

Abstract
Computational chemistry provides a versatile toolbox for studying mechanistic details of catalytic reactions and holds promise to deliver practical strategies to enable the rational in silico catalyst design. The versatile reactivity and nontrivial electronic structure effects, common for systems based on 3d transition metals, introduce additional complexity that may represent a particular challenge to the standard computational strategies. In this review, we discuss the challenges and capabilities of modern electronic structure methods for studying the reaction mechanisms promoted by 3d transition metal molecular catalysts. Particular focus will be placed on the ways of addressing the multiconfigurational problem in electronic structure calculations and the role of expert bias in the practical utilization of the available methods. The development of density functionals designed to address transition metals is also discussed. Special emphasis is placed on the methods that account for solvation effects and the multicomponent nature of practical catalytic systems. This is followed by an overview of recent computational studies addressing the mechanistic complexity of catalytic processes by molecular catalysts based on 3d metals. Cases that involve noninnocent ligands, multicomponent reaction systems, metal–ligand and metal–metal cooperativity, as well as modeling complex catalytic systems such as metal–organic frameworks are presented. Conventionally, computational studies on catalytic mechanisms are heavily dependent on the chemical intuition and expert input of the researcher. Recent developments in advanced automated methods for reaction path analysis hold promise for eliminating such human-bias from computational catalysis studies. A brief overview of these approaches is presented in the final section of the review. The paper is closed with general concluding remarks.
1. Introduction
Catalysis plays a pivotal role in all strategies for the establishment of energy- and atom-efficient sustainable chemical technologies. Currently, society is critically dependent on cheap oil, gas, and coal feedstocks,1,2 which are used for the production of almost all consumer goods ranging from transportation fuels and fertilizers to fabrics and pharmaceuticals. Dependency on these nonrenewable feedstocks increases the danger of global political and economic instabilities and is associated with the emissions of vast amounts of greenhouse gases, causing widespread concern about climate change. This situation clearly creates an unsustainable future. Novel technologies must be conceived and materialized in the near future to enable the direct use of alternative renewable feedstocks and to make the existing technologies more efficient and greener. Endeavors toward the viable use of chemical feedstocks should be accompanied by efforts to make catalyst utilization also more sustainable.3 This implies the transition to more durable and active catalytic systems, preferably made of earth-abundant elements. Accordingly, the research and development of new catalytic systems based on 3d transition metals is a very active and dynamic research area across the catalysis discipline (see examples in recent special issues on the topic in highly respected chemistry and catalysis journals).4,5
The chemistry of 3d metals has always held a central role in the fields of organometallic chemistry and molecular catalysis, with representative examples of molecules such as ferrocene and Mn2(CO)10. The fundamental concepts of ligand electronic and steric effects in homogeneous catalysis have been first formulated and developed for nickel carbonyl complexes.6 Although one could imagine that the earth-abundant elements would fulfill all the needs of chemists, many chemical reactions are currently feasible only when critical or scarce elements are employed in catalytic systems. The relatively high covalency of the organometallic bonds and the preference for conventional two-electron chemistry of the platinum-group metals are key to achieving both the high activity and durability of the respective catalyst systems, which, to a large extent, originate from the balance between the reactivity toward reactants crucial to initiate the catalytic cycle and the relative weakness of the bonds formed with the metal site needed for the propagation and completion of the catalytic cycle.7,8 These fundamental characteristics are at the core of the high activities, selectivities, and stabilities (that is, catalyst lifetime) often achieved with the catalytic systems based on noble metals. The 3d metals are intrinsically more reactive and are known to undergo processes such as facile single-electron redox processes and ligand redistribution, which give rise to the increased complexity of the respective catalytic cycles and open additional reaction paths resulting in decreased selectivity and limited catalyst lifetime. It is instrumental to understand the molecular details of these diverse reaction channels underlying the catalyst behavior to create a basis for the targeted development of the next generation of earth-abundant metal catalysts. Such an interdisciplinary challenge requires a consolidation of advanced chemical theory and computation, synthetic chemistry, and kinetic studies.9,10
In the last two decades, computational chemistry has evolved into one of the key components of catalysis research and established a place for itself in the catalysis toolbox next to such common laboratory techniques as infrared spectroscopy (IR), nuclear magnetic resonance (NMR), and X-ray diffraction (XRD). Computational modeling and molecular simulations have substantially contributed to the progress in the fundamental understanding of catalytic phenomena.8−13 Computations have become indispensable in providing an atomistic framework for the interpretation of spectroscopic data and elucidation of catalytic mechanisms. State-of-the-art quantum chemical methodologies and, particularly, the density functional theory (DFT) methods are well-suited for studying chemical reactivity, analyzing complex reaction paths, and modeling kinetics of catalytic reactions. Theoretical studies provide important guidelines for the development of new and improved catalytic systems. The widespread application of computational chemistry is facilitated by the availability of convenient quantum chemistry and molecular modeling software that enables the practice of quantum chemistry in the absence of advanced programming skills and dedicated theoretical training. Computational chemistry is currently routinely employed not only by theoreticians, but by a wide range of experimental catalysis groups who often use results of atomistic DFT modeling to support mechanistic proposals derived from the experiments. The direct correlation between the results of molecular modeling and experimental data has become a common practice supported by the great success of near-chemical accuracy that can potentially be achieved with the modern computational approaches.14,15 Conventionally, the accuracy in computational chemistry refers to the performance of a particular methodology in computing specific fundamental chemical properties with respect to experimental or highly accurate theoretical results. In practical calculations on catalytic systems, the overall accuracy also strongly depends on the quality of the model, that is, how well it accounts for the important chemical details of the specific reaction or property in question. There is a natural trade-off between the method (level of theoretical approximation) and model accuracies (level of chemical details included in the model). The intrinsic chemistry of 3d transition metals and the associated catalytic systems pose crucial challenges to both the attainable method and model accuracies in practical electronic structure calculations.
It this review, we will present a comprehensive overview of the key challenges and opportunities in computational catalysis with molecular systems based on 3d transition metals by discussing recent examples. Here we explicitly focus the discussion on the challenges associated with computational studies on “defined” molecular catalysts, without considering conventional solid state catalysis (e.g., catalysis by surfaces or nanoparticles) and the emerging field of single-atom catalysis that have been extensively covered in the recent literature.8,12,16−21 Older literature has extensively discussed theoretical studies on transition metal catalysts.22−24 A more recent review article selectively covered the progress of computational studies on homogeneous organometallic catalysis involving Ni, Pd, Ir, and Rh.11
This review begins with a critical overview of the methodological aspects and challenges of electronic structure theory that has been already applied or has the potential to be applied in studies on molecular catalysts based on 3d transition metals (section 2). Section 3 covers the main challenges in the field related to the problem of model accuracy by reviewing recent computational works that highlight the role of multisite reactivity and active site cooperativity in catalysis by defined 3d transition metal complexes. The mechanistic complexity of heterogenized molecular catalysts is illustrated by presenting relevant studies on catalytic reactivity of metal–organic frameworks (MOF) in section 3.3. The mechanistic impact of the solvents and various promoting additives commonly present in practical catalytic systems is also discussed. After covering recent studies on the computational catalysis by 3d metals in section 3, a brief overview is given in section 4 on the emerging methodologies that hold a promise of evolving into versatile tools that could enable the elimination of the expert bias from the computational analysis of reaction mechanisms. Finally, the last section 5 provides concluding remarks summarizing the key challenges and opportunities for computational catalysis by 3d transition metals.
2. Competition between Method and Model Accuracies in Computational Catalysis
Electronic structure theory has a key role in the elucidation of complex reaction mechanisms as it is the only theory that provides means for the description of the essence of catalysis: the breaking and the formation of chemical bonds. This section focuses on discussing the issue of accuracy of computational chemistry as applied to studies on catalytic systems based on 3d transition metals. In this context, the problem of computational accuracy can be viewed as being composed of two main components that are (a) the accuracy of the methodology employed to determine the electronic structure of the chemical system and (b) the accuracy or quality of the chemical model that is used to account for the elementary phenomena underlying the particular property of interest of the chemical system. Since the focus of the current review is on catalysis by 3d transition metals, the discussion centers on both accuracy aspects for methodologies applicable to molecular entities containing transition metals. Given the complexity of such catalytic systems, both in terms of the chemistry and electronic structure, it is often not possible to simultaneously reach the desired, highest levels of both model and method accuracy. Therefore, numerous approximations and simplifications to the definition of the model system and representation of the electronic structure have to be made. Examples of such methodological approximations include the choice of the density functional, the level of correlation that is used in post-Hartree–Fock methods, or the size and type of the basis set. Importantly, the choice of the theory level determines the completeness of the set of fundamental physical phenomena that one reliably accounts for the actual computation. In practical computational catalysis research, this emphasizes the role of adequate calibration of these schemes and the estimation of the errors that are introduced to the computational study. Therefore, in this section, we will provide recommendations for obtaining (semi)quantitative results with electronic structure theory methods. The model approximations are mostly related to the need to reduce the size of the model in the broad sense (i.e., the number of atoms, size of the configurational or reaction space probed in the simulation, etc.) to make the system tractable and reach the desired computational accuracy. Here, we will discuss practical approaches allowing for simplification of the effects due to the extended structure of the catalyst or the presence of solvent that affects the reactivity and behavior of the catalyst.
2.1. Accuracy of Quantum Chemical Methods
2.1.1. Density Functional Theory
Density functional theory (DFT) will be discussed first due to its broad applicability in the field of computational catalysis. Even if, historically, DFT was applied in chemically relevant problems much later than the other important family of quantum chemical methods, the wave function theory (WFT) methods, it has gained significant popularity due to its straightforward nature and applicability. DFT has been extensively covered in excellent textbooks25−29 and review articles;30−35 therefore, here we will limit ourselves only to a very brief introduction to the basics of the method to form a foundation for the subsequent discussion of the power and limitations of the related methodologies for studying realistic catalytic systems.
The foundations of DFT are the two Hohenberg–Kohn theorems, which state that the ground-state energy is uniquely determined by the electron density and that the energy can be obtained variationally.36 The second theorem dictates that if the exact expression of the density was known then an energy greater or equal to the true energy can be computed. A method analogous to the solution of the Hartree–Fock (HF) equations via the self-consistent field (SCF) method was derived by Kohn and Sham that formed the basis of current Kohn–Sham DFT (KS-DFT).37
KS-DFT that is currently the basis for most practical DFT methods considers a system of noninteracting electrons, while the true (complete) ground-state density is formed by the electrons that do experience electron–electron correlation. The Kohn–Sham operator, which is the DFT equivalent of the Fock operator, is then expressed as a sum of one-electron operators, the eigenfunction is a standard Slater determinant formed by individual one-electron functions, and the eigenvalue is the sum of one-electron eigenvalues. The function summing the kinetic and potential energy operators is a density functional (i.e., a function of the electron density), with the density itself being a function of three-dimensional spatial coordinates. The overall energy functional is then given as
| 2.1 |
where Tni[ρ(r)] represents the noninteracting model kinetic energy, Vnuc–e[ρ(r)] the Coulombic attraction term between nuclei and electrons, and Vee[ρ(r)] the classical repulsion term between electrons. The final two terms on the right-hand side of eq 2.1 represent corrections to the kinetic energy (adding in the effects of electron–electron interactions) and the nonclassical electron–electron interactions, respectively. These correction terms are typically collected together into a term denoted Exc[ρ(r)], known as the exchange-correlation energy functional. This term has a cornerstone role in the accuracy of DFT methodologies and proves to be the only term in eq 2.1 which cannot be determined exactly. When discussing density functional theory methods, the differences between methods lie in the form of the exchange-correlation functional that provides Exc:
| 2.2 |
The energy functional is expressed as the interaction between the electron density ρ(r) and the term εxc[ρ(r)] (energy density), which represents the sum of individual exchange and correlation terms and is defined per particle.
Perdew et al.38 proposed a systematic organization of the various DFT approximations that have been used for the derivation of the density functionals (Figure 1). The uncorrelated Hartree product (i.e., merely a nonantisymmetrized product of spin orbitals) that includes neither Coulomb nor Fermi correlation is placed at the bottom of this scheme.39 The various approximations are the “rungs” of the “Jacob’s Ladder of DFT” that lead to the heaven of chemical accuracy (errors within 1 kcal mol–1 for energies). An important feature of the ladder, as it was initially proposed by Perdew et al., is that every rung should be based on the previous rungs, and each step should satisfy specific and exact constraints. Thus, each rung should, in theory, improve upon the performance of the previous, but there is no robust theoretical framework to certify that increase of accuracy (vide infra).
Figure 1.

