifs. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9890–9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Lei HX; Chen CJ; Xiao Y; Duan Y The protein folding network indicates that the ultrafast folding mutant of villin headpiece subdomain has a deeper folding funnel. J. Chem. Phys 2011, 134, 205104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.