Schematic representation of the various DFT approximations by increasing accuracy and computational cost and decreasing simplicity. Some representative and popular functionals are given for each rung.
The first rung consists of the local density approximation (LDA) which considers that εxc can be calculated exclusively from the density. LDA has been successfully applied in solids and materials, but since it is based on a spatially uniform electron density, it typically gives poor results for molecular systems. However, it is mentioned here as it forms the basis for the subsequent rungs of Figure 1.
The generalized gradient approximation (GGA) corrects the locality of the uniform electron density by adding a gradient correction to εxc of the LDA. Similarly, meta-GGA functionals also consider the second-derivative of the density, including the kinetic energy density. Both GGA (such as BP86,40 BLYP,40,41 and PBE42) and meta-GGA functionals (such as TPSS43 and M06-L44,45) have been extensively and successfully used in many chemical applications.
The next rung of the Jacob’s ladder contains hybrid density functionals that are usually based on the adiabatic connection method. Hybrid functionals are nonlocal functionals of the occupied orbitals where the energy functional expression contains exchange terms from HF theory:
| 2.3 |
Among others, B3LYP,41,46 PBE0,47 and M0644,45 have been extremely successful in the field of chemistry and are widely used by many computational, as well as experimental, chemists. Note that the dependence of the density gradients in a pure GGA is different from that in a hybrid or higher-rung functional, which may affect the generality of the “Jacob’s ladder”.
The fifth rung, the final step before reaching chemical heaven, uses both occupied and virtual orbitals by applying a correlation scheme explicitly, such as second-order perturbation theory within the random-phase approximation (RPA)48−51 or the Møller–Plesset (MP2)52−54 formalisms. These functionals are also known as “double hybrid functionals”.
Significant improvements to DFT have solved some important drawbacks of the theory, such as the introduction of a local variant of exact exchange (local hybrids)55−57 or the separation of the electron–electron interaction into long-range and short-range interactions.58−60 For example, it is known that local and semilocal functionals fail to correctly describe the dynamic electron correlation and therefore fail to accurately predict the attractive part of the energy that asymptotically decays as 1/R6, which has resulted in the poor performance of DFT for noncovalent interactions and, in particular, dispersion forces. Successful methodologies that have been applied include semilocal functionals optimized for the description of noncovalent interactions, effective one-electron potentials that reproduce noncovalently bound systems, density functionals with a nonlocal kernel, and the addition of a (semi)empirical pairwise corrections accounting for noncovalent interactions.53,61−64 In particular, Grimme’s D3 correction63 with the Becke-Johnson damping function65,66 [abbreviated as -D3(BJ)] has been used extensively in DFT molecular calculations.
Although the accuracy is increased in principle as we move to higher rungs, the complexity of the underlying equations leads to an increase in the computational effort. For the calculation of reaction mechanisms and potential energy surfaces that include the computation of many intermediates and transition states (TS) of complex reaction systems, one is often forced to rely on a GGA or meta-GGA functional rather than more computationally demanding hybrid or double-hybrid levels of theory. Therefore, this trade-off between accuracy and applicability might affect the outcome of the calculation, that is, the computed reaction barriers and energetics of the elementary steps. Despite the possibility to formally construct the hierarchy of exchange-correlation functionals as described above, many popular DFT methods are not constructed by strictly following this hierarchy and contain optimized parameters helping to improve the performance of the method in obtaining specific quantifiable chemically relevant values. This gives rise to one of the main limitations of DFT which is the dependence of the computed energies and properties on the choice of the density functional. It is well-known that the accuracy of some of the most popular functionals is based on fortuitous error cancelation. Recent studies suggest that some of the modern functionals deviate from the theory and principles of DFT.67,68 This results in the correct answer being obtained for the wrong reason or based on ill-defined physical properties. This viewpoint has been later debated in a series of follow-up papers, keeping the discussion on the accuracy of DFT methods open.69−71 In addition, a functional that performed well on one specific application might not be transferable to another application. The same might hold for a specific reaction or molecular system.
Calibration and benchmarking are highly recommended prior to any applied computational catalysis study based on DFT. Crucially, one has to be critical about the transferability of such benchmark studies. For example, a method capable to provide accurate description of organic molecules or closed-shell late transition metals bearing strong donor ligands may not provide reliable results when applied to a paramagnetic 3d transition metal complex. Additional parameters that should be considered in benchmarking are the basis set, solvation effects, and/or relativistic effects. The latter are usually not of high importance for 3d transition metals; exploratory calculations can verify this argument. Similarly, DFT is less sensitive to the choice of a basis set than WFT; typically, a triple-ζ basis set is generally adequate; however, calibrating the choice of the basis set is recommended.
An efficient scheme that does not introduce significant errors involves geometry optimizations with a computationally less expensive functional (e.g., GGA or meta-GGA) and/or small basis sets followed by a single-point calculation with a computationally more demanding functional (e.g., hybrid) and/or larger basis sets. Such approximative schemes aim on more affordable geometry optimizations and transition state searches without loss of accuracy, but the choice of the dual level of theory should also be the subject of benchmarking. Representative examples will be discussed in section 3, where the notation SP-functional/SP-BS//Opt-functional/Opt-BS is used (SP = single point calculation, Opt = geometry optimization, and BS = basis set). For cases where different basis sets are used for specific atoms, the notation functional/BS(1)&BS(2) is used.
Every quantum chemist has her/his own preferences on the choice of a functional. Usually, the functional choice is based on intuition, experience, previous literature, as well as on comparison with experimental and/or higher-level computational results, but in-depth, comprehensive benchmark studies can help DFT practitioners select the proper functional for a specific application. In a recent study, Mardirossian and Head-Gordon examined 200 density functionals on a molecular database of nearly 5000 data points which included noncovalent interactions, isomerization energies, thermochemistry, and barrier heights, although without including transition metals or other multiconfigurational systems (see section 2.1.2).72 That study named the newly developed ωB97M-V,73 a range-separated hybrid meta-GGA with nonlocal correlation function for dispersion correction,74 as the functional with the smallest errors. In another benchmark study by Yu et al.,75 84 functionals were tested on a large range of applications, such as transition-metal reaction barrier heights, electronic excitation energies, semiconductor band gaps, transition-metal dimer bond lengths, and noncovalent interactions. One of the conclusions of that study is that Minnesota functionals M06-L, M06, MN15-L, and MN15 have the smallest mean unsigned error for multireference systems. This is an important conclusion for catalytic applications since 3d transition metal complexes usually have a multireference character (vide infra). A recent benchmark study evaluated 30 density functionals for 60 diatomic M-L bond energies (M = 3d transition metals from Sc to Zn, L = H, F, Cl, Br, O, and S, all diatomics are neutral).76 The authors included zero-point vibrational energy and relativistic corrections and compared the DFT energies with experimental data. The study reported that specific functionals are more accurate for specific M-L pairs than others (e.g., metal hydrides require large HF exchange, while halides need 0–10% HF exchange). The authors concluded that, overall, the PW6B95 and the MN15 and MN15-L functionals, and the double hybrid B2PLYP53 functional are the most consistent in terms of accuracy.
For the sake of completeness, the DFT+U method, a popular method for modeling inorganic solids with periodic electronic structure calculations,77,78 should be mentioned. The DFT+U approach introduces a correction functional based on the Hubbard model for treating the self-interaction problem of standard DFT, but it is usually considered as a semiempirical method since an interaction parameter has to be introduced in order to control the strength of the Hubbard correction.79−81 Even though DFT+U has originally been initially developed and successfully applied for computing electronic structures of solids and surfaces, it has recently been introduced for calculations on molecular species as well.82−84
Independent of the choice of functional, there are a few additional components that should be taken into account after the successful completion of a DFT calculation on a 3d transition metal complex. These components can help the user evaluate the computed energy and properties and decide if further examination with different density functionals and/or computational methods is needed. Those can be summarized in the following: (1) Spin states: since the energy levels of the various spin states in a 3d transition metal might be in a range of a few kcal mol–1 or, in some cases, a few wavenumbers (cm–1), the full spectrum of different spin states should be computed and compared with either experimental data or a higher level of theory. (2) Expectation value of the ⟨Ŝ2⟩ operator: almost all quantum chemistry program packages print at the end of a successful DFT calculation the expectation value of the ⟨Ŝ2⟩ operator for the Kohn–Sham determinant which should be compared with the expected S(S+1) value. If the two values deviate significantly (typically more than 0.2) then the computed state is described as spin-contaminated and results should be used with caution. (3) Open shell vs closed shell configurations: in polynuclear complexes, an unrestricted singlet calculation might converge erroneously to a closed-shell configuration.85,86 For example, the ground state of the copper acetate (or copper paddlewheel) complex, which is composed by two Cu(II) metal centers with d9 electronic configuration, is an antiferromagnetic open-shell singlet ground state with an exchange coupling constant of −149 cm–1.87−89 On the contrary, an unrestricted HF or DFT calculation might converge to an erroneous configuration with either a strongly sigma-bonded bis-Cu(II) system or mixed valence Cu(I/III) (d10 and d8 electron occupation, respectively), where both solutions have energies significantly higher than the antiferromagnetic ground state. This discrepancy can be solved by performing a broken-symmetry calculation starting with the high-spin (triplet) molecular orbitals as input. The broken-symmetry solution will be spin-contaminated, which means that it will not be a pure singlet or triplet state, but a mixture of both. Standard spin-projection techniques can be applied for recovering pure-spin states, consequently, for the calculation of exchange coupling constants.90 Another approach that can be used as a remedy to this issue is to perform a stability analysis which is usually based on reducing the symmetry of the orbitals or allowing a restricted wave function to become unrestricted.91,92 Both approaches aim to relax the electronic energy so a more stable wave function might be obtained. (4) Frequency analysis: for molecular geometries optimized with DFT, a frequency analysis can reveal if the geometry optimization converged to a (local) minimum of the potential energy surface or to a saddle point (one or more imaginary vibrational frequencies). For the latter case, the user should modify the structure according to the imaginary vibrational mode and restart the optimization.
To summarize, the correct utilization of DFT methodologies requires a careful evaluation of the ability of the method to correctly grasp the required physical phenomena that define the chemical properties of the catalytic system under investigation. Because of the huge dependency of the outcomes of the computations on the choice of the selected exchange-correlation functional, we observe that the major part of the current mechanistic DFT studies in computational catalysis are from the very beginning biased by the assumption of the transferability of the method accuracy to unrelated systems. The simple recommendations summarized above do not allow one to completely eliminate this human bias in computational research but only help to additionally validate the methodology. The development of a physically accurate, universal, and fast computational method is by all means one of the holy grails in modern chemistry.
2.1.2. Multiconfigurational Wave Function Theory
Similar to HF methodologies, the conventional KS-DFT is a single-reference method. The wave function of the system is expressed as a single Slater determinant (SD) that captures the Fermi correlation. For an N-electron system (closed-shell) we can write
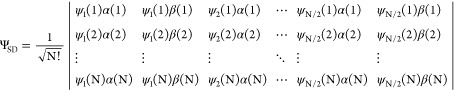 |
2.4 |
where N electrons occupy N/2 orbitals ψa(i) [or N spin orbitals ψa(i)σ(i), σ = α or β]. HF theory can qualitatively describe the electronic structure of closed-shell systems with a restricted wave function or high-spin open-shell molecules (unrestricted) at their equilibrium geometry. For quantitative results, the application of single-reference correlated methods such as post-HF methods (e.g., many-body perturbation theory, coupled-cluster theory) or KS-DFT (at least of GGA character) is needed.
The computational study of a catalytic reaction involves the correct description of the electronic structure of the transition metal catalyst, the dissociation and formation of chemical bonds, and in many cases, the description of electronic excited states. A single-reference method typically fails to capture all the effects of these cases and might yield erroneous results, for example, at the bond dissociation limit or for the full spin manifold of a polynuclear molecular catalyst. One solution is to use an unrestricted single-reference method (UHF or UKS-DFT), but the spin-contamination issue may yield wrong energies and properties. Such unrestricted methods are used extensively in applied computational catalysis, and in most of the cases quite successfully; however, their success is, to a large extent, the result of error cancelation. Additionally, they fail to capture the fine details of the electronic structure of complex molecular species. Because single-reference methods should be applied with caution when dealing with catalytic systems based on many 3d transition metals and since the aforementioned examples are central topics in the field of catalysis, the application of higher-level multireference methods is recommended as they pose elegant solutions to these issues.
The wave function of multireference or multiconfigurational (MC) methods is given as an expansion of SDs or configuration state functions (CSFs, symmetry adapted expansions of SDs), Ψn:
| 2.5 |
where cn are parameters that are determined variationally. The configuration interaction (CI) expansion of eq 2.5 forms the basis of CI and multiconfigurational self-consistent-field (MCSCF) theories.93 In CI theory, only the CI coefficients, cn, are variationally optimized. In the full CI (FCI) theory, a complete (full) wave function is used (i.e., all possible configurations are included in the CI expansion of eq 2.5). When FCI is used with a complete (infinite) basis set (CBS), it provides the exact Born–Oppenheimer nonrelativistic electronic energy in the absence of an external field. The FCI/CBS limit includes all electron correlation phenomena and does not differentiate between static (strong) and dynamic correlation, the usual categorization of correlation energy for practical purposes. However, the size of the CI expansion increases exponentially as the number of electrons and basis functions increase. Thus, FCI can be used only for small basis sets and for small di- and triatomic molecules with few electrons. For practical applications, the CI expansion is usually truncated based on the chemical considerations. This knowledge bias and the need of expert interference in the computational procedure represents one of the key challenges in the utilization of MC methods in applied computational chemistry and computational catalysis.
The truncated CI expansion should include all those configurations that arise from the degenerate or near-degenerate molecular orbitals (MO). This type of correlation energy that arises due to the MO degeneracy is usually termed as static, strong, or nondynamical correlation.
In MCSCF theory, the molecular orbital coefficients are also optimized in addition to the CI coefficients. The Hamiltonian is decoupled into two problems that can be solved separately, the CI step (microiterations) and the orbital optimization step (macroiterations).94 The most widely applied MCSCF method is the complete-active-space self-consistent-field (CASSCF) method. In CASSCF, the wave function is expressed as a FCI expansion within an orbital subspace that includes the most important valence orbitals and electrons (Figure 2a). A single-reference configuration obtained from HF, KS-DFT, or extended Hückel calculations usually provides the initial set of orbitals for CASSCF. The orbitals that do not participate in the CI expansion are either doubly occupied (inactive) or unoccupied (virtual or secondary). The orbitals included in the active space should be those that are responsible for the near-degeneracies. A usual notation that is followed by many computational chemists for the description of the active space is the CAS(n,m), where n is the number of electrons and m the number of orbitals included in the CI expansion. However, this notation does not explain which orbitals are actually included in the active space of the converged calculation, after minimizing the energy with respect to the rotation parameters (macroiterations or orbital optimization step).
Figure 2.
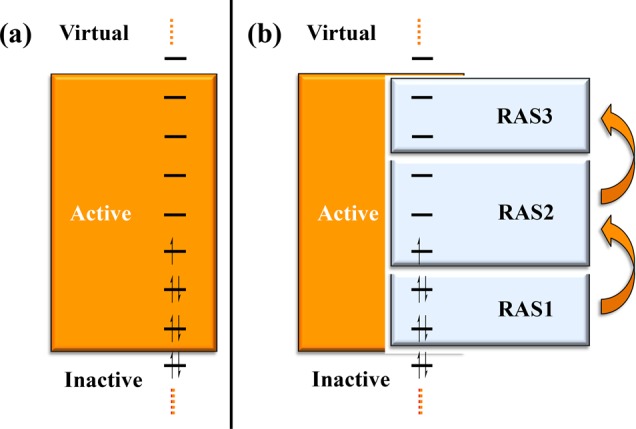
Schematic representation of the molecular orbital subspaces defined for (a) CASSCF and (b) RASSCF calculations. Inactive and virtual orbitals remain doubly occupied and unoccupied, respectively, and they do not participate in the formation of the multiconfigurational wave function. A CI expansion is formed from the orbitals in the active space. In the RAS scheme, the active space is divided into three subspaces (RAS1, RAS2, and RAS3) and a constrained CI expansion is formed.
The CASSCF method has proven to be a successful quantum chemical model as it can properly describe the electronic structure at nonequilibrium geometries, excited states, and mono- and polynuclear transition metal and actinide complexes.93 However, the method has three main limitations that constrain its applicability: (1) similarly to FCI theory, the size of the CI expansion increases exponentially with the increase of the electrons and orbitals included in the CAS space, (2) it lacks correlation energy from the inactive and virtual orbitals, and (3) it is not a black-box method since it requires the definition of the size of the CAS(n,m) as additional input. These three problems are discussed in the next paragraphs.
Perhaps the most crucial limitation of CASSCF is the size of the active space. Energy calculations with a CAS(16,16) (singlet spin states) can be routinely performed using standard computer clusters. Introducing symmetry considerations, which converts the Hamiltonian matrix into a block-diagonal format and significantly reduces the CI expansion (e.g., by a factor of 8 for a molecule with D2h symmetry), calculations with a CAS(18,18) are feasible. Recent developments in CI algorithms for high-performance computing raise this barrier to CAS(22,22) (singlet spin state).95,96 However, CASSCF calculations with such large active spaces need thousands of processors and an enormous amount of memory (a few TB), and therefore, they are not suitable for “routine” calculations. We emphasize that these values are given here for single-point energy calculations, while geometry optimizations, not to mention transition state search or frequency analysis, for any chemically relevant chemical systems are still prohibitively demanding.
The search for methods that can surpass the exponential scaling of the CI expansion is a field of active research. Solutions to this problem will allow the examination of challenging 3d transition metal catalysts discussed in this review that conventional CI methods cannot address, such as, for example, transition metal complexes with polynuclear cores. A straightforward approach is to impose restrictions on the CI expansion and exclude configurations that do not contribute to the state of interest (the so-called “deadwood”). A systematic approach that involves chemical insight (in other words, human intervention) for the exclusion of those configurations is the restricted active space (RAS) method (Figure 2b).97 In RAS, the full active space is divided into three subspaces: RAS1, RAS2, and RAS3. A full CI expansion is considered within RAS2, analogous to the CAS approach, complemented by a limited number of configurations that involves orbitals from the subspaces RAS1 and RAS3. RAS1 contains doubly occupied orbitals, and a restricted number of excitations (creation of holes) is allowed from this subspace to RAS2 and RAS3. RAS3 contains unoccupied orbitals and a restricted number of excitations to RAS3 (creation of particles) is allowed from RAS1 and RAS2. The generalized active space (GAS) scheme is a direct extension of RAS, where an arbitrary number of subspaces can be defined.98,99 A full CI expansion is considered for every GAS subspace, and similar restrictions on the number of inter-GAS excitations as in the RAS scheme can be imposed. Both RASSCF and GASSCF methods are variational and greatly reduce the size of the CI excitations but they require an a priori understanding of the molecular system under study for the definition of the subspaces and the number of interspace excitations.
Recent advances in alternative CI methods have pushed the boundaries of MCSCF methods beyond the limitations of standard methods. These approaches include tensor networks, such as the density matrix renormalization group (DMRG),100−102 stochastic methodologies for sampling the CI space, like the FCI Quantum Monte Carlo (FCIQMC) method,103 or the revival of selective CI.104−107 Extensions of the CI solver with an orbital-optimization step has resulted in CASSCF-type methodologies (e.g., DMRG-CASSCF,108 FCIQMC-CASSCF109), which are expected to surpass the traditional MCSCF methods. Applications of these methods on catalysis are discussed in the next sections.
The second limitation of CASSCF is the lack of dynamical correlation, which arises from instantaneous correlation of the electron motion due to their mutual repulsion. This type of correlation energy can be recovered by introducing a set of determinants formed by excitations from the zeroth-order wave function to the virtual space. As an extension for CASSCF, the multiconfigurational expansion of eq 2.5 is used as zeroth-order wave function. The missing dynamical correlation is usually added by perturbation theory,110 coupled-cluster theory,111 or DFT.112−115 From those approaches, the most widely applied are two variants of multireference second-order perturbation theories (MRPT2), the complete-active space second-order perturbation theory (CASPT2)116 and the N-electron valence state second-order perturbation theory (NEVPT2),117 which are used routinely on many studies on catalytic applications. New implementations of the domain-based local pair natural orbital NEVPT2 (DLPNO-NEVPT2) are expected to provide both accuracy and low computational requirements for computations on 3d transition metal catalysts.118
The third limitation of CASSCF, crucial from a methodological perspective, is the determination of the number of electrons and orbitals that will be included in the active space. Chemical intuition and knowledge of the molecular system or the electronic state of interest is needed prior to the performance of a multiconfigurational calculation, a feature that makes the method less attractive for applied quantum chemistry and mechanistic studies in catalysis. This bottleneck becomes more evident for cases where little is known for their electronic structure (e.g., transition metal complexes with noninnocent ligands, excited states). Many approaches have been suggested on the selection of orbitals that should be included in the active space,119 some of which are based on automated procedures.120−123
As a general rule for first-row transition metal complexes, the full 3d subshell manifold should be included in the active space since the molecular orbitals arising from the 3d atomic orbitals are degenerate or near-degenerate. Radial correlation may also be included by addition of the second d shell (4d), an approach that usually helps to obtain the desired orbitals inside the active space. Ligand field orbitals should also be part of the active space since they affect the relative stability of the 3d orbitals of the metal(s). For catalytic applications, additional orbitals should be considered for the CI expansion of MCSCF, such as orbitals involved in bond formation and bond breaking promoted by the transition metal. For polynuclear complexes, where there is no direct metal–metal interaction, superexchange should be considered, and the full spin manifold spanned by the coupling of two or more open-shell metals should be calculated. Those spin states may affect the energies and electronic properties of the catalytically active metal.
2.1.3. Single-Reference Wave Function Theory for Catalytic Applications
Coupled-cluster singles-and-doubles with perturbative triples (CCSD(T))124 has been denoted as the gold standard of quantum chemistry for almost three decades because it can provide highly accurate results for many molecular applications. However, because of the reasons explained above, single-reference post-HF methods (including such highly accurate CCSD(T) approaches) should be used with caution for transition metal chemistry and for studying bond breaking and bond formation. It is desirable to have a highly correlated scheme such as coupled-cluster theory since it provides systematically improved accuracy by increasing the size of the basis set or the correlation level. This feature holds also for multiconfigurational methods, while there is no straightforward systematic increase in accuracy for DFT.
One of the problems of (restricted or unrestricted) CCSD(T) is its nonvariational character, since it includes triple excitations computed with perturbation theory. This leads to non-negligible deviations from the FCI limit when it is applied on bond breaking processes.125 The T1(126) and D1 diagnostics127 calculated from the coupled-cluster amplitudes can be used as an estimate of the multireference nature of the system under study and the importance of the missing strong correlation. However, the well-established criteria T1 < 0.02 and D1 < 0.05 hold only for organic molecules and should be used with caution when they are applied on transition metal complexes.128 Alternative diagnostics based on simpler but more affordable concepts, namely the B1 diagnostic introduced by Truhlar and co-workers,129,130 provide similar results as the more theoretically robust T1/D1 diagnostics.
Another issue of conventional CCSD(T) is its dependence on the basis set size, which consequently leads to large memory and disk space requirements. This bottleneck has been successfully surpassed by explicitly correlated methods,131,132 exploration of the local nature of correlation,133,134 or combination of both. A few applications of those methods on first-row transition metals have been reported,135−141 but their use should be considered carefully when dealing with systems with strong multiconfigurational character. The completely renormalized coupled-cluster scheme of Piecuch and co-workers introduces strong correlation inside the coupled-cluster framework and has been applied successfully on applications involving transition metals.142,143
High-performance computing95 and linear-scaling methods144 offer alternative methods for speeding-up quantum chemical calculations for large molecules. Coupling of those methodologies with strongly correlated methods will allow carrying out highly accurate theoretical studies on molecular catalysts.118
2.1.4. Fragmentation and Embedding Schemes
The practical application of advanced and most accurate electronic structure methods is still limited to quite small molecular systems containing at most a few dozens of atoms. When translated to a practical catalytic system, such an ensemble corresponds to only a fraction of a catalyst system that may not capture some crucial chemical features, resulting in a very low model accuracy. Fragmentation schemes can extend the applicability of the standard quantum chemistry methods to larger molecular systems by dividing the system (molecule, cluster, or periodic structure) into smaller fragments. The computed results of each fragment are collected to evaluate the energy of the complete system. The challenge that each fragmentation scheme has to address is how to provide an exact definition of the nonadditive terms (i.e., the energies and properties that arise between the various fragments or subsystems). Recent reviews on fragmentation schemes have extensively covered the progress in this field.145−149 An important drawback of most fragmentation methods is that the orbitals and, thus, the wave function of the parent system cannot be fully recovered. Consequently, this hinders their applicability to cases where understanding of the electronic structure of a molecule (e.g., transition metal complexes) is mandatory. Therefore, applications of such approaches to transition metal catalysts are scarce. A few promising examples that include transition metals are the analysis of the excited states of an extended Zn-porphyrin chain,150 and the incremental scheme of Friedrich and co-workers (originally proposed by Stoll151) which has been applied to transition metal complexes, such as the TiCp2Cl2152 and the Zn4-cluster, an important building unit of many metal–organic frameworks (e.g., MOF-5).153
Methodologies based on subsystems treated at different levels of theory, such as hybrid QM/MM (quantum mechanics/molecular mechanics)154−158 and ONIOM (“our own n-layered integrated molecular orbital and molecular mechanics”)159 approaches are much more popular in computational catalysis and have been successfully applied in numerous studies on 3d transition metal catalysts. In both methods, the full system is divided in two subsystems, the “model system”, which is treated with an accurate and more expensive method (WFT or DFT), and the environment, which is treated with a lower level of theory, usually DFT or molecular mechanics (MM). In QM/MM, the total energy is calculated as the sum of three energy terms: model or primary subsystem (EQM), environment (EMM), and the interactions (EQM–MM) between the QM model system and the MM environment system. ONIOM follows a different approach for the calculation of the total energy. It considers the sum between the energy of the model with higher level of theory (Ehigh,model) and the energy of the full (real) system at the lower level (Elow,real) and subtracts the energy of the model at the lower level (Elow,model). Both QM/MM and ONIOM can apply DFT and/or WFT (single-reference or multireference) as higher level of theory to describe the active site of a catalytic system, while they treat its environment with a lower level of theory. For that purpose, they have been successfully applied in many studies on computational catalysis which have been covered in recent review articles.159,160
The computational challenge that these methods confront is the correct coupling between the two subsystems (primary and environment). Electrostatically embedded fragment methods are considered more accurate than methods based on mechanical embedding as they do not depend on electrostatic parameters for the coupling of the two subsystems, and they are capable of adjusting the electronic structure of the high-level subsystem to the charge distribution of the MM subsystem.157 A more accurate embedding scheme developed by Wang and Truhlar uses screened electrostatics rather than point charges.161 Subsystem embedding methods can be considered as an upgraded version of fragmentation methods that provide an exact property (e.g., density) of the full system.162−167 The theoretical framework is known as frozen-density embedding (FDE) theory and was developed by Wesołowski and Warshel.168 In subsystem DFT, the total density is represented as a sum of subsystem densities. The total density is calculated self-consistently by minimizing the total energy with respect to the subsystem densities, which simplifies the electronic structure problem into subsystem problems.163 Similar approaches have been developed for extending subsystem Green’s functions and subsystem density matrices (Green’s function embedding and density matrix embedding, respectively).165 Evaluation of such new methodologies on transition metal complexes are scarce, but since this is a field of active research, applications on catalysis are expected in the near future. For example, Fux et al. showed that subsystem DFT can reasonably describe donor–acceptor systems like ammonia-borane but fails for the π-backbonding of transition metal complexes.169
Introducing a WFT method for the (model) subsystem can circumvent that problem and provide an accurate description of local catalytic sites with correlated methods (WFT-in-DFT). The theoretical framework of a WFT method used in an embedding scheme was initially developed by Wesołowski170 and was later extended to molecules and materials by applying an embedding potential calculated by approximate functionals.164,171−177
An extension to those models is the self-consistent reconstruction of the embedding potential from the partition of the density.178,179 This step is no longer the most time-consuming step since the WFT subsystem calculation becomes the bottleneck of the WFT-in-DFT scheme. Miller and co-workers introduced the first fully self-consistent WFT-in-DFT scheme that uses an accurate embedding potential.180,181 This scheme can be coupled with CASSCF and has the advantage that it can treat covalent bonds between subsystem and environment. Goodpaster et al. have studied cobalt diimine-dioxime catalysts for hydrogen evolution with this WFT-in-DFT embedding scheme.182 The transition metal, its first coordination sphere, and atoms that are bridging the other atoms of the first coordination sphere of Co were treated at the CCSD(T) level, while the rest of the complex with DFT [CCSD(T)-in-DFT scheme, Figure 3 (panels B and C)]. The potential energy curve along the proton transfer coordinate obtained by DFT significantly deviates from that calculated with CCSD(T). The CCSD(T)-in-DFT embedding scheme can reproduce the pure CCSD(T) curve with significantly lower computational effort.
Figure 3.
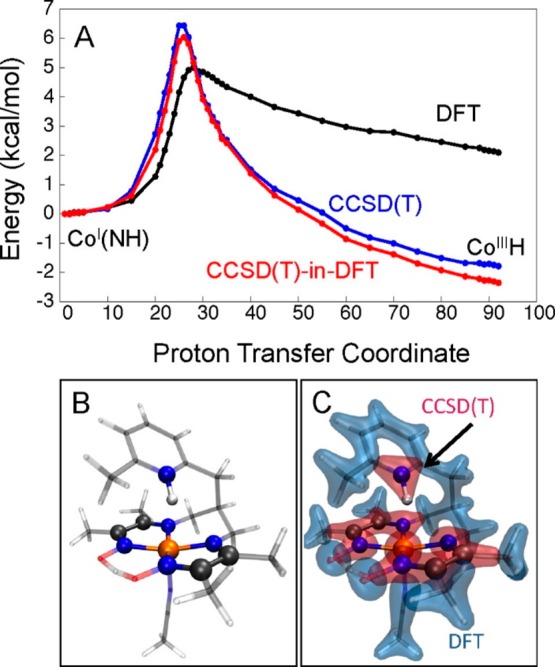
(A) Energy profiles for the intramolecular proton-transfer reaction in the co-diimine-dioxime catalyst with methyl substitution, obtained using CCSD(T) (blue), DFT (black), and CCSD(T)-in-DFT (red). (B) Partition of the catalyst into two subsystems treated at different level of theory. (C) Associated partitioning of the electronic density into subsystems that are treated using CCSD(T) (red) and DFT regions (blue). Reproduced from ref (182). Copyright 2016 American Chemical Society.
Similarly, Coughtrie et al.183 reported a multilayer scheme where internally contracted multireference coupled-cluster (icMRCC)184−186 within a CASSCF wave function was embedded in local PNO-CASPT2.187 This WFT-in-WFT scheme uses a small active space for the icMRCC, and subtractive multilayer ONIOM-type embedding is applied between the different layers. For demonstrating the power of this scheme, calculations on a nickel complex with 231 atoms and 4175 basis functions were performed. Despite the difficulties that arise from the highly demanding icMRCC, the approach of Coughtrie et al. holds promise for very accurate computations involving transition metals. As it becomes evident, more applications on large molecular catalysts are expected from the WFT-in-DFT and WFT-in-WFT schemes in the future, since multiconfigurational methods can now be applied at the active site subsystem.
In this subsection, we mainly focused on the method accuracy aspects of the utilization of the fragmentation and embedding schemes. We should mention that they also pose substantial limitations on the attainable model accuracy. In these methods, we assume that the key chemical phenomena requiring the highest-level method accuracy are mostly localized and can be represented by the small ensemble of “important” atoms, while secondary effects can be safely accounted for by less accurate methods. Such an assumption effectively leaves out from the computational analysis the reaction paths featuring multisite reactivity.
2.1.5. Excited States
Excited states of transition metal complexes are crucial for photocatalytic applications of transition metals where the radiation with light brings the chemical system onto the excited PES, enabling thus the catalytic cycle either via the generation of free radicals or by formation of specific, more reactive configurations.188 Besides, excited states might play a role in reactivity by providing additional reactive channels, such as the σ and π channels of the nonheme iron(IV)-oxo intermediate (verified by both spectroscopy and computations).189−192 From a WFT standpoint, multireference methods (CASPT2, NEVPT2, or MRCI) can qualitatively and quantitatively describe excited states of transition metals and can intrinsically treat conical intersections between electronic states. In addition to the methods discussed in the previous sections, state-averaged (SA) CASSCF193 and multistate (MS) CASPT2194 calculations can simultaneously optimize many roots (states) and elucidate excited states and accessible reaction pathways. An alternative efficient scheme for excited states of transition metal complexes is the DFT/MRCI method, which uses Kohn–Sham orbitals to construct CSFs and to modify an MRCI-type Hamiltonian.195,196 The main drawback of those methods remains their dependence to the definition and size of the active space size that limits their applicability.
Despite this limitation, such methods can accurately treat the ground and excited states of mono- and dinuclear complexes, but approximations should be introduced for polynuclear molecular complexes or extended chromophores. Linear response single-reference methods can circumvent some of those problems. Equation-of-motion coupled-cluster197 or second- and third-order wave function methods, such as the CC2/CC3,198 the algebraic diagrammatic construction [ADC(2) and ADC(3), respectively],199,200 and the second-order polarization propagator approximation (SOPPA)201,202 are used nowadays both for the calculation of excited states with high accuracy and as a reference in many benchmarking studies, but their applicability on transition metal complexes is limited.
An extension of the Hohenberg–Kohn theorems to time-dependent theory can give information to excited state properties via linear response. For chemical applications, the adiabatic approximation defines conventional time-dependent DFT (TD-DFT), which has been successfully applied on electronic spectroscopy and photochemistry of organic molecules and chromophores, among others.203,204 Main sources of error are attributed to erroneous long-range behavior of most density functionals, important for charge-transfer excitations, the lack of double and higher excitations, and limited accuracy for high spin states.205 The accuracy of TD-DFT is significantly increased when it is combined with functionals with 100% HF exchange or range-separated functionals. TD-DFT has been also applied successfully on the computation of electronic spectra of transition metal complexes.206−208 Similar to conventional DFT, there is a large dependence of the results of TD-DFT computations on the actual choice of the exchange-correlation functional,209 which may result in erroneous agreements between spectroscopic data and computational results on simplified models.210
Green’s functions with the Bethe-Salpeter equation can be considered an alternative methodology that can reach TD-DFT’s popularity in the near future.211 Two recent benchmark studies of GW and Bethe-Salpeter equation on the excited states of small molecules with transition metals show some promise for the application of those methods.212
2.1.6. Ab initio Molecular Dynamics
All methods discussed so far are suitable to examine the properties of isolated points on the potential energy surface (PES). Conventional quantum chemical calculations (WFT or DFT) are commonly used in combination with geometry optimization methods to locate local minima (reactants, reaction intermediates, or products) and saddle-points (transition states) at the 0K PES and compute their energies and all related properties derived from the solution of the electronic structure problem. This information can be related to the intrinsic reactivity of a catalytic system. In practice, however, the reactivity and catalytic performance is defined not only by the PES but also by the conditions at which the reactions are carried out, environmental effects associated with the medium where the reactions take place, and other “secondary” effects which are manifested by the condition-dependent free energies.
The dynamic evolution of the properties of solids, liquids, and interfaces at finite temperatures relevant to catalytic phenomena can be obtained by coupling the electronic structure methods with classical mechanics approaches, giving rise to the so-called Car–Parrinello molecular dynamics (CPMD) and ab initio molecular dynamics (AIMD) methods. In AIMD, trajectories from finite temperature molecular dynamics are generated from forces obtained from accurate, on-the-fly quantum chemical calculations (e.g., DFT).213−216 AIMD simulations can tackle complex problems that cannot be addressed by conventional force-fields or standard quantum chemical calculations. Key aspects of a catalytic reaction can be examined from AIMD simulations, such as the role of the solvent medium and fast dynamical motion of light atoms.217
AIMD simulations on enzymatic reactions involving proton transfer promoted by transition metal-based active sites have been reported,218,219 such as the dismutation of superoxide radical anions,220 the catalase reaction (peroxide bond cleavage) from catalase/hydrogen peroxidases,221−225 hydrogen production from [FeFe]-hydrogenase,226 antibiotic hydrolysis by metallo-β-lactamase enzymes,227,228 and Cu-mediated amyloid formation.229,230 Similar reactions involving proton transfer on biomimetic catalytic complexes and materials have been reported, such as hydrogenase model complexes,231 hydrogen production from water promoted by a hydrogenase-inspired catalyst [FeFe]/electrode complex [FeS2],232 or cobaloxime catalysts.233
From the computational perspective, the AIMD and CPMD approaches provide the possibility to directly probe entropic contributions to catalytic reactivity and acquire the conformational freedom of the reactive complexes that is necessary for accurate free energy calculations. Besides, the explicit consideration of solvent and the extended molecular structure of the catalyst system commonly employed with these methods substantially improve the model accuracy of the calculation and create an opportunity to study reaction channels that could not be otherwise probed with static approaches.23,234,235 However, these methods are highly demanding since they require long simulation times and sampling of many trajectories and can only be used to accurately evaluate selected reaction steps of the catalytic networks. Additional limitations to the applicability and the predictive power of the AIMD and CPMD methodologies arise from the classical description of the atomic trajectories and the adiabatic approximation to the potential energy surface (PES). These assumptions may have a detrimental effect on systems where quantum effects associated with the dynamics of light atoms (e.g., hydrogens) are important or where the reactivity is contributed by the coupling of multiple Born–Oppenheimer PES.236−239
2.1.7. Zero-Point Vibrational Energy and Thermal Corrections
The inclusion of finite temperature effects and estimation of entropic contributions in the analysis of extended reaction paths commonly encountered in catalytic processes requires a faster and implicit way to correct the electronic energies for such effects. To compare with the experimental values, the 0 K energies have to be first corrected for the zero-point vibrational energy (ZPVE). ZPVE is introduced by computing vibrational normal modes within the harmonic approximation, which consist of a valid approximation at the equilibrium distances of intermediates and transition states. Moreover, the finite temperature and entropic corrections can directly be computed using statistical mechanics. These thermal corrections allow the calculation of the molecular partition function and the estimation of enthalpies (ΔH) and standard free energies (ΔG). For the calculation of force constants and, subsequently, vibrational frequencies, the determination of the first (gradient) and second (Hessian) derivatives of the energy are needed. This step involves the transformation of nuclear coordinates to mass-weighted coordinates, which is valid on tightly converged geometries, when applied to structures computed at the same level of theory as the geometric optimization. Calculation of force constants should also be used as an additional validation of optimized geometries since an imaginary frequency means that the converged geometry is a saddle-point of the full potential energy surface. Note that despite the fact that such thermochemical corrections are currently routinely computed following the normal-mode analysis by most quantum chemical packages, they are formally valid only for gas-phase systems. Their direct application to estimating reaction and activation free energies for processes even in highly dilute solutions, not to mention the concentrated multicomponent reactive systems commonly encountered in practical catalysis, should be done with certain care.136,240,241
2.1.8. Implicit Solvation
For homogeneous or heterogeneous catalysts operating in liquid phase or in solution, the effect of the solvent molecules should be included in the modeling of the reaction since it can directly or indirectly affect the evolution of a catalytic reaction. Therefore, for the accurate modeling of the catalyst and the quantitative calculation of reaction intermediates and reaction barriers, the interactions between solvent molecules and catalyst, reactants, and intermediates should be taken into consideration.
Two different approaches that couple the quantum-chemical description of the catalyst with the solvent environment can be applied. The first is explicit solvation, where an extended atomistic model of the catalyst together with its environment of solvent molecules are considered explicitly. Practically, one explicitly adds a necessary number of solvent molecules around the active site of the catalyst to achieve a representative solvation shell or to ensure the availability of specific secondary chemical interactions that can potentially facilitate the investigated reaction channels (e.g., proton shuttle). The catalytic complex and the solvation shell are then treated either at the same level of theory or by using embedding schemes with the higher level applied to the reactive site and the lower level used for the solvent molecules (e.g., QM/MM, ONIOM). The explicit solvation of the reactive ensemble increases the model accuracy at the expense of the increased model size and higher complexity of the reaction channels to be explored. The implicit solvation models approximate solvent effects by a continuum mean-field potential that addresses the interactions between solvent and solvated system.242−244 This implicit solvation reduces the computational effort not only by reducing the size of the model, but also more importantly, by reducing the degrees of freedom of the problem by substituting the solvent molecules with the solution of a classical electrostatic problem (Poisson problem). Implicit solvation models such as the conductor-like screening model (COSMO),245 the polarizable continuum model (PCM),246 and the parameter-dependent PCM-type solvation models (SMx)247 have become standard tools for quantum chemists and they have been successfully applied in numerous computational studies on catalysis. Conventional computational schemes utilizing these models allow computing free energies in standard diluted solution.
The extension of the computational analysis to complex reaction paths in the liquid phase under more realistic conditions could be achieved with the concept of free energy surfaces (FES). Recently, the role of FES in condensed-phase chemistry as the analogue of potential energy surface used to describe the reactions in the gas-phase has been discussed by Truhlar and co-workers.248,249 For dilute solutions, the existence and type of stationary points on FES (i.e., the intermediates and transition states) do not depend directly on concentrations in solutions. However, the key reactivity parameters (i.e., the free energy differences between the reagents), intermediates, and transition states depend directly on concentrations. Depending on the system, the free energy correction to account for the realistic composition of the reaction medium can reach several kcal mol–1, a value comparable to the characteristic energy barriers for elementary steps in related catalyst systems. A conceptually similar approach, largely inspired by ab initio thermodynamics methods widely employed to predict active site composition and evolution in heterogeneous catalysis,250−252 has been utilized by Filonenko et al.253 to optimize the performance of a Ru-CNC pincer catalyst in CO2 hydrogenation through the analysis of DFT-computed free energy profiles for competing catalytic and deactivation reaction channels. Liu et al.254 have recently extended this approach to analysis of realistic solvents by combining the concept of concentration-dependent free energy surfaces with the realistic description of the solvents with the COSMO-RS method.255 This approach has been successfully employed to reveal the role of base promoters in ester hydrogenation by Mn–P,N complexes (see section 3.1 for more details).
2.2. Quality, Accuracy, and Comprehensiveness of the Chemical Models
Another key aspect of computational catalysis involves the choice of the molecular model. For a complete computational study on a reaction mechanism, multiple reaction pathways should be examined. Competing pathways should be evaluated and will be accepted or rejected based on thermodynamic and kinetic criteria. Each pathway might include numerous intermediates and transition states, and the 3d transition metal used at the active site can also have a high-, intermediate-, or low-spin. In many cases, the reaction might be promoted by two or more transition metals, which raises the number of possible spin states of the molecular model, from the highest-spin possible until low-spin states (closed-shell or open-shell). Some of those spin states might be difficult to converge or need special treatment, such as cases with a superexchange mechanism, where there is a facilitated nonlocal-exchange interaction between ligand-bridged metals. For a thorough study, all possible spin channels should be computed for all possible intermediates and transition states, for multiple reaction pathways. This is important since spin-crossover or two-state reactivity schemes might accelerate the reaction.256 To effectively study all possible reaction schemes, minimization of the computational effort that is needed for each molecular complex included in the study is desirable. In this section, computational approaches that can speed-up reactivity studies without significant loss of accuracy will be presented.
2.2.1. Simplification of the Catalyst System
Compared to heterogeneous catalysis where the nature and state of the catalytic sites is often unknown, the construction of a model for molecular homogeneous catalysts is more straightforward. The direct structural information on the catalyst precursor from single crystal X-ray diffraction data can be readily used as a starting point for constructing models of potential active complexes and reaction intermediates. To speed up computations, in practical mechanistic analysis it is quite common to simplify these molecular structures by replacing bulkier side groups far from the active center with some smaller counterparts of similar chemical nature. In general, atoms located at the first or second coordination sphere of the transition metal should not be removed, since they affect the electronic structure of the metal. Similarly, functional groups that are close to the active site of the catalyst should remain intact because their steric effects will affect the various reaction intermediates and transition states. This approach reduces the total number of atoms but should be verified by calculations with the full and the truncated ligand. Properties such as charges, energies between different spin states, and reaction barriers can be applied for the calibration of the reduced model. For multiconfigurational calculations in particular, full and truncated models should both provide the same CI vector and the same fractional occupation numbers for the active orbitals.
The choice of molecular models becomes more challenging for heterogeneous environments, such as metal–organic frameworks (MOFs) with catalytically active undercoordinated framework or extraframework transition metals or zeolites modified with transition metals. When molecular models are constructed from periodic structures, additional considerations should be taken into account; no artifacts should be introduced from the reductionism of the model.257 The first is to maintain the electronic structure of the active site intact. The truncated model should not affect the partial charges or the spin density of the transition metal complexes or the other atoms that participate in the catalytic reaction (e.g., oxo/oxyl, hydroxo, etc.). The second is to retain a neutral overall charge. A charged model implies that the full material will have an infinite positive or negative total charge. The third is to maintain the confinement effects of the micro- or/and mesoporous nature of the substrate. Such long-range interactions play a crucial role for the catalytic performance since they usually affect the reaction barriers of the key reaction steps.258,259
These reductionisms are valid for isolated cluster models of zeolites or MOFs. In cases where the catalytically active metal is not purely isolated from other transitional metals (catalytically active or not), the electronic structure of the site under study might be affected by a superexchange mechanism. For example, consider the Fe-MOF-74, a two-dimensional material that has as a backbone of its framework infinite-size iron-oxo chains.260 For ferromagnetically coupled transition metals, a valid reduction of the complexity of the model is the substitution of the neighboring metals with closed-shell atoms of the same size and charge (e.g., Mg2+ or Zn2+ for M2+ cations, or Al3+ for M3+). This approach should be applied with higher caution for polynuclear models with antiferromagnetic couplings. In both cases, additional calibration of the model is needed, and an estimated error of the approximate model should be reported. The same considerations hold as well for the modeling of homogeneous polynuclear molecular catalysts.
Even when the direct structural information is available regarding the catalytic sites, the formulation of molecular models adequately representing the catalytic systems may be a challenge. The assumption of single-site reactivity generally dominates classical catalysis theories and creates a foundation for the simplification of catalyst models by reducing complexity of the structural motifs distant from the main site. These fundamentally constrain the mechanistic paths considered in the computational study and do not allow investigating paths enabled through cooperative and multisite reactivity effects. This is a common problem persistent even for homogeneously catalyzed reactions with well-characterized catalyst precursors. The mechanistic complexity and the associated aspects of model accuracy in homogeneous catalysis by 3d transition metal complexes will be discussed in detail in section 3 with the show-case examples of hydrogenation (section 3.1) and cross-coupling catalysis (section 3.4). The concepts of multisite (cooperative) catalysis is the focus of sections 3.1 and 3.2 discussing, respectively, the mechanistic roles of metal–ligand and metal–metal cooperation (dual metal catalysis). Special attention in those sections will be devoted to the comparison of the mechanistic concepts developed for the noble and 3d transition metal catalysts as well as to the discussion of their mutual transferability. Recent examples of computational studies on molecular heterogeneous catalysis based on 3d transition metals in porous materials are presented in section 3.3, with particular emphasis on the choice of accurate and reliable molecular models.
2.2.2. Conformations
A fast, accurate, and reliable screening of the chemical space can speed-up the completion of reactivity studies and accelerate the computational prediction of novel reactions and functional catalysts. Such high-throughput computational screening methodologies can be extended to three different directions: (1) screening of reactants for the discovery of new reactions, (2) scanning for reaction conformers, and (3) screening of molecular catalysts. Reaction network analysis which covers the first and second directions is briefly reviewed in section 4. The third direction, which targets the catalyst optimization, is discussed here.
High-throughput computational screening, graph theory, and machine-learning approaches have significantly contributed to drug261−266 and material discovery.267−270 The focus of cheminformatics and conformational searches of molecules has traditionally been on organic species for drug discovery and small molecules composed of hydrogen and second-row atoms. As it becomes evident, the automated generation of databases of molecules containing heavier atoms, such as transition metal, lanthanide, or actinide complexes, for an in silico screening of molecules is an emerging field with great potential for many applications. One of the bottlenecks of such approaches is the reliability of the molecular structures. Quantum chemical calculations at the DFT level can be very time-consuming when geometry optimizations of thousands of transition metal complexes are needed. On the other hand, molecular mechanics cannot provide the desired accuracy due to lack of reliable force fields. For rapid generation of molecular conformers, empirical models based on distance geometry271−274 or stochastic algorithms have been proposed.275−278 Those methods have been incorporated in many commercial or free software.279 However, structures generated by these methods are still subject to further geometry optimization with more robust approaches, and their applicability on transition metal complexes is limited.
An automated fragment-based evolutionary algorithm for the generation of molecular libraries that takes into account the synthesizability of organometallic complexes was developed by Jensen and co-workers.280−282 The authors have generated a large database of potential Ru-carbene complexes (Grubbs’ second-generation catalysts) for olefin metathesis by gathering molecular fragments from already synthesized and stable molecules. On the basis of group contribution considerations, they developed compatibility matrices and were able to restrict the generation of unrealistic functional groups and molecules.281 This approach is based on simple rules and has the potential for further development with more robust algorithms that can be easily generalized.
Kulik and co-workers have developed a powerful open-source computational toolkit (molSimplify). The methodology is designed to generate structures and compute molecular descriptors for transition metal complexes. The program follows the “divide and conquer” strategy that implies a separate description of the organic ligand and the metal site. molSimplify utilizes the artificial neural network method to automatically predict the geometries of transition metal complexes and predict their electronic structure-related properties based on the steric and electronic descriptors implemented in the code.283 This computational tool may be efficiently employed to aid in the design of new inorganic materials and transition metal-based catalysts.283,284
Doney et al. developed an automated alkylation reaction optimizer for N-oxides (AARON) for predicting enantioselectivities for bidentate Lewis base catalyzed alkylation reactions.285,286 This algorithm requires optimized geometries of intermediates and transition states from DFT and was able to screen a database of 59 potential catalysts for asymmetric propargylation of benzaldehyde, resulting in 12 molecules with higher enantioselectivity.
The aRMSD (automatic root-mean-square deviation), developed by Wagner and Himmel, is a computational tool for structural analysis of molecules and has the potential to be applied for studies on catalysis.287 aRMSD offers an analysis of structural similarity and diversity which can evaluate structure–function relations.
Another important aspect of the field of cheminformatics is the representation of structures for data-driven algorithms, but again, little has been done for the description of transition metal complexes. The simplified molecular-input line-entry system (SMILES)288 and IUPACs international chemical identifier (InChI)289 are perhaps the most common human-readable formats for molecular encoding. Their string-based representation of molecules is based on connectivity, type of bonds, branching, aromaticity, stereochemistry, and isotopes. Clark suggested an extension for addressing nonorganic molecules by introducing zero bond orders and addition of an atom property to control the number of inferred attached hydrogen atoms.290
Significant progress has been observed in this field since data-driven approaches were combined with conventional methods. The COSMOS (not to be confused with the COSMO and COSMO-RS solvation models) model of Baldi and co-workers considers libraries of fragment and torsion angles for the prediction of small molecules, including simple transition metal complexes.291,292 However, the reported root-mean-square deviation of 1.68 Å for organometallic complexes limits the direct applicability of such schemes on catalytic applications.292
A novel geometry optimizer based on Gaussian process regression applied on a molybdenum amidato bisalkyl alkylidyne complex was reported by Denzel and Kästner.293 One of the advantages of their algorithm is that its performance is improved from steps that either overpass the minimum or predict structures with higher energy than the previous steps.
3. Mechanistic Complexity in Catalysis by 3d Transition Metals
3.1. Metal–Ligand Cooperativity
Catalysis by transition metal complexes relies on the active metal centers to which ligands are attached. The surrounding ligands shape the reaction environments, and in traditional homogeneous catalysis, they do not directly participate in the catalytic reactions but rather influence the electronic structure of the central transition metal atom. In the past years, there has been a growing interest in developing catalytic systems featuring a synergistic or cooperative chemical action between the metal and the ligand, enabling a more selective and efficient substrate activation. Such metal–ligand cooperation (MLC) has become an important concept in catalysis by transition metal complexes. The cooperation between the metal center and the so-called noninnocent ligands enables novel catalytic reactivity facilitated by concerted substrate activation and dual-site-driven catalytic transformations.294−297
The MLC-type catalytic systems have played key roles in the rapid advancement of the field of (de)hydrogenation transition metal catalysts witnessed in the last decades.298−301 The classical bifunctional Noyori-type catalysts based on noble metals302 are well-established, and they show excellent activity in hydrogenation of carbonyl-containing compounds including ketones, esters, and other carboxylic acid derivatives.301 In recent years, there has been rapid development of 3d transition metal catalysts particularly based on Mn and Fe elements in view of their low toxicity and high abundance.299,303,304 The current strategies for the synthesis of new 3d transition metal catalysts still rely on the well-established ligand library developed for noble metals. However, there is an intrinsic activity difference between the 3d transition metals with their noble counterparts, and tailored ligands are necessary for the rational design and the development of novel efficient 3d transition metal catalysts, which requires a clear mechanistic understanding of different catalytic systems. In the following, we will discuss the recently revised hydrogenation mechanism of Noyori-type catalysts and compare it with the recent computational studies on the catalytic hydrogenation by 3d transition metals (mainly Mn and Fe).
The metal–ligand cooperativity in Noyori-type hydrogenation catalysts originates from the metal/NH acid–base synergy effect.305,306 The conventional Noyori mechanism proposes that during the catalytic reaction, the NH group of the chelating ligand first accepts and then delivers a proton via its N–H bond cleavage and formation (E–H moiety in Scheme 1a, where E stands for the electron-donating, in this case, nitrogen atom), and such metal–ligand cooperativity involves both a hydrogenation (TSa) and an H2 activation step (TSb and TSc), as shown in Scheme 1a.306 This concept has been supported by delicate experimental characterizations and gas-phase computations, and it is also in line with the observation that the alkylation of the N−H moiety leads to a dramatically decreased catalytic reactivity.
Scheme 1. Catalytic hydrogenation of ketonic/aldehydic substrates by bifunctional Noyori-type catalysts: (a) Conventional Noyori mechanism (NH group deprotonates and reprotonates); (b) Revised Dub’s mechanism (NH group serves to stabilize transition states). Reproduced from ref.306 Copyright 2017 American Chemical Society.

Recent computational studies by Dub et al.306−309 with full catalyst models utilizing both implicit and explicit solvation provide evidence that this mechanistic picture needs to be substantially revised. For example, the concerted transition states TSa and TSc (Scheme 1a) as located from gas-phase computations could not be identified when the dual solvent models were introduced. The authors309 have emphasized the importance of employing appropriate computational methodologies such as the inclusion of (implicit/explicit) solvent effect and the necessity of performing an intrinsic reaction coordinate (IRC) analysis, which is sometimes neglected in theoretical reports. Accordingly, a revised mechanism has been postulated, in which the key proposal is that the N–H bond of the ligand does not cleave but serves to provide stabilization for the transition states via hydrogen bonding interactions (Scheme 1b). In such a mechanism, the chelating ligand is cooperative with the metal center but is chemically innocent. The hydrogenation of ketonic substrates from metal-hydrido complex via TSd (Scheme 1b) forms an anionic intermediate, and the proton to neutralize the generated anion is either from the σ-H2 ligand (catalytic cycle I) or a protic solvent molecule (catalytic cycle II). In comparison with the direct H2 dissociation via TSb (Scheme 1a), the H2 cleavage energies via TSe and TSe’ (Scheme 1b) decrease by 40–80 kJ mol–1.309
Van Putten et al. have reported a nonpincer-based Mn–P,N catalyst (Figure 4) highly active in ester hydrogenation and carried out a detailed investigation of the associated reaction mechanisms.310,311 In addition to the proposed catalytic cycle, the chemical processes underlying the stage of catalyst activation and a potential deactivation path were included in the computational analysis. This Mn system was found to exhibit similar chemistry to that postulated by Dub for Ru catalysts, where the main difference was that the ligand appears to be chemically noninnocent in Mn–P,N. DFT calculations accounting for both the local and bulk solvent effects indicated the importance of the metal–ligand cooperativity; in such a Mn system, this manifests itself in the deprotonation/reprotonation of the chelating NH ligand in the reaction steps of both H2 cleavage and carbonyl group reduction. Importantly, the heterolytic dissociation of H2 by the deprotonated Mn–P,N catalyst shows a very distinct energy profile in comparison with the noble metals, which is hampered by the unfavorable formation of the σ-H2 complex. The subsequent H2 cleavage usually proceeds with a very low barrier (<10 kJ mol–1) irrespective of whether it proceeds via the direct dissociation or a base-assisted reaction path. In contrast, the noble-metal based catalysts usually show a facile H2 coordination with the H2 dissociation step being the activated process. The initial mechanistic analysis, based on a combination of experimental and computational results, led to a proposal that a stable alkoxide adduct (4c, Figure 4) formed in the course of the reaction is the main cause of in situ catalyst inhibition. Importantly, the successful catalysis with Mn–P,N requires substantial concentration of an inorganic base present in the reaction medium, and this effect could not be rationalized based on the analysis of DFT-computed free energy profiles. Supporting DFT studies commonly focus on the analysis of the reactivity of the main catalytic complex, and little attention is often devoted toward the mechanistic role of the promoting substances that are necessary to achieve practical catalytic runs. The role of the base promotor was investigated in detail in ref (311). It was shown that the base promotor can play an important mechanistic role in facilitating key reaction steps, but more importantly, the presence of the base substantially influences the solvent properties and activities of key reaction components affecting thus dramatically the predicted free energy profiles (Figure 4).
Figure 4.
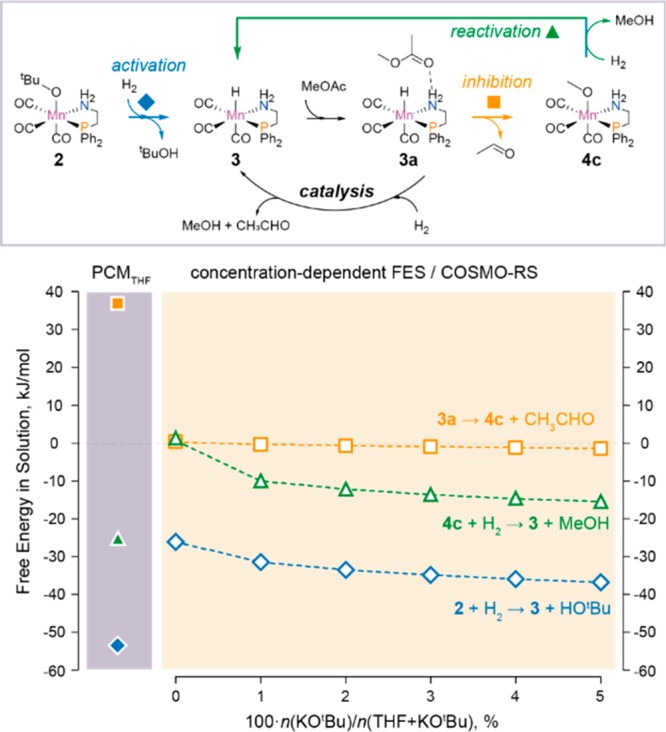
Key reaction steps of the catalytic ester hydrogenation by a Mn-P,N homogeneous catalyst, and the respective reaction free energy changes showing a strong nonlinear dependency of the thermodynamic parameters on the composition of the reaction mixture (base concentration). The graph presents a comparison of the free energy changes at the reaction conditions (373 K and 50 bar H2) computed using the implicit PCM method for THF solvent (PCMTHF) and the combination of concentration-dependent FES with realistic COSMO-RS model that account for such effects as hydrogen solubility, change of solvent properties as a function of the composition of the reaction medium, presence of promotors, and other components of the system (data from ref (311)).
In 2016, Beller and co-workers312 reported the first application of Mn pincer complexes for the catalytic hydrogenation of nitriles, ketones, and aldehydes. The experimental data were accompanied by DFT calculations helping to rationalize the activity of Mn complex (Figure 5). The mechanistic analysis was carried out at the B3PW91/TZVP level of theory applied to gas-phase molecular models. For the nitrile hydrogenation, a concerted mechanism was proposed, in which the transfer of the Mn-bound hydride and a proton from the ligand’s NH scaffold occurs simultaneously, in line with the concept of metal–ligand cooperativity. However, for the hydrogenation of benzaldehyde, the calculations suggest that the catalytic reaction follows a stepwise mechanism, in which an alkoxide intermediate is first generated via a hydride transfer from the Mn center, followed by the proton transfer from the NH moiety of the ligand. The group of Gauvin313 reported a similar stepwise mechanism for the acceptorless dehydrogenative coupling (ADC) of alcohols, which is a microscopically reverse reaction of the hydrogenation of ester process (Figure 6). The ADC of ethanol to form an adduct 2-H–CH3CHO proceeds via a sequential proton and hydride transfer with a highest free energy barrier of 21.6 kcal mol–1 at the B3PW91/SDD(Mn)&6-31G(d,p) level of theory. The further H2 elimination by direct routine has a free energy barrier of 21.5 kcal mol–1, which is close to the value of 20.2 kcal mol–1 reported by Beller and co-workers.312 The ethanol-assisted H2 elimination was found to lower the activation barrier by 3.8 kcal mol–1, consistent with the promoting effect of O-containing alcohol or alkoxy salt on H2 activation as proposed for other metal systems.309
Figure 5.
(a) Proposed reaction mechanism for nitrile hydrogenation catalyzed by a Mn pincer complex [E = P(isopropyle)2]. (b) Optimized structure of transition state for the hydrogenation step of acetonitrile. Reproduced from ref (312). Copyright 2016 American Chemical Society.
Figure 6.

Free-energy profile for the acceptorless dehydrogenative coupling (ADC) of ethanol catalyzed by a Mn pincer complex. Free energies ΔG and enthalpies ΔH (in parentheses) were computed at the B3PW91/SDD(Mn)&6-31G(d,p) level of theory. Direct and ethanol-assisted H2 elimination are indicated as orange and blue lines, respectively. Reproduced from ref (313). Copyright 2017 American Chemical Society.
The catalytic dehydrogenation of ammonia borane (AB) by a N-heterocyclic phosphenium Mn complex was recently reported by Gediga et al.314 The computational study identified two possible catalytic cycles (Scheme 2). In cycle I, the activation of AB proceeds as a cooperative transfer hydrogenation of the Mn = P bond, which renders it as metal–ligand cooperativity. Cycle II implies that the AB dehydrogenation is solely promoted by the sites at the chelating ligand, where the substrate transfers the H+/H– pair to the phosphorus and nitrogen sites of phosphenium unit, respectively. The role of the transition metal center in this mechanism is indirect. The computed overall activation barriers for cycles I and II are 42.2 and 41.4 kcal mol–1, respectively, at the level of ωB97xD/def2-SVP (PCM with THF as a solvent). In view of the limited accuracy of computational methods and the similarity of the activation barriers of the two mechanisms, the authors cannot exclusively conclude on the preferred reaction path but still suggest that the ligand-centered mechanism is in better accord with the experimental observations.
Scheme 2. Catalytic Dehydrogenation of Ammonia Borane (AB) with N-Heterocyclic Phosphenium Mn Complex 2Me (R = Me).
Cycles I and II illustrate the metal-ligand cooperative and ligand-centered mechanism, respectively. Reprinted with permission from ref (314). Copyright 2017 Wiley-VCH.
The variation of metal centers could dramatically alter the catalytic activity and the reaction mechanism. Kirchner and co-workers315 observed that the isoelectronic Mn and Fe hydride pincer complexes show distinct catalytic reactivity toward alcohol-amine coupling (Scheme 3). For the Mn catalyst, the alcohol-amine coupling selectively yields imines, while the Fe catalyst gives monoalkylated amines. The reaction paths by the Mn catalyst were analyzed by DFT calculations at the M06/6-311G(d,p)(SMDtoluene)//PBE0/SDD(Mn)&6-31G(d,p) level of theory (Scheme 3a), and a bifunctional mechanism involving metal–ligand cooperativity was proposed. The first step is the generation of σ-H2 complex BMn with a highest free energy barrier of 33 kcal mol–1 along the reaction paths, and this process is accompanied with the deprotonation of the NH ligand and the assistance of an ethanol molecule nearby (not shown). The further H2 release from BMn forms a 5-coordinated intermediate AMn, which can react with alcohol to regenerate the initial catalyst and to release an aldehyde. Such a reaction step proceeds via a single concerted transition state (32 kcal mol–1), which is similar to the mechanistic concept proposed by Beller and co-workers.312 An alternative path for the dehydrogenation of alcohol is via the alkoxide complex CMn and further inner-sphere β-hydride elimination to recycle complex 1, which is less favored with a barrier of 38 kcal mol–1.
Scheme 3. Divergent Cataltyic Reactivity of (a) Mn and (b) Fe Pincer Complexes for Alcohol-Amine Coupling.

Reprinted with permission from ref (315). Copyright 2016 Wiley-VCH.
The Mn catalyst promotes the alcohol oxidation and imine production upon water release, but it is inactive for further imine hydrogenation. The key difference between Mn and Fe systems is that the Fe catalyst (AFe, Scheme 3b) is capable of performing both alcohol oxidation (oxidation cycle) and further imine hydrogenation (reduction cycle). In the reduction cycle, it is proposed that the reaction starts from a 5-coordinated AFe to bind the imine substrate, and the adduct complex DFe can proceed via an intramolecular hydride transfer to release the vacant site. The following H2 adsorption generates σ-H2 complex EFe, in which the heterolytic cleavage of H2 occurs to produce amine (proton acceptor) and regenerate the Mn-hydrido complex AFe (hydride acceptor). In such a hydrogenation mechanism, the ligand remains chemically intact. A similar mechanism was proposed by the same group for the hydrogenation of aldehydes based on DFT calculations at the M06/6-311G(d,p)(SMDethanol)//B3LYP/SDD(Fe)&6-31G(d,p) level of theory (Scheme 4).316 The authors found that the ligand-assisted heterolytic cleavage of H2 has a quite high activation barrier (34.1 kcal mol–1), while the protonation of coordinated alkoxide to yield the final product proceeds with a much lower barrier (16.0 kcal mol–1). This behavior is very different from other computational studies, which normally report that the H2 cleavage or formation directly involves metal–ligand cooperativity in Fe pincer complexes catalyzed (de)hydrogenation reactions.317−321 Such cooperative effects have been extensively discussed in a review by Li and Hall.322
Scheme 4. Catalytic Cycle for the Hydrogenation of Aldehyde Catalyzed by a Fe Pincer Complex.

Reprinted from ref (316). Copyright 2014 American Chemical Society.
3.2. Metal–Metal Cooperativity
Metal–metal cooperativity refers to interaction of multiple metals within a molecular complex for the enhancement of a specific property. For catalytic applications, this can refer to the promotion of a reaction step by the presence of multiple metal sites.323 This can be achieved by two different possible mechanisms. In the first, the metal centers act synergistically, where two or more metals interact with a reactant for the efficient reduction of a reaction barrier. The second mechanism involves a catalytically active transition metal supported by one or more metals. In that case, the supporting centers affect the electronic properties of the active site and enhance its catalytic behavior. Polynuclear complexes can give access to reaction intermediates or selectivities that are not available to mononuclear species. These effects are a result of unique electronic structure (oxidation states or spin states) and ligand environment.324−329
Many examples of such mechanisms exist in nature, such as the nickel–iron and iron–iron hydrogenases that oxidize molecular dihydrogen,331 the iron–molybdenum cofactor present in the nitrogenase enzyme performing nitrogen fixation,332−334 or the manganese metallo-oxo cluster which performs water oxidation within the photosystem II protein complex.335 These inorganic active sites contain multiple metals that work in a cooperative effort, such as providing extra coordination sites for binding substrates or helping to manage electron counts. In an effort to search for more viable catalysts, science has taken a cue from nature and has attempted to develop biomimetic compounds to reproduce the efficiency of the cofactors of these biological enzymes.
Transition metals find such a prominent role in catalysis due to their large reservoir of electrons and their ability to create complex structures with their d orbitals, creating covalent bonds with organic ligands. Metals can also form direct and stable metal-to-metal bonds with one another using their d orbitals.330 With the characterization of the [Re2Cl8]2– structure (Figure 7), metal–metal interactions became an extensively researched subject in an effort to elucidate the nature of the Re2 bond.336,337 In 1965, Cotton proposed a bonding model for the interaction between the two Re metals, which involved the concept of a δ-type bonding interaction in a scheme that introduced a quadruple bond.338,339 For more general reviews concerning the role of direct metal–metal bonds in catalysis or specific heterobimetallic pairs for particular reaction types, please refer to the reviews of Uyeda330 and Braunstein.329 Due to the complexity of the electronic structure of these systems, they are ripe for study with high-level computational methods.
Figure 7.
Molecular structure of [Re2Cl8]2– and a qualitative molecular orbital diagram. Adapted from ref (330). Copyright 2016 American Chemical Society.
3.2.1. Enzymatic Systems
Nature is often used as a source of inspiration toward new catalysts. Active site cooperativity including the metal–ligand synergy outlined in the previous subsection and the metal–metal cooperativity that is the focus of this part of the review are quite common mechanisms and are realized in enzymatic systems to enable highly selective and efficient chemical transformations. Numerous computational studies on enzymatic active sites have provided insight into the electronic and steric effects on important biological processes involving metalloenzymes.340 Similarly, computational work on synthetic model complexes that mimic the enzymatic reactivity has provided a better understanding of mechanistic and electronic structure properties that are difficult to probe from spectroscopic techniques. Combination of experimental and computational data has contributed to the design of homogeneous and heterogeneous biomimetic catalysts for challenging reactions of industrial interest. A well-known example is the Fe(IV)-oxo sites of the heme- and nonheme enzymes, which have been successfully introduced in materials and model molecular complexes for C–H oxidation.341−344
The hydrogenases are a diverse group of polynuclear metalloenzymes that catalyze the conversion of H2 into protons and electrons via the heterolytic dissociation of the dihydrogen molecule, as well as the reverse reaction.346,347 Their active site is composed of [Fe], [FeFe], or [NiFe] metal ions coupled to FeS clusters that establish electron transfer chains (Figure 8).348 The elucidation of the catalytic reaction promoted by the hydrogenases is a key point for a “hydrogen economy” where dihydrogen is used as a clean alternative fuel. Knowledge of the mechanistic details will provide synthetic directions for the development of tailor-made biomimetic catalysts for hydrogen production.349
Figure 8.
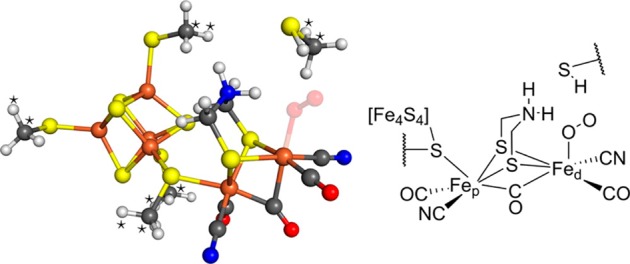
[FeFe] hydrogenase active site with the [4Fe-4S] cluster DFT-optimized structure used by the Reiher group. Orange = Fe; yellow = sulfur; red = oxygen; gray = carbon; blue = nitrogen; and light-violet = hydrogen. Reprinted from ref (345). Copyright 2014 American Chemical Society.
Computational studies that span from highly accurate electronic structure theory methods350 to molecular dynamics and coarse-grained analysis351 have contributed to the understanding of the enzymatic environment and the reaction channels of the hydrogenases. In 2007 and 2014, two detailed reviews on computational studies of both [NiFe] and [FeFe] hydrogenases covered the progress of this field.352 Here, only key articles and recent progress are discussed.
Ryde et al. have applied DFT calculations on the raw crystallographic data of the [FeFe] hydrogenase for a quantum refinement of the atomic composition of the [FeFe]/[4Fe-4S] site.353 There proved to be conflicting accounts concerning the composition of the bridging dithiolate ligand with proposals including CH2(CH2S–)2 and NH(CH2S–)2 as plausible ligands.354 After evaluating five different model complexes, they concluded that a nitrogen-bridged structure provides the best match to the raw crystallographic data. QM/MM calculations by Greco et al. elucidated the interplay between active site and environment on the electronic and magnetic properties for both activation and reaction steps.355,356 Thomas, Darensbourg, and Hall performed a computational study defining the active site of the [FeFe]-hydrogenase in its resting state with a mixed valence Fe(II)Fe(I) model.357 The study included analysis of the frontier molecular orbitals using DFT for comparison with the neutral Fe(II)Fe(II) species. The geometry of the resting state features a rotated geometry in which a carbonyl ligand moves into a bridging position. Analysis showed that the lone unpaired electron in the species lies on the iron site featuring the rotation. Additionally, Chang published a study in 2011 using DFT for the elucidation of different configurational isomers of the [FeFe]-hydrogenase enzyme, mostly concerning the geometrical changes upon oxidations and reduction as well as the positioning of various diatomic ligands (cyanide and the bridging carbonyl in particular).358 Long et al. have also published a computational study concerning the proton transfer process in the same hydrogenase system using QM/MM simulations to determine the free energies of competing pathways for the mechanism.359 In 2013, the Reiher group published a study on the effects of electric fields on the active site of the [FeFe]-hydrogenase enzyme which catalyzes the formation of H2.360 In this particular study, DFT calculations were performed on a 96-atom model of the active site to simulate the effect an electric field would have on the structural parameters of the [4Fe-4S] cubane as well as the energetics of the studied mechanism. In a follow-up article, the same group investigated different O2 activation paths by the distal Fe of the [FeFe] site.345 The possible O2 activation has been proposed as a deleterious reaction which generates superoxo species that damage the enzyme. A combined nuclear resonance vibrational spectroscopy and DFT study showed that the hydride intermediate corresponds to the state prior to H2 formation for the reverse proton reduction.361 Sode and Voth performed multistate empirical valence bond (MS-EVB) reactive MD simulations362,363 to examine the proton transport step.364
A recent benchmark study considered 24 density functionals and six basis sets for the relative stability of oxygenated isomers of diiron models of the [FeFe] hydrogenase.365 Energies calculated with approximate CCSD were used as reference, and it was shown that the BP86 and PBE0 functionals give results in best agreement with the coupled-cluster energies. Such studies are extremely important for the evaluation of the quantum chemical level of theory that can provide accurate results for these or analogous molecular systems.
Similarly, quantum chemical calculations have been performed on the [NiFe] hydrogenase active site. QM/MM calculations considered the protonation of the four cysteine ligands located in close proximity to the [NiFe] core.366 A more favorable hydrogen bond formation was found for Cys-546, that allows an easier, energetically more favorable protonation. A detailed examination of the different redox states by means of theoretical spectroscopy revealed the role of the CN and CO ligand stretching frequencies as fingerprints of the structural composition of the [NiFe] site.367 A DFT study on intermediates of the hydrogen evolution cycle on a FeNi model that included the first and second coordination spheres of the hydrogenase predicted a metal–metal bond between Ni and Fe that might play a role in the reaction mechanism.368 Large-scale DFT calculations on the aerobic and anaerobic oxidation of the [FeNi] active site elucidate the reaction pathways that block the reactivity of those species for biotechnological applications.369 Benchmarking calculations were performed on mononickel and FeNi models in an effort to compare DFT calculations to results from WFT methods such as CASSCF/CASPT2, RASSCF/RASPT2, and CCSD(T).370 These calculations help the characterization of the enzymatic active site and provide additional understanding about how the active site may behave in the presence of H2. Follow-up studies were performed to investigate the binding of H2 to the active site by means of multiconfigurational DMRG-CASPT2, CCSD(T),371 and multiconfigurational DFT calculations.350
Diiron and NiFe model complexes that mimic the enzymatic activity of the [FeFe] and [NiFe] hydrogenases, respectively, have been also the target of computational studies. The mechanistic pathways for hydrogen evolution catalyzed by the bioinspired Fe2(adt)(CO)2(dppv)2 (adt = azadithiolate, dppv = diphosphine) which facilitates the formation of a doubly protonated ammonium-hydride intermediate have been computationally investigated.372,373 The mechanistic study of the H2 evolution revealed the role of the ammonium hydride interaction, the effect of the ligand environment, and the intramolecular electron transfer between the two Fe cations. The catalytic hydride transfer promoted by a biomimetic Ni–Fe complex has been studied with Mössbauer spectroscopy and DFT calculations.374,375
The Solomon group has also performed quantum mechanical studies of polynuclear species. In 2002, the group published a study including DFT calculations on the μ4-sulfide bridged tetra-copper active site of N2O reductase.376,377 Additionally, the same group performed calculations to understand the mechanism and electronic structure of multicopper oxidases (MCOs) which catalyze the reduction of molecular dioxygen to water.378 More recently, they have studied the activation of peroxide by the diiron AurF enzyme active site, where they applied DFT to study the catalytic cycle. This work identifies the redox-active orbitals of the active site and uses TD-DFT to study the electronic transitions for comparison with magnetic circular dichroism (MCD) data.379
It is evident that polynuclear copper sites have a ubiquitous role in enzymology, and the metal–metal cooperativity plays an important role in the ability for these enzymes to operate effectively. A recent study considers the hydroxylation of methane by the tricopper [Cu(II)Cu(II)(μ-O)2Cu(III)(7-N-Etppz)]1+ complex (7-N-Etppz: 3,3′- (1,4-diazepane-1,4-diyl)bis[1-(4-ethylpiperazine-1-yl)propan-2-ol]) that mimics the active site of the particulate methane monooxygenase (pMMO) enzyme.380 In this work, DFT calculations were carried out to investigate the C−H bond activation process over the tricopper active site of pMMO represented by a truncated active site model, in which the histidine groups bound to the copper sites were replaced with amino groups. Computations reveal three possible mechanisms for the hydroxylation reaction. The radical and nonradical mechanistic pathways at different spin states for the [Cu(II)-Cu(II)(μ-O)2Cu(III)(7-N-Etppz)]1+ complex have also been examined.
Several groups have studied the FeMo-cofactor of the nitrogenase enzyme that fixes molecular dinitrogen as well. With eight metal atoms present within the cofactor, the in-depth understanding of the mechanism by which nitrogenase functionalized dinitrogen to ammonia remains challenging for both experiment and theory. It was not until 2002 that an interstitial central ligand was found to be present at the center of the active site,381 and it took until 2011 to properly support that this interstitial element is a carbon,382 while the oxidation states of the irons were elucidated only in 2016.383 With such a large system of metal–metal cooperative effects in conjunction with little insight into a plausible mechanism, studying the reactivity of FeMo-cofactor proves to be a daunting task. Typically, smaller model systems are used to study particular active sites such as in the calculations on both the FeMo- and FeV-cofactors performed by Grunenberg in 2017 to investigate the potential force constants of the interstitial carbide atom.384 Scott et al. published a study on the effects of CO-inhibition on the Mo-nitrogenase active site using both spectroscopy and DFT.385 Similarly, Siegbahn et al. claimed that the central carbon in the FeMo-cofactor becomes protonated during the nitrogen fixing process. Siegbahn posited this mechanism based on DFT calculations which shares striking similarities to the mechanism posed by spectroscopic experiments.386
Reactivity and electronic structure calculations on full metalloenzyme complexes are inhibited by the sheer size of the enzyme. Therefore, calculations are required at the active sites only and, therefore, can sometimes neglect some of the subtler effects of the surrounding environment. For a more exhaustive list of quantum mechanical studies of metalloenzymes, Blomberg et al. published a review on this subject in 2014.387
3.2.2. Multichelating Ligands for Bimetallic Catalysts
Multichelating ligands provide a preferential environment that facilitates the coordination of two or more metals. Cooperativity between 3d transition metals promotes many electron processes, allowing for easy oxidation and reduction via the formation or breaking of metal–metal bonds, respectively. This flexibility on oxidation states allows for interesting reactivity and catalysis. For instance, in 2015, a study was reported on the reactivity of a dicobalt species ligated with a trisphosphino(triamido)amine ligand (Figure 9) with molecular dinitrogen and Me3SiCl and KC8 under ambient conditions for the formation of tris(trimethylsilyl)amine.388 The interaction between the two cobalt atoms and how it enhances the catalytic reactivity of the active Co center was elucidated by CASSCF/CASPT2 calculations. Multiple viable reaction mechanisms were computed with DFT, and the most thermodynamically and kinetically favorable path was proposed as the full N2 silylation mechanism. This study was complemented by experiments to elucidate the mechanism and the electronic structure of the dicobalt system. Calculations determining the ground state spin of the precatalyst were compared with the magnetic data derived from the experiment. Further studies were later performed by exchanging the penta-coordinated cobalt from the precatalyst with Al, Ti, V, and Cr in order to see how the cooperativity affects the dinitrogen activation.389 This work involved a combination of DFT and WFT calculations. DFT was used for geometry optimization and calculation of CM5 charges,390 while CASSCF and CASPT2 calculations were performed on the optimized structures to compare the fine details of electronic structure of these species. Once again, this study is an excellent example of an interplay between theory and experiment as computations were used to support the electrochemical data and to understand relationships between the activity and cooperativity of different metals. There are several other examples where computational methods, specifically those involving multireference calculations, were used to characterize the electronic properties of binuclear species synthesized in the same research group.391−393 Those studies allow for more guided analysis of catalytic activity wherein, by understanding the interaction between the pair of metals and its relationship to the catalysis, one can gain a heuristic that can guide later synthesis. Such works show two powerful aspects of electronic structure theory in regard to the understanding of metal–metal cooperativity: (1) DFT calculations of the proposed mechanistic pathway to support experimental findings and (2) multireference calculations to understand the underlying electronic structure of the systems and aid the characterization of the compound.
Figure 9.
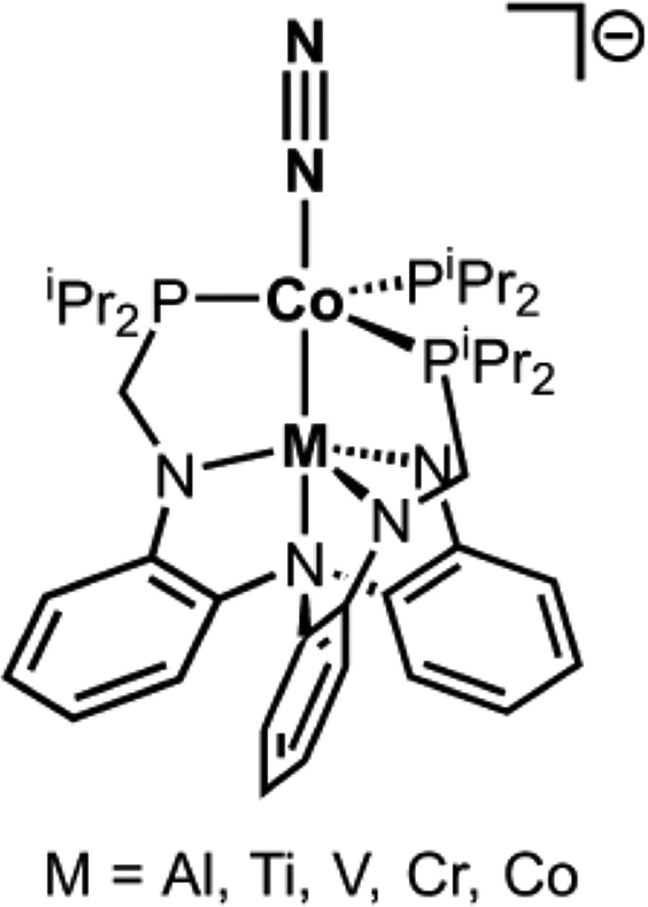
Isostructural series of the trisphosphino(triamido)amine ligated bimetallic complex of Lu et al. with varying supporting metal.391
The Thomas group studied the effects of metal–metal cooperativity as well. One of the heterobimetallic pairs in their work is a complex comprised of a Ti/Co core which can promote N–N bond cleavage of hydrazine or methyl hydrazine.394 DFT calculations were performed to elucidate the Ti−Co metal bond, as represented pictorially in Figure 10.395−397 Experimental results were supported by computations that improve the understanding of the electronic structure of the catalytic species and specifically the nature of the Ti–Co interaction.
Figure 10.
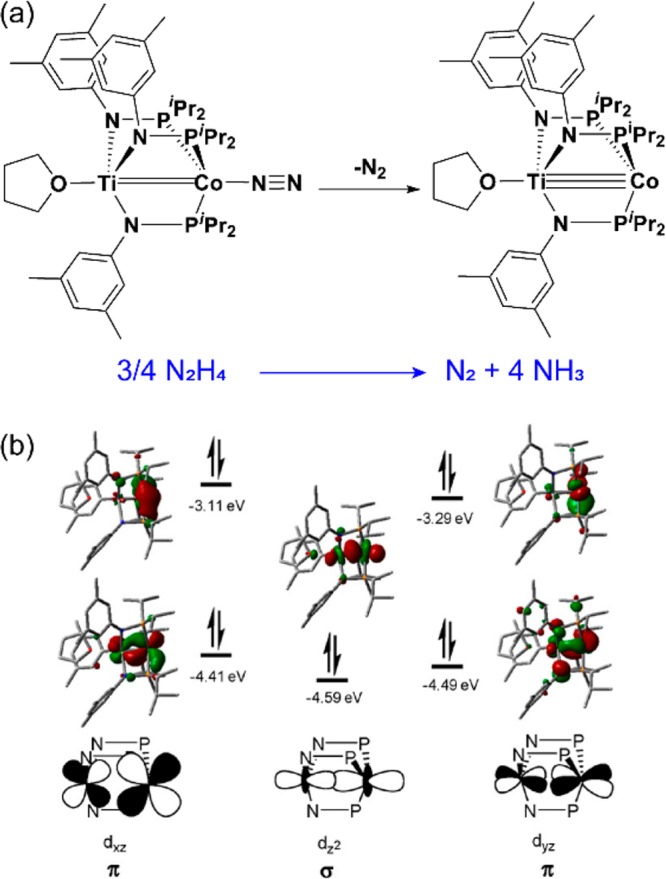
(a) Nitrogen evolution over the Ti/Co molecular complex with (b) the pictorial representation of frontier molecular orbitals. Adapted from ref (394). Copyright 2015 American Chemical Society.
The Holland group has also used computational methods to analyze catalysts able to cleave strong covalent bonds. One such study involves a diiron hydride complex that cleaves the N–N double bond of azobenzene (PhN = NPh).398 Besides, they investigated a Co−O2 system capable of C−H oxidation via an oxo-bridged heterobimetallic intermediate (with either low-valent iron or cobalt).399 Both of these studies included computationally calculated mechanisms for the proposed pathways.
The research group of Ess has studied by DFT the significance of the binuclear interaction between Pd and Ti on allylic amination reactions.401,402 The electron-withdrawing dative intrametallic interaction lowers the reaction barrier of the rate-determining reductive amine addition by enhancing the Pd(II) to Pd(0) reduction. Both WFT and DFT were used to understand the interaction between Pd and Ti by studying monometallic species in the absence of the Ti promotor. Synthetically, the TiCl2 group was replaced by an ethylene moiety, allowing thus to directly assess the role of the Ti in the allylic amination. The same group has recently reported the [4 + 2] cycloaddition mechanism and explained the selectivity for alkyne cyclotrimerization catalyzed by the dinickel complex of Uyeda et al.400,403 The reaction involves three steps: oxidative C–C coupling, migratory insertion, and metallacycloheptatriene reductive elimination (Figure 11). The cooperative effect of the dinuclear site provides selective cyclotrimerization, whereas mononuclear Ni catalysts yield complex product mixtures. DFT computations revealed a spin-crossover mechanism involving Ni–vinyl nonclassical intermediates that leads to a fast reductive elimination step.
Figure 11.

Reaction profile of the acetylene cyclotrimerization catalyzed by the dinickel complex. Three different spin states were considered (MECP: minimum energy crossing point). All energies in kcal mol–1. The DFT optimized geometries of the C–C coupling reaction step (3 → 4) are shown at the bottom. Aryl ligands and ligand hydrogen atoms are not shown for clarity. Adapted from ref (400). Copyright 2017 American Chemical Society.
In 2015, Kilpatrick et al. reported a DFT study on the reductive activation of CO2 using a dititanium catalyst and obtained an energy profile for the binding of CO2 and disproportionation of Ti2Pn2O(CO).404 The combination of computational results together with spectroscopic and X-ray crystallography data enabled the identification of key reaction intermediates. Liu et al. reported a computational study on the cooperative effects of titanium–chromium catalysts in the polymerization/copolymerization of ethylene.405 Similarly, DFT was used to study the energetics of the different reaction paths and the effect of the proximity of the metal centers on the polymerization reaction.
3.2.3. Water Splitting
The evolution of O2 through the splitting of water is an important complex process with a key role in a hydrogen economy. The electrolysis of water determines why there is a strong need for the search for catalysts to perform the reaction. The two half reactions are as follows:
| 3.2.1 |
| 3.2.2 |
With the anodic reaction being a four-electron process, there exists a considerable free-energy change of 359 kJ mol–1 encompassing an endergonic reaction.406 In addition to not being a thermodynamically favored reaction, a considerable kinetic barrier exists for the process. A large overpotential (more than ca. 400 mV to overcome) for electrochemical water oxidation exists due to the number of intermediates necessary to couple the two oxygens and release the four protons and electrons.407
In natural photosynthetic processes, oxygen evolution occurs via the photosystem II (PSII), also known as water-plastoquinone oxidoreductase complex. The active site contains a Mn4CaO5-cluster (often referred to as the oxygen evolving complex or OEC, Figure 12). Three of the manganese ions, the calcium, and four of the five oxygens form a cubane-like structure, while the fourth Mn is tethered to the outside of the cubanelike structure by μ-oxo bridges.408−410
Figure 12.
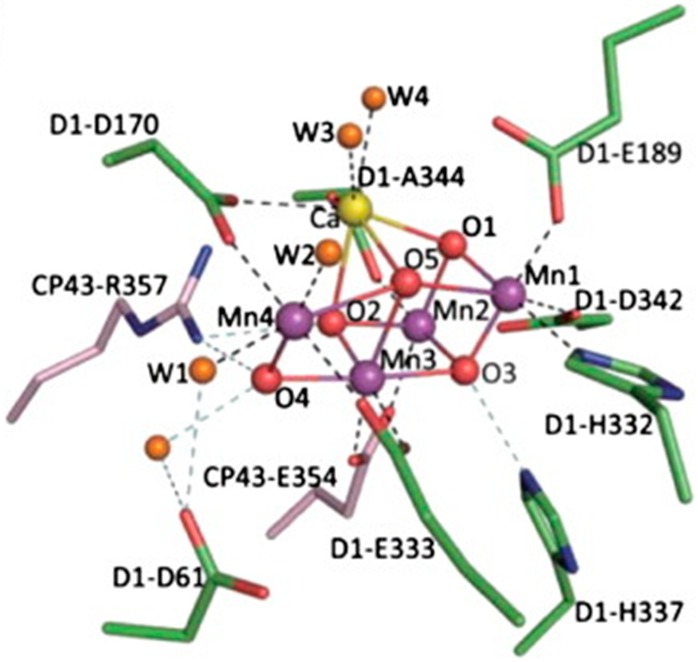
Active site and nearby residues in oxygen-evolving photosystem II. Reprinted with permission from ref (409). Copyright 2011 Springer Nature Limited.
In the mechanism of biological water splitting, the OEC oxidizes water by being sequentially oxidized four times by a tyrosine residue, creating a potential sufficient enough for oxygen evolution to occur. For a more complete description of the photosynthetic pathway, please refer to the review of Blakemore et al.407 Several groups have performed calculations directly on the active site of PSII. In a study published in 2013, Cox et al. elucidated the electronic and geometric structure of the OEC.411 They compare quantum chemical calculations from models that predict EPR parameters along with DFT to understand the geometric and electronic structures of each metastable state of the system to correlate structural and spectroscopic data.411 Additionally, Terrett et al. published a study characterizing the magnetic exchange interactions within the OEC cluster, comparing the hyperfine tensors along with the spin projected hyperfine values of a particular metastable state with experimental EPR values.412
3.2.3.1. Artificial Photosynthesis
The splitting of water is an important reaction in sustainable catalysis and renewable energy on many different levels. Global climate change has motivated the search for cleaner, renewable energy sources. Currently, the world’s total demand for energy is on the order of 15 terawatts, while the sun can provide over 50 terawatts of energy (theoretically capable of providing roughly 101,000 terawatts).413 Current methods for photovoltaic technology involves converting the energy provided by the sun into an electrical potential.407 However, since the sun is only present for half of the day and its energy is spread over a large area, finding clean, low-cost, and efficient methods for storing this energy is becoming increasingly important. One such method is the splitting of water by solar energy into hydrogen and oxygen gases. The dihydrogen can be stored and consumed when necessary without creating any harmful byproducts, in fact regenerating the initial water at the point of use.414Figure 13 shows a schematic diagram of a photovoltaic electrolysis cell developed by Jia et al. in 2016 that is capable of achieving over 30% solar-to-hydrogen efficiency over a continuously operating 48 h period.414 The advantages of using solar energy to split water are self-explanatory: using standard electrolysis still requires electrical input energy typically generated from nonrenewable resources. Additionally, standard electrolysis typically implores the use of platinum electrodes, an expensive, nonrenewable resource. On the industrial scale, water splitting techniques can be used to generate large amounts of H2 gas for either cleaner burning alternative fuels to traditional fuel feedstocks or as hydrogen sources for other large-scale reactions. For instance, industrial ammonia production from molecular dinitrogen requires an ultrapure source of H2 which is currently obtained from fossil fuels, giving this reaction a large carbon footprint, although it itself requires no carbon.415
Figure 13.
Schematic design of the photovoltaic-electrolysis device by Jia et al.414
3.2.3.2. Molecular Catalysts for Water Splitting
The first molecular, homogeneous catalyst for water splitting was synthesized by Meyer and co-workers in 1982.416 The complex is a ruthenium polypyridyl species that performs a series of proton-coupled electron transfer (PCET) steps.417,418 The mechanism evolves by performing progressive oxidations of the ruthenium atom, creating a shift in the pKa values of the ligated water molecules, thereby increasing their proton donating power. This results in the successive activation of the aqua species to a hydroxo and then an oxo species, making each successive oxidation easier.407 Since the synthesis of this ruthenium species, considerable effort has been expended to create molecular catalysts capable of promoting a cleaner and easier water splitting, but certain complications lie in the use of molecular complexes. For example, the number of oxidizing equivalents must be correct in order to avoid partially oxidized products (such as hydroxyl radicals or peroxide). Additionally, in electrocatalytic species, there is a prevalent concern for ligand oxidation and degradation, especially for organic-based ligands. Therefore, there is a great deal of interest in bi- or polynuclear molecular catalysts which can interact through metal–metal bonding, creating a built-in redox mechanism for the metal centers without the need for an external oxidant. Having multiple metal centers can also ease the combination of oxygen atoms into O2 by creating more sites for the water molecules to bind and split. Special attention must also be made for the characteristic metal centers of these molecular catalysts; both ruthenium and iridium have proven to be among the most active metals for water splitting in both the heterogeneous and homogeneous environments.407 Therefore, the search for cheaper metals such as iron or cobalt is mandatory for the development of sustainable and more cost-effective methods for catalyzing the oxidation of water. Here, we shall survey relevant literature concerning computational studies of water splitting reactions, specifically those with more than one 3d transition metal center.
Computational methods can help elucidate the catalytic pathway a particular reaction is undergoing as well as to help understand the electronic nature of the metals involved in polynuclear species. As stated before, the redox interactions of the metal centers are important for the oxidation of water and being able to study the electronic structure at each step can provide vital insight into the mechanism. For a general overview of the capabilities of DFT in understanding catalytic water splitting, we refer the reader to the article of Mavros and co-workers.419
Cobalt species play a special role in the field of water oxidation as an earth-abundant transition metal species capable of performing water oxidation with a high degree of success. Gimbert-Suriñach and co-workers published a study concerning the characterization of the reaction intermediates in a dicobalt species using spectroscopy and DFT computations.420 This study concerns the dinuclear μ-peroxido [(CoIII(trpy))2(μ-bpp)(μ-OO)]3+ (trpy = 2,2’;6’:2″-terpyridine; bpp– = bis(2-pyridyl)-3,5-pyrazolate). Figure 14 shows the DFT-computed catalytic cycle while Figure 15 shows the experimental and simulated EPR spectra and the SOMO of an end-on superoxido intermediate. Computational results agree well with experimental evidence, providing a detailed characterization of the reaction intermediates. Additionally, the study showed that the dicobalt complex can serve both water oxidation and oxygen reduction.
Figure 14.
Calculated water oxidation catalytic cycle. The arc represents the bpp– bridging ligand. Other ligands are not shown for sake of clarity. Reprinted from ref (420). Copyright 2016 American Chemical Society.
Figure 15.

Left shows experimental (black) and simulated (red) EPR spectra of the end-on superoxido intermediate (with oxygen-17). Right shows the SOMO of the same species (oxygen-16). Reprinted from ref (420). Copyright 2016 American Chemical Society.
The Nocera and Chen groups published a study characterizing the cofacial Co(IV) metal centers of the doubly oxidized Co4O4 cubane portion of the Co-OEC active site.421,422 Broken symmetry DFT calculations were performed on the Co4O4(py)4(OAc)4 molecular model complex (py = pyridine, OAc = acetate)423 for the elucidation of the ground state electronic structure of the cubane system, confirming the presence of a localized, antiferromagnetically coupled Co(IV) dimer. Both electrochemical studies and DFT predict the oxidation of Co(III)4 to Co(III)2Co(IV)2 rather than a single site double oxidation to Co(III)3Co(V). These results show that the Co4O4 cubane provides an amenable model for the oxidic cobalt OER catalyst. In their 2016 study, Stich and co-workers performed electronic structure calculations to determine the magnetic properties of dioxygen-bridged cobalt dimers relevant to the oxidation of water. These studies provided an electronic structure description of the Co(III)-superoxo observed during the water oxidation reaction by Co4O4.
Kanady and co-workers studied a cuboidal Mn3MOn (M = Mn, Ca, Sc; n = 3, 4) series of complexes as biomimetic analogues to the PSII active site.424 However, rather than evolving molecular dioxygen, the species performs an oxygen atom transfer. In an effort to understand the underlying mechanism of this reaction, DFT calculations were performed with Poisson–Boltzmann solvation to study the oxygen atom transfer from the manganese–metal clusters to PMe3. Computational and experimental results agreed that the Mn(III)2Mn(IV)2O4 structure had the greatest rate of oxygen atom exchange while the M = Ca and Sc congeners were found to be unreactive.
Extensive computational research has been performed on the field of catalytic water splitting. This important and challenging process was used here to provide illustrative examples on the metal–metal cooperativity in molecular catalysis by 3d transition metals. A comprehensive overview of the computational and mechanistic aspects of homogeneous water oxidation catalysis has recently been published by Liao and Siegbahn.425 Besides, Sala et al. reported an important account concerning the mechanism of water splitting by transition metal complexes.426 Recent excellent reviews by Blakemore, Crabtree and Brudvig,407 Patzke and co-workers,427 and Meyer et al.428 provide a comprehensive overview of the state-of-the-art in the field of molecular water oxidation catalysis. For further (and more general) information regarding the state of the art in water oxidation by molecular compounds, we refer the reader to the aforementioned sources.
3.2.4. CO2 Reduction
CO2 reduction into higher-energy products is a process closely related to water splitting as it represents an important component of perspective technologies for the efficient conversion and storage of solar energy and electricity into renewable “solar fuels”. The different approaches to the efficient CO2 reduction including the thermo-, photo-, and electrochemical methodologies have been the subject of several recent reviews.429−435 The “dark” thermocatalytic homogeneous reduction of CO2 with molecular H2 to formates or methanol300,436−438 usually employs catalytic systems featuring the acid–base metal–ligand cooperativity functions discussed in section 3.1. The electrocatalytic reduction of CO2 is an alternative approach that has attracted significant attention in the recent years as it provides a path to a controllable process within compact electrocatalytic systems. Coordination complexes of earth-abundant 3d transition metals constitute an important class of highly efficient molecular electrocatalysts for CO2 reduction,439−441 and their utilization for electrochemical conversion of carbon dioxide has been a subject of several recent reviews.442−446 The important fundamental advantage of the electrochemical process is the possibility to establish reaction channels involving two-, four-, six-, and eight-electron reduction processes, providing a controlled path toward variety of carbon-based products.447,448 The main persistent challenge in the field is related to the development of high-performance and highly stable electrocatalysts.
The utilization of multinuclear complexes providing multiple redox-active sites for CO2 reduction is one of the promising catalyst design paths considered in the field,446 since the redox tuning through the formation of multimetallic ensembles is considered a fruitful strategy for activity optimization of electrocatalytic systems.449 For example, the synergistic effects of a dinickel electrocatalyst for the selective reduction of CO2 to CO have been demonstrated in a recent combined experimental and theoretical study by Cao et al.450 The DFT-computed redox potential for Ni2II,II/Ni2I,I (−1.15 V vs NHE) coheres well with the experimental one obtained from cyclic voltammetry (−1.18 V). The calculations reveal a lower activation barrier for the rate-determining CO2-to-CO reduction step over the dinickel catalyst compared to that provided by its mononickel counterpart. A similar synergistic effect in a cryptand dicobalt photocatalyst have been highlighted by control experiments and DFT calculations.451 Besides, dicopper complexes have also been successfully applied as catalysts for CO2 reduction to oxalates.452,453
3.2.5. Where is the Field Heading?
As synthesis of novel catalytic compounds continues and computational methods improve, where does this field stand to go? Presently, more and more studies are being performed on polynuclear systems, whether on magnetism, spectroscopy, or catalysis. In 2013, Chan and Neese published a study using DMRG to study the [2Fe-2S] and [4Fe-4S] clusters (Figure 16).454 With newer methodologies and more efficient implementations of electronic structure theory methods, larger polynuclear complexes requiring more extensive active spaces can be studied with a high degree of accuracy. Benchmark studies which present the strengths and the limitations of those methods on molecular systems involving transition metals aid other research groups to adopt them and utilize them on relevant applications.455 It is expected that in the near future, more robust and accurate computational studies on polynuclear systems will be performed, not only for catalytic activity but also for magnetism, spectroscopy, and characterization as well.
Figure 16.

(a) [2Fe-2S] (top) and (b) [4Fe-4S] clusters. Adapted with permission from ref (454). Copyright 2014 Springer Nature Limited.
3.3. Catalysis with Metal–Organic Frameworks
Metal–organic frameworks (MOFs) are highly ordered, porous crystalline hybrid materials composed of inorganic secondary building units (SBUs), which are comprised of metal ion nodes or clusters nodes. The inorganic building blocks are connected by organic linkers, and together they form one-, two-, or three-dimensional frameworks.456 MOFs are often regarded as an important link between molecular and heterogeneous catalysts. They are extensively studied as catalysts for a wide range of applications due to their isolated active sites resembling those in homogeneous catalysts incorporated into a well-defined crystalline porous matrix. Additionally, MOFs retain high thermal stability and large pore size and surface areas, while their properties can be tuned by choosing proper inorganic and/or the organic building blocks. Additionally, altering the organic linkers connecting the metal nodes can modify the MOF pore size and shape, active site accessibility, metal coordination, hydrophilicity of the pores, density of catalytic sites, and electronic properties, all of which can significantly affect catalytic activity. Theoretical studies on MOFs have contributed on the elucidation of their electronic and catalytic properties by providing insight on reaction mechanisms at an atomistic level.457,458 Recent review articles are recommended for a detailed overview of catalytic reactions using MOFs.459−466 Here, we will review recent literature concerning electronic structure studies on MOFs comprised of first-row transition metal active sites with a focus on oxidation and reduction reactions. The theoretical perspective on stability and role of surface defects will also be discussed.
3.3.1. Scope and Scale
Modeling catalytic activity in MOFs explores how molecular structure impacts reactivity, providing better understanding of mechanisms and catalyst design. The intrinsic reactivity of rather isolated metal sites embedded in the extended crystalline structure of MOFs can accurately be described using the methodologies commonly employed in studies on molecular systems by representing the active site and its local environment using a cluster approach. The steric and confinement effects due to the microporous environment inside the MOF pores require the extension of such cluster models by using embedding schemes such as ONIOM or QM/MM described in section 2. Fully periodic DFT simulations can also be carried out on MOF structures having relatively small unit cells. For an extensive coverage of computational methods applied to studies of MOFs, we direct the reader to the comprehensive reviews of Fang et al.467 and Getman et al.468 The main advantage of the QM/MM hybrid methods is the possibility to apply very high-level DFT or even WF levels of theory to the atoms of the active site directly involved in the catalytic reaction with the remaining part of the system being approximated by interatomic potentials.469−471 A similar strategy could be used to refine the results obtained with fully periodic DFT calculations by combining multiple levels of electronic structure theory such as MP2 and DFT (QM/QM).472
Selecting an appropriate representative structural model for a MOF is an essential step in modeling reactivity of these crystalline solids. The well-defined structure of MOFs can be represented with periodic repeat units, which often contain hundreds of atoms. Theoretical calculations can be performed either on the full structure by applying periodic boundary conditions or on molecular cluster models by choosing to study only a fragment of the material. Figure 17 illustrates different models of Fe-MOF-74 (or Fe2(dobdc) or CPO-27-Fe) as reported by Lee et al.474 The left model contains an extended structure that is representative of a periodic system where the right two clusters consider representative fragments from the bulk material to study the electronic properties with DFT (88-atom model, originally reported in ref (475)) or higher-level wave function theory (19-atom model). In selection of the appropriate model size, there is an inherent trade-off between the level of theory used and system size. For example, Bernales et al. started with a 70-atom formate cluster for the Zr6 node of NU-1000, shown in Figure 18a, and condensed it to the smaller model given in Figure 18c.473 The geometry of the 70-atom cluster was optimized with DFT while the smaller cluster was used for higher level multiconfigurational calculations (CASSCF/CASPT2). Truncating the system size allows a higher-level of theory to be used but inherently neglects some of the features of the chemical system that can potentially be crucial for the reactivity. The use of hybrid QM/MM or QM/QM allows recovering the long-range effects but may not remediate the loss of the model accuracy due to the assumption of a single-site nature of the catalytic environment of the inorganic material.257 For more details on cluster modeling in computational catalysis with MOFs, we refer the reader to Rogge et al.466 and Odoh et al.457
Figure 17.
(Left) An extended structure for Fe-MOF-74 that represents the periodic MOF structure. (Right) Smaller representative clusters of Fe-MOF-74. Atoms Fe, O, C, and H are represented as green, red, gray, and white, respectively. Reprinted from ref (457). Copyright 2015 American Chemical Society.
Figure 18.

A representation of the (a) DFT modeled formate-cluster, (b) selected atoms for the smaller multireference clusters (the retained atoms are shown with balls and sticks, removed atoms are wires), and (c) the final small-cluster model used for CASSCF and CASPT2 calculations. Reprinted from ref (473). Copyright 2016 American Chemical Society.
3.3.2. Oxidative Catalysis
MOFs have been recommended as scaffolds for biomimetic active sites, such as iron(IV)-oxo intermediates similar to those found in heme and nonheme enzymes, which can oxidize strong C–H bonds. However, the proposed biomimetic active sites are often subject to decomposition,477,478 but it has been suggested that incorporating the system onto a solid support would help reduce this self-degradation mechanism. Conventionally, such systems were engineered using zeolites as the confinement matrices for stabilizing reactive transition metal complexes.251 In particular, the copper and iron-containing zeolites have been considered the heterogeneous analogues of enzymatic and molecular oxidation catalysts.343,479,480 However, the practical zeolite-based catalysts usually exhibit heterogeneity of the extraframework transition metal sites with a substantial fraction of the introduced species being catalytically inactive or contributing to undesirable reaction paths. For Fe-zeolite systems, the catalytically active sites were proposed to be the coordinatively unsaturated Fe(II) cations.479,481 Inspired by the success of the zeolite-based catalysts, the introduction of Fe(II) into MOF structures has also been attempted, although most synthetic methods did not produce coordinatively unsaturated sites that are needed to form the Fe(IV)-oxo active species.482−484 Xiao et al. used N2O to oxidize Fe2(dobdc) and a magnesium-diluted Fe0.1Mg1.9(dobdc) for the formation of the nonheme Fe(IV)-oxo active sites necessary to catalyze the selective oxidation of ethane to ethanol inside the pore of the MOF.485 The electronic structure of the biomimetic site and the catalytic reaction promoted by the Fe0.1Mg1.9(dobdc) for ethane functionalization was the subject of several computational studies.476,485 Periodic DFT calculations using the PBE+U42,486 functional were performed for optimization of the full structure of the pure Mg material Mg2(dobdc), since it shares more similarities with the diluted Fe0.1Mg1.9(dobdc). The periodic structure was condensed to an 88-atom cluster that contained three coordinated cations (Figure 19, upper row). The central Mg2+ was substituted by a Fe2+ cation for the efficient study of a single Fe active site (Figure 19, lower row). The Fe-containing 88-atom model based on an optimized periodic structure allowed an adequate representation of the first and second coordination sphere of the reactive site, while it included the electrostatic effects between reactants and organic linkers of the MOF. Cluster DFT calculations with the M06-L functional and multiconfigurational CASPT2 calculations were performed to determine the ground spin state of the nonheme Fe(IV)-oxo group. Although most mononuclear synthetic nonheme model complexes have S = 1 ground spin states, it has been shown that oxidation is energetically more accessible via the high-spin S = 2 Fe(IV)-oxo pathway, increasing the interest in synthesizing Fe(IV)-oxo sites with a quintet ground state.487,488 All different levels of theory agreed on the quintet ground state with an Fe–O bond length of 1.64 Å, which is consistent with previous high-spin nonheme Fe(IV)-oxo units.489−491 Verma et al. employed the same methods to study the reaction mechanism of the oxidation of ethane over the mixed-metal Mg–Fe MOF.476 The M06-L functional was used to calculate the three main steps of the catalytic cycle: (1) N2O oxidant activation over Fe(II) to form the Fe(IV)-oxo site, (2) H-atom abstraction from alkane to form a Fe(III)-hydroxo intermediate, and (3) ethyl radical rebound around the Fe(III)−OH moiety to yield ethanol product and regenerate the initial Fe(II) site. SA-CASSCF/MS-CASPT2 calculations elucidated the relative stability of the two possible reaction channels of the Fe(IV)-oxo site (σ and π channels).492−494 At the same time, Hirao et al. computed the reaction mechanism by the same MOF using QM/MM calculations.495 The reaction barriers obtained using the different computational approaches were in very good agreement.
Figure 19.

An 88-atom model cluster was obtained from the geometry of Mg2(dobdc) MOF optimized at the periodic DFT level (upper row). Substitution of the central Mg2+ cation by Fe2+ for the formation of a Fe-containing 88-atom model (lower row). Reprinted from ref (476). Copyright 2015 American Chemical Society.
The coordination environment of the Fe(IV)-oxo centers such as those found on Fe-MOF-74 strongly impacts their catalytic activity. It has been argued that increased catalytic performance is observed for the high-spin S = 2 ground state Fe(IV)-oxo site which is stabilized by a weak ligand field.496 A follow-up study based on high-throughput computational screening of the Computation-Ready Experimental (CoRE) MOF database497 that contains more than 5000 already synthesized materials identified five more materials that can potentially stabilize the high-spin Fe(IV)-oxo intermediate.498 The computational methodology included two steps: (1) the screening of the CoRE database for the identification of Fe2+ cations with exposed sites in their coordination sphere and (2) DFT calculations of the reaction barrier that leads to the Fe(IV)-oxo formation (i.e., the N–O dissociation of the oxygen-atom donor N2O). The latter was considered as a descriptor of the catalytic activity based on the previous study on the oxidation of ethane to ethanol by Fe0.1Mg1.9(dobdc). Similarly, Liao et al. studied hypothetical mononuclear Fe(IV)-oxo models with differing coordination environments that can be introduced as building units of MOFs and reported C–H activation barriers promoted by the Fe(IV)-oxo unit.499
A conceptually similar strategy of stabilizing reactive Fe species in mixed-metal MOF matrices has been recently employed by Osadchii et al.500 to design MIL-53(Fe, Al) catalysts for the selective methane oxidation with H2O2 in the liquid phase. Periodic DFT calculations revealed that site-isolation is crucial for stabilization of the microporous matrix under the reactive conditions. Single- and dual-site O-bridged Fe complexes embedded in the octahedral structure-forming AlOx chains were found to be sufficiently stable toward leaching in an oxidizing H2O2 environment, while at the same time allowed a low-energy reaction path to establish the selective methane oxidation to methanol (Figure 20). In line with the experimental observations, pure Fe-MIL-53 materials undergo a facile reaction with H2O2 that ultimately results in a complete degradation of the crystalline matrix. A follow-up dedicated computational study on this system501 showed that the high selectivity of the oxidation reaction over MIL-53(Al,Fe) to mono-oxygenated CH3OH and CH3OOH products is ensured by the limited coordination freedom of the isolated Fe species within the extended AlOx chains, which effectively prevents the overoxidation paths prior to product desorption from the active sites.
Figure 20.

(a) Proposed mechanism of methane to methanol oxidation with H2O2 over the dimeric Fe site in MIL-53(Al,Fe) and (b) the DFT-computed minimum-energy reaction path diagrams (including spin-transitions indicated with stars) for the catalytic process over different Fe-containing MIL-53 models. Reprinted from ref (500). Copyright 2018 American Chemical Society.
Stability of MOFs in oxidative environments is an important aspect that, to a large extent, determines their practical applicability.502,503 The structure of the MOFs as well as the nature of both the metal and ligands are crucial parameters determining the stability of the hybrid material. For example, Mg-MOF-74 is structurally robust in the presence of water vapor, whereas under similar conditions Ni-MOF-74 undergoes hydrolysis resulting in considerable damage to the structure.504 Similarly, Mn-MOF-74 is also susceptible to hydrolysis that ultimately yields complete structural deterioration.505 However, upon introduction of defect sites with missing organic linkers, the stability could be substantially improved so that the material could be used as a catalyst for the oxidation of alkyl aromatics.506,507 Electronic structure calculations can shed light onto the factors that control the stability of these materials under reactive conditions. For example, Khramenkova et al.508 employed a cluster model approach to investigate stability of dimeric Mn(II)-carboxylate MOF building blocks toward the direct and oxidation-induced hydrolysis. The computational results suggested that although materials containing such structure-forming units can show a pronounced stability in aqueous environments, their structural integrity can be readily compromised in the presence of reactive oxidizing species such H2O2 or even O2.
The Cu-modified NU-1000 [Zr6(μ3-O)4(μ3-OH)4(OH)4(OH2)4]8+ MOF has been identified as a promising catalyst for the selective methane oxidation under mild reaction conditions, although only a fraction of the introduced Cu ions contributed to the catalytic performance.509 In order to screen the effect of different metal dopants on the catalytic performance, Pahls et al. performed DFT calculations on the activity of different Cu-M (M = Fe, Co, Ni, Cu, Zn) bimetallic functionalized inorganic nodes of NU-1000 with 1,3,6,8-tetrakis(p-benzoate)pyrene for the conversion of methane to methanol.510 The activation free energies for the C−H activation over Cu(II)-M(II) range from 47.2 to 19.6 kcal mol–1, with the following order: Fe > Co > Ni > Cu > Zn. The activity of the reaction is shown to be linked to the oxyl character of the ligand, and therefore, the free energies were compared with the spin densities of oxygen atoms. Higher spin densities were correlated with lower activated free energies, which suggests species with more spin density located on oxygens have a stronger oxyl character and provide lower reaction barriers.
The NU-1000 family has also been used in dehydrogenation catalysis.511,512 Similar to the aforementioned study, the reactivity of first-row transition metals supported on a NU-1000 node were computationally evaluated for the acceptorless dehydrogenation of cyclohexanol by Ortuño et al.513 The reactivity was evaluated with the M06-L functional and the SMD514 solvation model using a 149-atom cluster model of the MOF node that contained a single reactive 3d transition metal site. Calculations predicted Co(II) and Ni(II)-containing catalysts to be most active in the title reaction with reactivity ca. 2000 times higher compared to the parent Fe(II)-containing catalysts at 298 K.
On the basis of the work of Ye and Liu,515 Ketret et al.516 performed a DFT mechanistic study on M3(btc)2 MOFs (btc = benzene-1,3,5-tricarboxylate, M = Cr, Fe, Co, Ni, Cu, and Zn) to evaluate their suitability to oxidize CO with NO2. This study was based on the efficient CO oxidation catalyzed by several MOFs, including Cu3(btc)2.485,517−519 The M3(btc)2 node was evaluated through a cluster containing a metal–metal paddlewheel-type structure with four btc linkers by means of broken symmetry DFT520 using the M06-L functional. The reaction mechanism was studied by accounting for different spin states of Cu and Fe. The mechanistic analysis was extended to related systems containing Cr, Co, Ni, and Zn sites, and the computational screening suggested the Cr3(btc)2 as the most active catalyst for the oxidation of CO by N2O.
3.3.3. Reductive Catalysis
Ni-containing zeolites are active catalysts for small olefin activation and oligomerization with isolated nickel sites being considered as the active species.521,522 The incorporation of similar catalytic functionalities into MOFs provides a better control over the location and coordination environment of the Ni centers, and could produce promising alternatives to the established zeolite-based systems. Ni-MOF-74 has been successfully employed as a catalyst for propene oligomerization.523 In a search for further improved MOF-based catalysts, Li et al. investigated the activity of single-site Ni species on NU-1000 MOF in ethylene hydrogenation and oligomerization.524 First, nickel deposition in the node was evaluated to determine the distribution of the nickel over the nodes. Experiments showed that a practical catalyst contains on average four Ni atoms per node. Calculations clearly evidenced that although multiple Ni ions can be stabilized on same nodes, there is a strong thermodynamic preference for the site-isolation. It is ca. 10 kcal mol–1 more favorable to coordinate Ni atoms to different faces of the same node than to form Ni pairs. Thus, the reaction mechanism of ethylene hydrogenation was studied using a node with Ni deposited on a single face of the node. The postulated mechanism starts with the generation of a nickel hydride active site and subsequent removal of two waters via a a highly activated (Eact = 43.2 kcal mol–1) and endothermic (ΔE = 40.3 kcal mol–1) two-step process. The activated Ni complex promotes ethylene hydrogenation with a rate-determining step activation energy of only 4.9 kcal mol–1.
Another important reaction enabled by MOF-based catalysts is the reduction of NO, an atmospheric pollutant commercially removed from exhaust by vanadium catalysts with high operating temperatures and high environmental toxicity.525 Experimentally, the MIL-100-Fe MOF shows excellent performance in selective catalytic reduction at lower temperatures than the commercial vanadium catalysts with adequate stability.526 However, the mechanistic details of the catalytic reaction are not fully understood. Zhang et al.527 proposed a mechanism for the reduction of NO on the MIL-100-Fe MOF that involves an intermediate oxidation step to convert NO to NO2, which subsequently reacts with NH3 in a selective catalytic reduction process (Figure 21). DFT calculations at the PBE-D2 level of theory42,63 were carried out on a cluster model containing three iron centers and extended organic linkers truncated to benzene groups. The direct reduction of NO by NH3 was found to be very unfavorable, confirming the need for the intermediate oxidation of NO to NO2 as suggested by previous studies.528,529 The NO oxidation occurs via a direct coupling with the Fe–O2 intermediate to form a nitrate species, which upon the reaction with another NO molecule yields two NO2 molecules. This process is overall exothermic (−46.6 kcal mol–1), with the largest reaction barrier arising from the cleavage of the O2 bond (24.4 kcal mol–1). The reduction of NO2 to N2 and H2O occurs via two unique mechanisms: the first involves the interaction of Fe as a Lewis acid with O2, which generates a Brønsted acid on an adjacent Fe-coordinated oxygen. Interestingly, the Brønsted acid catalytic site reacts with NH3 to generate NH4+, which subsequently produces N2 and H2O and regenerates the Lewis acid site. The largest reaction barrier was found to be the formation of the NH2NO intermediate at approximately 37 kcal mol–1 and was determined to be the rate-limiting step. The authors note the reaction is also hindered by competition at the metal site between reactants, where O2 is adsorbed at the active site (for NO oxidation) much weaker than NH3 (reduction step), as has been reported for other Fe catalysts as well.530,531
Figure 21.
Proposed reaction mechanism of the NO oxidation (red cycle) selective catalytic reduction of NO (blue cycle). Reprinted with permission from ref (527). Copyright 2018 Royal Chemical Society.
The utilization of MOFs as the catalysts for the conversion of CO2 to other value-added chemical products has received significant attention in the last years.532,533 Maihom et al.533 performed a computational study on the reactivity of a Cu-alkoxide functionalized MOF-5 for CO2 hydrogenation to formic acid. The Cu-MOF-5 includes Zn4O clusters connected by 1,4-benzenedicarboxylate (BDC) linkers where a benzene ring in the linker has a Cu alkoxide moiety. The calculations were performed on a truncated system that included two Zn4O clusters connected by a single Cu-alkoxide functionalized BDC linker. The structural rigidity of the crystalline matrix was approximated by fixing the terminating hydrogen atoms of the cluster to their initial positions. The effects of electron donating and electron withdrawing group functionalization on the linkers on the reactivity of the Cu sites was investigated. It was found that adding NH2 (electron donating) functional groups to the catechol ring decreased the largest reaction barrier of CO2 reduction from 24.2 to 20.1 kcal mol–1. Similar metalated catecholate linkers have been examined computationally for their performance in gas separation processes.534 In particular, certain metal–catecholates were identified that can activate N2, a procedure which holds great promise for ammonia synthesis.
3.3.4. Role of Structural Defects in Catalysis by MOFs
Although MOFs are commonly regarded as well-defined crystalline materials represented by an infinite periodic repetition of atoms in space, this idea of an “ideal crystal” is not truly accurate due to structural irregularities and deformities present in real crystals.467 Such defects can be local (e.g., metal or linker vacancies and dislocations) or larger structural defects such as voids within the crystal matrix and the presence of a different phase within the material.466,535 Most computational efforts used to evaluate the catalytic activity of MOFs consider a defect-free material as determined through experimental crystal structures. However, such models generally neglect how the performance of the material may be altered by the presence of structural defects. These defects can significantly modify the catalytic activity and, in some cases, can even lead to enhanced reactivity.536,537 For an in-depth discussion of the effects of structural defects on MOFs, we refer the reader to the reviews of Fang et al.467 and Ren et al.538 This subsection will only highlight computational studies for the evaluation of defect-sites on MOFs for catalysis applications.
As previously mentioned, the modified NU-1000 MOFs show applicability in a vast array of catalytic reactions utilizing a plethora of metals. To understand the catalytic activity of these hybrid materials, the detailed atomistic insight into the local structure and nature of the reactive species at the Zr6O8 nodes is necessary.539 Platero-Prats et al.540 performed ab initio molecular dynamics (AIMD) and DFT calculations on NU-1000 to model the localized metal site distortions observed at experimental conditions that were linked to increases in reactivity.541 The study was performed on the Zr6(O)8 node of the NU-1000 MOF, where the metal nodes were capped with formate anions and optimized with DFT to generate starting models for subsequent dynamics simulations. It was found that at low temperatures, only proton transfer can occur between adjacent H2O and −OH ligands of the node. As temperature increases, H2O ligands can dissociate from the metal node, and at even higher temperatures, ligand coordination can be affected (Figure 22). After observing water dissociation in the dynamics simulations, the dehydrated Zr6(O)8 nodes were geometrically optimized via cluster and periodic DFT to determine how the stability of the node was affected. The free energy of the dehydrated nodes was found to be between 36 kJ mol–1 (periodic PBE) and 67 kJ mol–1 (M06-L single node) lower than the hydrated node. These calculations provide theoretical evidence for structural transitions found at mild reaction conditions that can significantly impact electronic structure and reactivity of Zr and Hf-based MOFs.
Figure 22.
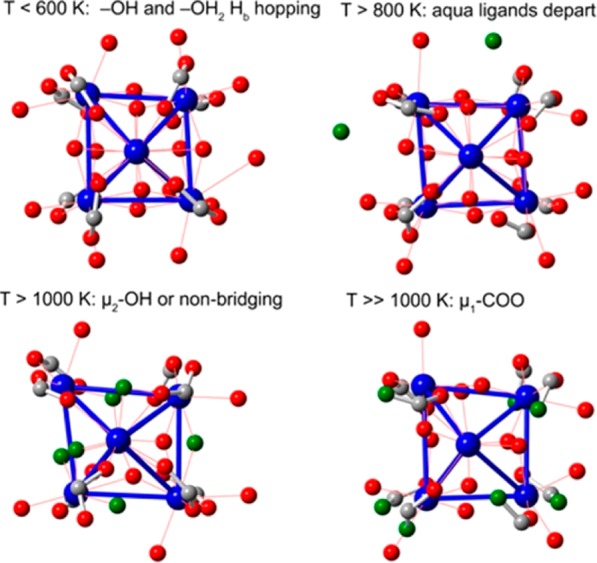
Snapshots from the molecular dynamics trajectories simulating the distortion of the Zr6(O)8 metal node of NU-1000. Reprinted from ref (540). Copyright 2016 American Chemical Society.
The dehydration of ethanol to diethyl ether over undercoordinated defect sites at the Zr6(O)8 metal node site within UiO-66 and UiO-67 MOFs was computationally studied by Yang et al.542 A combined experimental and DFT study was carried out to elucidate the identity and nature of the defect sites. These studies have concluded that the ligand vacancies in MOF structures are capped primarily by aqua and hydroxyl groups.543,544 However, DFT calculations show that capping the vacancies with formate is more thermodynamically favorable. Infrared spectroscopy confirmed the computational prediction and thus, additional treatment would be necessary to prepare aqua or hydroxyl caps. Several mechanisms were evaluated for the formation of diethyl-ether, considering one and two site vacancies and several reaction pathways. During the reaction, the formate groups can be replaced by ethoxy groups, and the most stable reaction pathway is established when two adjacent defect sites act cooperatively in the catalytic reaction.
In addition to metal sites, organic linkers can have defects that significantly affect the electronic structure of the system. Experimental studies indicate the importance of ligand defects for the catalytic activity.545−548 To demonstrate the impact of linkers on the catalytic properties of MOFs, Fang et al.549 studied a Cu3(btc)2 (btc3– = 1,3,5-benzenetricarboxylate) MOF, also known as HKUST-1, to determine how incorporation of defective linkers alters the interaction of the Cu sites with CO. The system was studied using a QM/MM model where the QM system includes the paddlewheel and adjacent aromatic rings, and the extended periodic structure was treated with the MOF-FF force field.550 Calculations were performed on the unmodified structure, and the one formed by the replacement of the btc linker by pyridine-3,5-dicarboxylate (pydc). It was found that the incorporation of the pydc linker leads to a Cu2+/Cu1+ paddlewheel structure opposed to the defect free Cu2+/Cu2+ paddlewheel, which is corroborated by a calculated redox potential that is 2.1 eV lower for the defective paddlewheel. For CO adsorption, the defect-free MOF can bind a CO at each Cu site with subsequent interaction strengths of −10.7 and −9.4 kcal mol–1. CO binds stronger on Cu1+ sites (−28.1 kcal mol–1), while the Cu2+ site binds the second CO with an interaction of −9.6 kcal mol–1. Due to the stronger interaction of CO at the defect-linker structure Cu1+ site, it was experimentally determined that this CO does not react with O2 to form CO2. Further computations on potential linker defects showed that significant electronic structure and reactivity changes are made upon defect formation in MOFs.
3.3.5. MOF Catalyst with Metalloporphyrin Active Sites
Many examples of heterogeneous catalysts based on coordination polymers and MOFs featuring a metalloporphyrinic ring as an “organic” ligand have been reported so far. These systems deserve a separate discussion here as their computational examination becomes demanding due to the size of the molecular model or unit cell that should be used.464,551 Those materials can be considered as biomimetic catalysts and can perform oxygen evolution reactions (OERs). The electronic structure and reactivity of metal-porphyrins have been thoroughly studied,552 but their catalytic performance can be enhanced when they are introduced inside MOFs.553 Mandal et al. have examined such materials with periodic DFT to evaluate the electronic, sorption, and magnetic properties of the binuclear M1-porphyrin/M2-node MOF (M1, M2 = Fe or Co).554 Those materials exhibit a complex electronic structure that involves a ferromagnetic coupling between ligand and node metals. The oxygen generation of the heterobimetallic MOFs is two times higher than that of metalloporphyrins and was associated with spin-polarized Dirac bands that induce higher electron mobility, an important aspect for an efficient four electron OER.
Roy et al.555 have calculated the initial catalytic enhancement of the acyl-transfer reaction between 3-pyridylcarbinol and N-acetylimidazole by a Zn-porphyrin incorporated in the framework of a Zn-acetate-based MOF.556 The specific MOF has attracted attention since the initial rate of an acyl-transfer reaction is enhanced by a factor of 236. The rate was calculated by introducing to a kinetic model computed binding energies and dissociation constants of reactants and products by means of DFT and equilibrium statistical mechanics.
3.3.6. Summary
Most of the computational studies on MOF-based catalysts discussed above focused on the analysis of a single proposed catalytic cycle. By evaluating reaction energy diagrams for a selected mechanism over varied active sites in a single type MOF material, proposals were made regarding the potentially superior catalytic materials. We should note here that catalytic performance is not solely defined by the kinetic parameters of the desirable reaction route but by complex reaction networks combining other reaction channels besides the target catalytic path, resulting in nonselective conversions or catalyst degradation. As an example, we report the work of Li et al.,512 who used theory to compute the relative energies of the desired oxidative dehydrogenation reaction and the undesired hydrocarbon oxidation. The computational analysis of such processes is much less common in the literature. Yet, we argue that such information is crucial for the understanding of catalytic phenomena and for guiding the development of improved catalyst systems. Having said this, one should realize that the deactivation and nonselective chemistry is in general much more versatile than the selective catalytic conversions and at the same time much less understood. This makes the computational studies of these processes not only much more demanding in terms of computational resources but also very challenging in view of the lack of reliable initial hypotheses regarding the preferred reaction channel and underlying mechanisms.
3.4. Mechanistic Complexity of Multicomponent Reaction Systems: An Illustrative Case of Cross-Coupling and C–H Functionalization Catalysis
Previous sections presented selected examples of multisite effects in molecular catalysis with 3d transition metals that contribute to increased mechanistic complexity. Most of the catalytic reactions in solutions are intrinsically multicomponent. Despite the availability of information regarding the structure and nature of the starting compounds in the homogeneous catalytic systems (e.g., precatalyst or other experimentally refined intermediates), these can be dramatically different from those formed after the activation step or in the course of the catalytic reaction. Therefore, the mechanistic proposals need to account for the multicomponent nature of the systems and the evolution of catalytic species. Below we will discuss these concerns, as well as issues regarding the model and method accuracy for studying catalytic reactions by 3d transition metals on the example of cross-coupling reactions. The questions of transferability of the mechanistic concepts derived for noble metal-based systems to those based on cheaper 3d transition metals will be emphasized.
The emergence of cross-coupling reactions, Buchwald-Hartwig amination and C–H functionalization revolutionized organic synthesis in the last five decades. Although the role of these reactions in industrial synthesis has not yet reached the levels anticipated by academic researchers, it still proves to be very significant.557−561 These reactions deliver the synthesis of complex molecular structures that are simple and efficient in terms of yields and selectivity.562 These cross-coupling and functionalization reactions are typically catalyzed by Pd complexes. The reason for the dominance of Pd is the subtle balance between stability and activity of the catalysts.563 Palladium complexes are closed-shell species that can easily be characterized with NMR spectroscopy and have low ligand lability while may deliver TON’s up to ∼105 in the so-called “homeopathic” regime.564,565 The lighter group 10 element Ni is considered as overreactive,563 and the rational development of Ni catalysts has stalled due to the lack of bench-stable catalytic systems.566
Nowadays, however, there are Ni analogs for the vast majority of cross-coupling and functionalization Pd catalysts,563,567 while the development of Fe and Co catalysis is an emerging field as well.568−571 The transition from Pd- to Ni- and Fe-catalysts is appealing due to high and, moreover, qualitatively unique reactivity of 3d transition metals in, for example, activation of R-X-molecules with sp3-organic electrophiles and (pseudo)halides (X-) as carbamate-, sulfamate-, carbonate-, Cl-, and even F-groups.566,572−578 Catalysis offers the increase of chemical synthesis output, reducing the corresponding material and energy costs at the same time. However, the development of 3d metal-based analogs of Pd, Pt, and other noble metal catalysts offers a viable path to more sustainable organic synthesis.579 Nickel in its elemental form has 2000 times lower cost than Pd,574 and the NiCl2 salt, often used as a catalyst precursor, is 58 and 322 times cheaper than PdCl2 and PtCl2 analogs, respectively.563 The issues of Pd catalyst leaching and subsequent environment contamination,580 as well as platinum group metal toxicity which leads to the low margin of 10 ppm in pharmaceuticals,581 are well-recognized. Although the transition to Ni and Co compounds for catalysis may be problematic due to toxicity, Fe toxicity should not be an issue.582−584
Due to the above-mentioned interest for the transition to 3d metal homogeneous catalysis, there is a growing number of reviews on synthetic aspects of C–C bond formation and functionalization reactions catalyzed with Mn, Fe, Co, and Ni.568,582,585−590 However, these reviews do not comprehensively cover the computational studies performed on this topic. Notable exceptions are the reviews on homogeneous organometallic catalysis including Ni,11 and on Ni-catalyzed cross-coupling reactions with esters as pseudohalide coupling partners.591
As was discussed in section 2, good mechanistic models in a computational catalytic study should capture not only the essential physics (governed by the method accuracy) but also the relevant chemical phenomena on the molecular level as well (model accuracy). State-of-the-art quantum chemical calculations are believed to be on par with experimental methods regarding their accuracy and may help in correcting possible errors in experimental results.592 However, it may happen that computational modeling correctly reproduces the experimental observations due to error cancellation by the employed methods (“physics”) or due to an incomplete mechanistic model (“chemistry”), or both. Therefore, agreement between computational and experimental results may be misleading if computations reproduce the experiment on a wrong basis.257 This subsection specifically aims to provide a guide for performing computational studies of 3d metal-catalyzed reactions by discussing in detail the recent progress in theoretical studies of cross-coupling reactions, the Heck reaction, direct C–H arylation, and Buchwald-Hartwig amination. Special attention is paid to the phenomena that deviate from the conventional single-cycle mechanisms with electronically closed-shell intermediates and two-electron chemistry.
Here, Ni- and Fe-based metal-complex catalysts are considered as well as rarer Co, Cr, and Mn species, which promote catalysis via similar mechanisms. In section 3.4.1, we discuss the general mechanism of cross-coupling, Heck, amination reactions, and how the individual mechanistic steps may change upon the transition to 3d transition metal catalysts. In section 3.4.2, an overview of the general theoretical models which help to estimate the catalytic efficiency or reactivity in cross-coupling and functionalization reactions is presented. In section 3.4.3, we highlight the nonclassic mechanistic events that lead to the necessary shift from the single-cycle catalytic models. We also tangentially highlight the computational and experimental results of the studies regarding Pd catalysts that are relevant to the understanding of 3d metal-catalyzed cross-coupling and functionalization reactions.
3.4.1. Cross-Coupling, Heck, C–H Arylation, and Amination Reactions
The conventional mechanism of homogeneous cross-coupling and functionalization reactions is a sequence of well-defined organometallic microreactions (Figure 23a). The classic cross-coupling catalytic cycle (well-known form the homogeneous Pd-catalysis, Figure 23b) begins with the oxidative addition (OA) of an organic electrophile R-X to a transition metal complex, which proceeds with a transmetalation step and ends with the reductive elimination (RE). In the case of 3d metal catalysis, the transmetalation and OA steps may often proceed in reverse order (Figure 23c). The catalytic cycle in amination reactions includes OA and RE steps as well (Figure 23d). This mechanistic modularity makes cross-coupling and functionalization reactions especially appealing for rational catalyst design through computational modeling; however, one should keep in mind the possible complications associated with spin-transitions and off-cycle intermediates (vide infra). Despite the fact that many catalytic systems have reserved their place in the synthetic organic chemistry toolbox, the mechanistic details underlying their high efficiency are often quite unclear. For example, cobalt-catalyzed cross-coupling reactions are already a widely employed synthetic method,589 though many studies report excellent yields with Co-catalysts lacking a clear mechanistic model.593−595 Manganese complexes with N-heterocyclic carbene ligands show suboptimal catalytic activity in, for example, Kumada cross-coupling of phenylmagnesium cloride and bromocyclohexane,596 and clarification of the mechanism through quantum chemical modeling could be of help in further development of superior Mn-based catalysts.
Figure 23.

First-row transition metals (Cr, Fe, and Ni) in coupling reactions. (a) The key mechanistic steps and (b) the generalized mechanism of cross-coupling reactions. Representative mechanisms proposed for the (c) Fe-catalyzed coupling of Grignard reagents with organic halides,603 (d) the amination reaction catalyzed with an Ni-NHC complex,604 and (e) the Ar–Ar’-coupling reaction catalyzed by Cr-complexes.605.
3.4.1.1. Common Catalyst Precursors, Ligands, and Proposed Catalytic Species
Both Ni and Pd precatalysts in oxidation states from (0) to (II) and in the form of salts or complexes are readily available.566,597 Notably, L[Ar-NiII-X]L-complexes (L = (bidentate) amine or phosphine ligand, Ar = o-Tol) are used as precatalysts598−601 while also being a key intermediate species in the catalytic cycles of cross-coupling-, amination-, and C–H arylation reactions (Figure 23). In the case of iron precatalysts, stable Fe(II) and Fe(III) salts or their complexes are used.569 Transition metal salts with halide or acetylacetonate anions are a common choice.
Normally, reduction of the transition metal cation M(II) or M(III) in salt precursors to a M(0) or M(I) state is necessary to initiate a catalytic process. The reduction may proceed through the formation of M(+n)R2-species as a result of a double transmetalation process (Figure 23a) and subsequent reductive elimination of organic molecule (R-R) from the M(+n)R2-species. Another well-studied Pd precatalyst case is the formation of the active catalytic species through Pd(II) salt reduction by a phosphine (PR3) ligand resulting in the formation of phosphine oxide (O = PR3).563,601 Precatalyst activation via base-assisted allyl dissociation was considered in the computational study of the Suzuki-Miyaura cross-coupling catalyzed with [M(NHC)(allyl)Cl] (M = Ni, Pd) complexes. The activation of the Ni precatalyst proceeded with energy barriers 2.0–15.0 kcal mol–1 higher than in the Pd case.602
The choice of the ligand is one of high importance in the design and optimization of transition metal-based catalysts for cross-coupling and functionalization reactions. The costs of specialized ligand synthesis are often comparable to the costs of Pd salt precursor566 which hinders the economic efficiency of 3d transition metal catalysts. Ligand-free Pd- and 3d metal-catalysts (often based on metal nanoparticle catalyst precursors) were therefore proposed. The utilization of such catalytic systems may give rise to the so-called dynamic catalysis with highly complex mechanisms (vide infra). Strong-field ligands such as cyclopentadienyls, phosphines, and N-heterocyclic carbenes are common choices in catalyst design for C–C bond formation and functionalization reactions. These ligands stabilize the low-spin states of transition metal complexes,566,606 leading to catalytic processes proceeding over the low-spin potential energy surface (PES). Progress in homogeneous organometallic catalysis elucidated the role of spin transitions and high-spin complexes in C–C bond formation and functionalization reactions, which resulted in the use of nitrogen-containing heterocycles such as bipyridine (bipy), terpyridine (TPy), and NNN-pincer ligands. This is especially true for iron and cobalt catalysts of cross-coupling reactions because transitions between spin states are often unavoidable and may profoundly alter the reactivity (vide infra).
During the last decades, the tremendous evolution of phosphine ligands such as monodentate PR3 (R = Ph, t-Bu, Cy, etc.) and bidentate (Xantphos, dppe, ferrocene-containing dppf, etc.),590 enabled them to be considered as classic ligands in homogeneous catalysis.566 Their properties can be fine-tuned for optimized activity of the corresponding catalytic systems. The metal to PR3-ligand ratio in a catalytic system also needs attention since excessive ligand concentration hinders their catalytic performance. Compared to Pd(II), Ni(II) centers bind phosphines less strongly (24.5 kcal mol–1 for Ni–P-bond energy versus 32.1 kcal mol–1 for Pd–P-bond energy in [L2M(II)X2] complexes), while for M(0)-species, the stability ordering is reversed (39.7 kcal mol–1 for Ni–P-bond energy versus 33.6 kcal mol–1 for Pd–P-bond energy in [L2M(0)] species).607 This is somewhat undesirable for the transition from Pd to Ni in catalysis because of the above-mentioned superior activity of low-coordinated complexes and the lower stability of [(PR3)2Ni(II)X2] precursors.
Special care should be taken in computational estimations of catalytic activity of phosphine-based systems since both bis- and monoligand catalytic species may be formed under reaction conditions. Moreover, the active catalytic species may be ligand-free as in the Heck reaction, where HX acid (formed in the course of the reaction) leads to the quaternization of the phosphines for the formation of counterions to X[ArPdX]2X2– and [ArPdX2]− anions.608 Phosphine ligand dissociation through oxidation to phosphine oxides and PR3-transformations through R-moiety substitutions are also well-known processes which should be considered in a detailed mechanistic analysis.609 Thus, ideally, the set of modeled catalytic species in a mechanistic study of Ni-catalyzed cross-coupling reactions should include bis-, monoligand, and ligand-free catalytic species to ensure the comprehensiveness and adequacy of the chemical model.
N-Heterocyclic carbenes (NHCs) are another common ligand type in homogeneous catalysis. Here, steric and electronic effects can easily be separated since electronic effects are induced by the center carbene ring, and steric hindrance may be increased, if needed, by the bulky substituents bound to the center ring.610 Additional fine-tuning of the electronic effects can be achieved by functionalization of the carbene ring such as, for example, in imidazolic IPr-ligands by Cl-group addition. Moreover, the introduction of amine-groups in the bulky substituents of the carbene ring caused subtle steric and electrostatic interactions in the coordination sphere.611 The steric effect of bulky substituents in the Suzuki cross-coupling reaction was evaluated at the RI-B97-D/cc-pVTZ//RI-B97-D/6-31G* level of theory (cc-pVTZ-PP basis for Pd, isopropanol solvation modeled with PCM approach).610 No evidence for the steric hindrance in the case of bulky but flexible alkyl groups bound to the NHC core was found, but noncovalent interactions (“steric attraction”, Figure 24) between the ligands and reactant contributed to the stabilization of the transition states.
Figure 24.
Concept of steric acceleration of chemical transformations. Adapted from ref (610). Copyright 2013 American Chemical Society.
The possible deactivation of catalysts stabilized with NHC-ligands should also be discussed. The decomposition of X[(NHC)PdR](pyr) (R = vinyl, ethynyl, Ph, Me, pyr = pyridine) species that are key intermediates in the cross-coupling and Buchwald-Hartwig amination reactions was shown to proceed through the NHC-R reductive elimination step (Figure 25) verified both computationally (at the PBE0/6-311+G(d)&SDD level of theory) and with real-time mass-spectrometry experiments.612 The activation barriers increased in the order of R = vinyl < ethynyl < Ph < Me, and the exceptional instability of X[(NHC)Pd(vinyl)]L was confirmed by real-time ESI-MS. A minor effect of the halide substituent X was found (3 kcal mol–1 difference between X = Cl vs X = I cases), while the bulky t-Bu-substituents bound to the carbene ring increased the NHC-R reductive elimination barrier by 6.4 kcal mol–1 compared to the case of the conventional Mes-group. This NHC-R-coupling led to the ligand-free Pd catalytic process in the Heck reaction with the involvement of Pd nanoparticles.613 The same NHC-R-coupling may be envisioned for X[(NHC)NiR]L intermediates, though the reductive elimination of organic species is considerably less facile in Ni(II) than in Pd(II) complexes (vide infra). The deactivation of Ni-NHC catalysts, however, readily proceeds by hydrolysis leading to NHC-H and Ni(OH)2 formation. The traces of water in organic solvents induce the hydrolysis of [(NHC)2NiX2] complexes. The stability of the complexes is determined by the nature of the halide group (stability decreases in the order X = Cl > Br > I) and that of the main ligand.614
Figure 25.

NHC-Pd catalyst deactivation through NHC-R reductive elimination modeled at the PBE0/6-311+G(d)&SDD level of theory (bulk solvent effects modeled with the SMD method). Adapted from ref (614). Copyright 2015 American Chemical Society.
Ligand-controlled stereoselectivity was observed in 3d metal catalyzed cross-coupling reactions, and two computational studies of Ni-catalyzed Suzuki coupling are exemplary in this regard. The reaction involving benzylic pivalates and the aryl pseudohalide as the coupling partner was modeled at the M06/6-311+G(d,p)&SDD//B3LYP/6-31G(d)&LANL2DZ level of theory (bulk solvent effects modeled with C-PCM),615 while the coupling with benzylic carbamates was modeled at the B3LYP/6-31G+(d)&LANL2DZ level of theory (bulk solvent effects modeled with C-PCM as well).616 The reaction with NHC-ligated (SIMes) Ni complexes led to inverted stereoconfiguration in the coupling product relative to the aryl pseudohalide, while the [Cy3PNi] species promoted the stereoretention. In both cases, the classic Ni(0)/Ni(II) mechanism was considered exclusively, and the OA step controlled the stereoselectivity. The OA step proceeded via an SN2-type TS (NHC-ligand, inversion) or via the classical concerted OA mechanism (PR3-ligand, stereoretention). A detailed discussion of model TS structures for the OA step is given further in text.
Cross-coupling reactions, direct C–H arylations, and functionalizations follow catalytic cycles where the metal center undergoes consecutive oxidations (OA steps) and reductions (RE steps). Thus, different oxidation and spin states should be considered for the understanding of the metallic center reactivity as well as for performing a thorough computational study. For Pd, this implies even-electron catalytic cycles on a closed-shell singlet PES as Pd(0)/Pd(II) and Pd(II)/Pd(IV), with the latter being common in C–H functionalization reactions. While Ni follows conventional even-electron Ni(0)/Ni(II) catalytic cycles in, for example, cross-coupling and amination reactions, the odd-electron Ni(I)/Ni(III) states are easily accessible and generally should not be disregarded in modeling studies. At the same time, the Ni(IV) oxidation state is quite uncommon.11,563 Cross-coupling reactions with Fe catalysts may proceed through a Fe(I)/Fe(III) cycle resembling classic Pd catalytic cycles or may involve nonclassic Fe(II)/Fe(III) or Fe(I)/Fe(II)/Fe(III) mechanisms depending on reaction conditions.571,603,617
To summarize, the structure of catalytic species may differ considerably from the precatalyst structure. Ligand nature may affect the activity and stereoselectivity of a reaction. It is highly desirable that monoligand-, bis-ligand-, etc. species are considered in a catalytic model along with the possibility of ligand dissociation. In the case of 3d metals, both even-electron (closed-shell) and odd-electron catalytic cycles may be operational. These factors make computational catalytic studies of 3d transition metal catalyzed cross-coupling, direct C–H arylation, and amination reactions a challenging task since multiple catalytic cycles should generally be evaluated to obtain in-depth, meaningful, and predictive modeling results.
3.4.1.2. Key Elementary Reaction Steps
In this subsection, the common elementary steps in cross-coupling, C−H functionalizations, and amination reactions are discussed in detail. OA is the key step in these reactions and, as in the case of Pd-catalysis, is also the rate-determining one.11 In this elementary step, an organic (pseudo)halide R-X binds to a transition metal center M(n), forming R-M(n+2)-X species. OA of arylhalides Ar-X to Pd proceeds through a three-center transition state in a synchronous manner; however, the process involving 3d transition metal species is often mechanistically more complex on the molecular level (Scheme 5). For this reason, only the three-center synchronous process is often called OA; however, we will designate all processes in Scheme 5 as OA when it introduces no ambiguity.
Scheme 5. Five Mechanisms of the Oxidative Addition Step. SET is the single electron transfer, and AT is the atom transfer. Adapted with permission from ref (603). Copyright 2015 Wiley-VCH.

The mechanism of the OA step is substrate-dependent. For example, it was shown by combination of competitive kinetic experiments and DFT computations that while arylhalides are added to iron species through a classic three-center mechanism in Kumada-type coupling, alkyl halides addition proceeds through an atom-transfer pathway.603,618 Atom-transfer-type OA was also suggested for Fe-catalyzed Suzuki coupling with alkyl halides.619 Arylhalide addition to a conventional Ni catalyst [Ni(dppf)] proceeds through a standard three-center mechanism. Notably, the nonstandard SN2-type (SNAr-type) OA mechanism was proposed in the case of alkyl halides and heteroarylhalides addition to [Pd(PPh3)2] on the basis of kinetic experiments and DFT calculations.620,621 Thus, one should undoubtedly expect the conventional mechanism in case of the Ar–X addition only.
Considering the relative reactivity of transition metal M(II) centers in OA, Ni complexes are more reactive than Pd ones, and two computational studies of the Heck reaction are exemplary in this regard. In the first one, the activities of the [M(PH3)2] (neutral pathway) and [Cl-M-PH3]− (anionic pathway, M = Ni and Pd) were estimated at the B3LYP/LANL2DZ+p//B3LYP/LANL2DZ level of theory (bulk solvent effects modeled with PCM), where only the closed-shell PES and the conventional three-center OA transition states were considered. For the neutral pathway, the addition of CH2=CHCl to [Ni(PH3)2] had a 5.4 kcal mol–1 lower activation barrier in vacuum compared to [Pd(PH3)2], while for the anionic case [Cl–Ni–PH3]− vs [Cl–Pd–PH3]−, the barrier in vacuum was 1.4 kcal mol–1 higher. However, the inclusion of bulk solvent effects (DMF solvent) lowered the barrier of the OA to [Cl–Ni–PH3]− by 1.6 kcal mol–1.622 In the second study, the activity of Ni and Pd catalysts with NHC-type ligand (NHC = N,N′-dimethyldihydroimidazol-2-ylidene) was evaluated. The molecular geometries were optimized at the M06/6-31G(d,p)&CEP-31G level of theory, and the energies were corrected by single point calculations with the same M06 functional and a basis set of triple-ζ quality with solvent effects modeled with the SMD method. Both mono- and bisligand Ni-NHC-complexes underwent OA of chlorobenzene with considerably lower barriers (by 3.2 and 5.3 kcal mol–1, respectively).623
The common reactivity order of organic halides (ArI > ArBr > ArCl) in oxidative addition to [Ni(dppf)] along with the reactivity of pseudohalides (pivalates, sulfamates, triflates, etc.) was determined in an experimental study.625 Ni complexes have distinguished reactivity in OA of pseudohalides compared to Pd,574,577 and a review article on the corresponding computational studies is available.591 It should be noted that pseudohalides such as carbamates, sulfamates, and pivalates may undergo OA through the characteristic five-center transition states.577,591,626 Computational data on the comparative reactivity of, for example, [Ni(PMe3)]627 and [Ni(dppf)]628 species in pseudohalide OA may be found elsewhere.
In Ni-catalyzed cross-coupling reactions, both Ni(0)/Ni(II) and Ni(I)/Ni(III) catalytic cycles are common, and the two-electron variation of the metal oxidation state is caused by OA and RE steps inherent to the catalytic cycles (Figure 26). The reductive elimination step is facile if the Ni center is in the 3+ oxidation state according to the classic experimental work of Morrell et al.629 and recent computational data.630 The superior activity of Ni(0) compared to Pd(0) in OA and the more facile reductive elimination from [R2Ni(III)Ln] species compared to the less oxidized [R2Ni(II)Lm] (and vice versa in the case of the OA step with Ni(0) vs Ni(I) species) may be rationalized in terms of electronegativity of the metallic center. Nickel is more electropositive than palladium, thus Ni(II) formation is more favorable than Pd(II) while Ni(III) is overoxidized, and its formation through the OA step is unfavorable. At the same time, the reductive elimination of organic species from the overoxidized Ni(III) complexes is facile. The Ni reactivity dependence on the oxidation state may clearly be demonstrated via the comparison of Ni(0)/Ni(II) and Ni(I)/Ni(III) catalytic cycles in chlorobenzene amination modeled at the B3LYP-XDM/6-311+G(2d,2p)//B3LYP-XDM/6-31+G*&6-31G* level of theory (Figure 26).624 In Suzuki-type coupling of MeBr with EtB(Me)2 modeled at the B3PW91/6-311+G(2d,p)&SDD//B3PW91/D95v(d)&LANL2DZ level of theory (dioxane solvation was modeled with the IEF-PCM method), a Ni(0)/Ni(II) and Ni(I)/Ni(III) cycle comparison showed that the former is highly improbable due to the RE step barriers as high as 37.7 and 57.5 kcal mol–1 for singlet and triplet states, respectively.630
Figure 26.
Chlorobenzene amination as modeled at the B3LYP-XDM/6-311+G(2d,2p)//B3LYP-XDM/6-31+G*&6-31G* level of theory: (a) Ni(0)/Ni(II) cycle, fast OA and slow RE; (a) Ni(I)/Ni(III), slow OA and fast RE. Adapted with permission from ref (624). Copyright 2017 Wiley-VCH.
The elimination of HX molecules, formally a reductive elimination step, proceeds through a different mechanism in which proton abstraction by a base molecule and subsequent X– dissociation from transition metal species occurs. Due to the above-mentioned low electronegativity of Ni compared to Pd, HX elimination is considerably less thermodynamically and kinetically facile in the Heck reaction catalyzed by [Ni(PH3)2] and [Ni(NHC)2] species rather than the Pd analogues, as was demonstrated in recent computational studies.622,623 These conclusions are independent of the neutral or cationic pathway followed for the Heck reaction.
The key step of the direct C–H arylations is the activation of Ar–H bond. This mechanistic step has formal similarity with Ar–X oxidative addition to transition metal centers. The activation of Ar–H bond by Ni, Pd, and Pt NHC-complexes was calculated at the M06/6-311G(d)&SDD//M06/6-31G(d)&LANL2DZ level of theory with bulk solvent effects modeled with C-PCM method.631 The authors showed that while Pd and Pt easily form the [(NHC)Ar-M-H(alkyl)] hydride complex, Ar–H activation on the Ni-NHC complexes proceeds through the direct aryl-to-alkyl transfer, with propene being the proton acceptor and the coupling partner, which is a consequence of the Ni–H bond weakness and the lower aryl-to-alkyl distance in the Ni species. It should be noted that in C–H activation by 3d transition metals, the so-called two-state reactivity phenomenon is common which makes the computational modeling of these rather challenging (vide infra).
At the molecular level, the [L–M]–Ar–X-species formation and the so-called ring-walking steps precede the OA step (Figure 27). Here, M–Ar–X binding occurs through the η2-coordination of Ar-moiety to the transition metal, rationalized with the Dewar-Chatt-Duncanson model,632,633 and the [L–M]–Ar–X species are now recognized as important pre-OA intermediates.11,634,635 The difference in affinity to olefinic double C–C bonds of group 10 metals is worth noting. Among [M(PH3)2] complexes (M = Ni, Pd, and Pt), [Ni(PH3)2] has ca. 2 and 1.5 times higher affinity to ethylene compared to Pd and Pt counterparts (BP86/TZP level of theory).636 The outstanding Ni affinity to olefins also holds in bis-NHC-complexes such as [Ni(H-Im)2], which has ca. 4 and 5 times higher affinity to ethylene than the Pd- and Pt-containing counterparts, respectively. The differences in ethylene affinity are due to the activation strain (i.e., rigidity, vide infra) caused by the binding to [M(H–Im)2] species.637 The ring-walking in Ar–X–[Ni(dppp)] intermediates (Ar–X = 1-bromo-2-methylbenzene, 2-bromopyridine, 2-bromo-3-methyl-thiophene, and 2-bromopyrrole, dppp = 1,3-bis(diphenylphosphino)propane) was extensively investigated with DFT calculations at the B3LYP-D3/TZ2P&LANL2TZ-(f)&LANL08d level of theory and kinetic isotope effect experiments.634,635 The different ring-walking mechanism upon Ar–X variation was reported; for example, the [Ni(dppp)] fragment weaved along the edges of the benzene and pyridine rings and cut through the interior of the thiophene ring. All Ar–X–[Ni(dppp)] intermediates had high thermodynamic stability in solution, and the ring walking step ended with the highly exergonic OA event (Figure 27).
Figure 27.

Ring-walking of Ni(dppp) and the corresponding oxidative addition of 1-bromo-2-methylbenzene (solid red, X = C–CH3) and 2-bromopyridine (dashed blue, X = N); energies are given relative to the separated catalyst and substrate and were computed at the B3LYP-D3/TZ2P&LANL2TZ-(f)&LANL08d level of theory. Adapted from ref (635). Copyright 2017 American Chemical Society.
Another important mechanistic step in C–C bond formation reactions is the β-hydride elimination. The facile β-hydride elimination is also necessary for the efficient Heck reaction catalysis. This step is known to proceed slower with Ni(II) complexes than with Pd ones.563 In accordance with the results of the computational studies on the Heck reaction discussed above, both [(vinyl)2Ni(PR3)2] and [(styril)(Ph)Ni(NHC)2] species undergo β-hydride elimination with formation of butadiene and 1,2-biphenylethene ligands with activation barriers higher compared to their Pd counterparts.622,623 Similar stability toward β-hydride elimination was shown for Ni(I) species. Ethylene elimination from [EtNi(I)L] complexes (L = 2,6-bis((R)-4-isopropyl-4,5-dihydrooxazol-2-yl)pyridine) was found to be endergonic by 20.8 kcal mol–1 and proceeds with a large barrier of 27.1 kcal mol–1 according to calculations performed at the B3LYP-D3/6-31G&LANL2DZ level (6-31G basis set was used for atoms that were not directly involved in the modeled reaction, 6-31G* on the light atoms, and LANL2DZ effective core-potential basis set for the transition metal).638 Migratory insertion is another step in the Heck reaction that proceeds with lower barriers in the Ni-catalyzed process.622,623
On the other hand, β-hydride elimination is an unwanted event leading to the formation of olefinic side-products in cross-coupling reactions as Suzuki or Negishi coupling. The elimination may be facilitated by β-agostic interactions. A study of trans effect on the strength of β-agostic interactions in Ni(II) complexes was conducted at the DFT level of theory and included thorough CCSD(T)/CBS reference calculations with the Douglass-Kroll-Hess quasi-relativistic Hamiltonian. Strong π-acceptor ligands weakened the interaction in “1-trans” intermediate, while only a modest effect was observed in “2-trans” intermediates (Scheme 6).639
Scheme 6. Effect of Agostic Interactions in β-Hydride Elimination Step in Suzuki Coupling within Ni(0)/Ni(II) Cycle.

Adapted from ref (639). Copyright 2014 American Chemical Society.
Cross-coupling and functionalization reactions with Pd catalysts are represented by a chain of well-known mechanistic steps and thus are an appealing object for modeling and catalyst design through computations. However, caution should be taken upon the transition to 3d metal catalysts. The kinetic feasibility and thermodynamics of the individual steps may vary significantly. Moreover, the mechanism of the steps may change as well. Good practice in predictive catalytic process modeling would involve a proper sampling of alternative pathways. One should always keep in mind that the feasibility of each pathway can often be qualitatively rationalized a priori via simple models (e.g., the OA to Ni(0) vs Ni(I) species).
3.4.2. Models for the Estimation of Reactivity and Catalytic Efficiency
In this section, we highlight models which are helpful in the understanding of catalytic reaction mechanisms as well as catalytic efficiency and are directly related to the field of transition metal-catalyzed homogeneous reactions. The first is the energetic span model that was originally developed to understand (1) the catalytic efficiency in cross-coupling and Heck reactions proceeding through neutral and anionic mechanisms, and (2) the effect of linker chain in bidentate phosphine ligands.640 The model is based on the transition from kinetic k-representation of catalytic cycles (useful when experimental data are available) by means of the Eyring’s transition state theory to E-representation that is the straightforward result of quantum chemical computations (Figure 28a).
Figure 28.

Conventional catalytic models in k-representation and TOF calculations by means of the energetic span model applied to different mechanistic cases: (a) simple single-cycle catalytic model; (b) catalytic process with an off-cycle intermediate (resting state); and (c) catalytic cycle with an off-cycle “dead end” state (catalyst poisoning), in this case, TON may be calculated by means of the energetic span model as well. Note that the energy levels corresponding to intermediates and transition states should be free energies and not electronic energy values. Adapted from ref (641). Copyright 2011 American Chemical Society.
The error in quantum chemical calculations (associated with the intrinsic method accuracy for the calculations of the electronic energies, solvation energies, and errors arising from computation of thermochemical corrections and entropy estimations) leads to very substantial accumulating errors in TOF estimation. However, relative TOF values obtained from this approach may be of qualitative value and semiquantitative accuracy due to error cancellation typically inherent to the computations.
The model shows the deficiency of the well-accepted concept of the rate-determining step in catalysis. The flatter the free energy profile of a catalytic cycle, the lower the energetic span and the higher the corresponding TOF value. Thus, the concepts of TOF-determining intermediate and TOF-determining transition state become more valid and maximize the energetic span of a cycle. Catalyst resting state formation is easily accounted for by the introduction of a reversibly formed off-cycle intermediate in the model (Figure 28b). Notably, catalyst deactivation may also easily be incorporated in the energetic span model through the inclusion of an overly stable off-cycle intermediate (“dead end”) in the model, and thus TON values may easily be calculated (Figure 28c). This illustrates the distinction between the real catalytic activity on the molecular level and the apparent activity observed experimentally as over-reactive catalytic species forming exclusively high-free energy intermediates throughout the cycle (and thus having the flat cycle free energy surface with small energetic span) may easily bind a catalyst poison molecule or undergo a chemical transformation to a stable species of low free energy. The catalyst may therefore prove to be fast but short-lived (or overly active) and, thus, of little usage.641
As we have mentioned above, the energetic span model was developed when considering the cross-coupling and Heck reaction catalysis and has since been employed in a significant number of studies in homogeneous Pd-catalysis (see the excellent review of Kozuch640 for details). The theoretical study of Suzuki coupling with Pd-catalytic system based on popular SPhos ligand (2-(2′,6′-dimethoxybiphenyl)-dicyclohexylphosphine) should also be mentioned. There, the energetic span model was applied to analyze the performance of the catalytic system, and the InPhos ligand (having 1-methylcycloxexyl moieties instead of cyclohexyl ones as in SPhos) was proposed as having superior activity on the basis of the theoretical modeling.642 Unfortunately, to the best of our knowledge, the so-designed InPhos ligand has not been applied yet in homogeneous catalysis.
This model has also been applied in theoretical studies on Ni-based catalysts. Computations on simplified complexes at the B3LYP/LANL2DZ+p//B3LYP/LANL2DZ level of theory provided relative TOF values of 1 to 147 for the neutral Ni(0)/Ni(II) and Pd(0)/Pd(II) catalytic cycles, respectively.643 However, the mechanistic model employed in this study to determine these TOF values might have been incomplete since the often observed odd-electron Ni(I)/Ni(III) cycle was neglected. The energetic span model consistently reproduced the experimental enantioselectivity of the reaction by comparison of the stereoselectivity and TOF-determining transition states.644 This approach was also applied in a number of theoretical studies of Ni-catalyzed cross-coupling and amination reactions.630,645,646
The consideration of the nonstandard free energies corresponding to the concentration-dependent free energy surfaces outlined in the previous sections is often disregarded in computational catalysis studies. Therefore, a more phenomenological approach has been proposed based on the consideration of simplistic concentration-corrections to the free energies. In the first approximation, if we consider the ith intermediate in a catalytic cycle, its nonstandard Gibbs free energy Giø is related to the standard Gibbs free energy Gi through the equation 3.1 (Ci is the ith intermediate concentration):
| 3.1 |
The reaction free energy profile thus becomes a concentration-dependent stepwise descending plot (Figure 29). One can therefore postulate that for a catalytic process to occur, each following intermediate must have lower or equal nonstandard Gibbs free energy (i.e., the free energy profile must be strictly nonascending). Thus, the equilibrium will be established and the catalytic process will effectively stop when the corresponding (nonstandard) free energy profile becomes flat. This notion may be incorporated in the energetic span model and used to gain additional mechanistic and kinetic insights. For example, some intermediates in a catalytic cycle may be in a thermodynamic quasi-equilibrium due to, for example, rapid interconversions or rapid formation-depletion processes. Varying intermediate concentration, we may also suppress the formation of certain off-cycle intermediates as resting states or irreversibly forming dead ends.640
Figure 29.
Catalytic reaction free energy profile in standard vs in nonstandard Gibbs free energies. The process is driven in the direction of nonstandard Gibbs free energy decrease. Reproduced with permission from ref (640). Copyright 2012 John Wiley and Sons.
The theoretical formalism of the energetic span model is in steady development. On the basis of the results of accurate quantum chemical calculations, kinetic modeling of homogeneous transition metal-catalyzed reactions may be performed, incorporating recently developed features. For example, kinetic modeling of olefin hydrogenation catalyzed with Au(III) complexes647 was performed with the AUTOF program.640 The model was applied in the field of homogeneous electrocatalysis to model proton reduction with Fe complexes648 along with the closely related degree of rate control formalism.649,650 If a microreaction network of a catalytic process is functioning in a steady state regime (irrespective of its complexity and the number of intermediates included), the energetic span model may be coupled with graph theory formalism (by means of the spanning trees concept) to calculate the corresponding TOF value.651
Another useful theoretical approach, which was used in computational studies of cross-coupling reactions, is the activation-strain model. In accordance with the model, relative energy ΔE of a molecular complex undergoing a chemical reaction along the corresponding reaction energy profile (including the activation energy ΔE‡) may be decomposed in strain energy ΔEstrain(ζ) related to reactants structural rigidity and their interaction energy ΔEint(ζ):
| 3.2 |
where ζ is the reaction coordinate. For example, the energy ΔE along the reaction profile in the oxidative addition process becomes (Figure 30):
| 3.3 |
The activation-strain model was applied to investigate the physics of processes mediated by d10-metal ML2-complexes as C–H, C–C, and C–X bond activation reactions. Ligand effects such as bite angle and the aliphatic linker length effect in bidentate phosphines were investigated with the activation-strain model as well (see refs (652) and (653) for the review of the reported studies). The extended activation-strain model was used to study anti-Hammond behavior in R–X bond activation under anion assistance.654
Figure 30.
Reaction profile of the oxidative addition to a transition metal complex. Here, the energy is decomposed via the activation-strain model; see the equation 3.3, R–X = CH3X, [M] = [ML2]. Reproduced with permission from ref (652). Copyright 2017 Wiley-VCH.
The catalytic activity of 3d metal catalysts in the cross-coupling and functionalization reactions can be understood through electronic structure computations with the above-mentioned models. Such an analysis requires a guess of the process mechanism and preoptimized geometries of reaction intermediates. In the case of Fe- and Ni-catalysts, the individual mechanistic steps may differ from the textbook cases of Pd-catalyzed reactions as we have discussed in the previous subsection. However, 3d metal-catalyzed cross-coupling and functionalization reactions are often complicated by the “non-classic” phenomena which make the qualitatively different mechanistic models necessary.
3.4.3. Nonclassical Phenomena in Homogeneous Catalysis with 3d Transition Metals
In this subsection, the challenging phenomena that need to be accounted for in computational studies of 3d metal-catalyzed reactions with representative examples from the cross-coupling, Heck, C–H arylation, and Buchwald-Hartwig amination reactions will be highlighted. Particularly, these phenomena imply consideration of multiple catalytic cycles or a set of important off-cycle intermediates to ensure high model accuracy in the calculations. The consideration of such highly branched reaction path networks instead of individual catalytic cycles substantially increases the complexity of the model. Briefly, we discuss the phenomena of spin state changes in catalytic systems, nanoparticles involvement in catalysis, catalysis by charged species, and photoredox catalysis.
3.4.3.1. Two-State Reactivity
Potential energy surfaces of different spin states in 3d metal systems are often located close in coordinate space, giving rise to near-degenerate spin states.564,655,656 This increases the methodological complexity as the accuracy of DFT methods in computations of high-multiplicity systems is functional-dependent.208 Another important issue in the context of this section is that the inclusion of multiple PES corresponding to different spin states becomes necessary for an adequate mechanistic analysis,564 which consequently increases the computational cost.
The reactivity of transition metal complexes is often affected by spin crossover effects (spin-flip) resulting in the so-called two-state reactivity phenomenon.657,658 If the transition state on the PES of a high-spin catalytic intermediate lies above the transition state on the low-spin PES then spin acceleration may occur (Figure 31). Often, multiple crossing points between the low-spin and high-spin potential energy surfaces may be present, and the ones with the lowest energy are called minimum energy crossing points (MECPs). If MECPs are below the high-spin transition state (as in Figure 31), spin acceleration occurs. On the other hand, the opposite situation with MECPs located above the transition state is also possible, and this leads to spin-blocking phenomena.564
Figure 31.
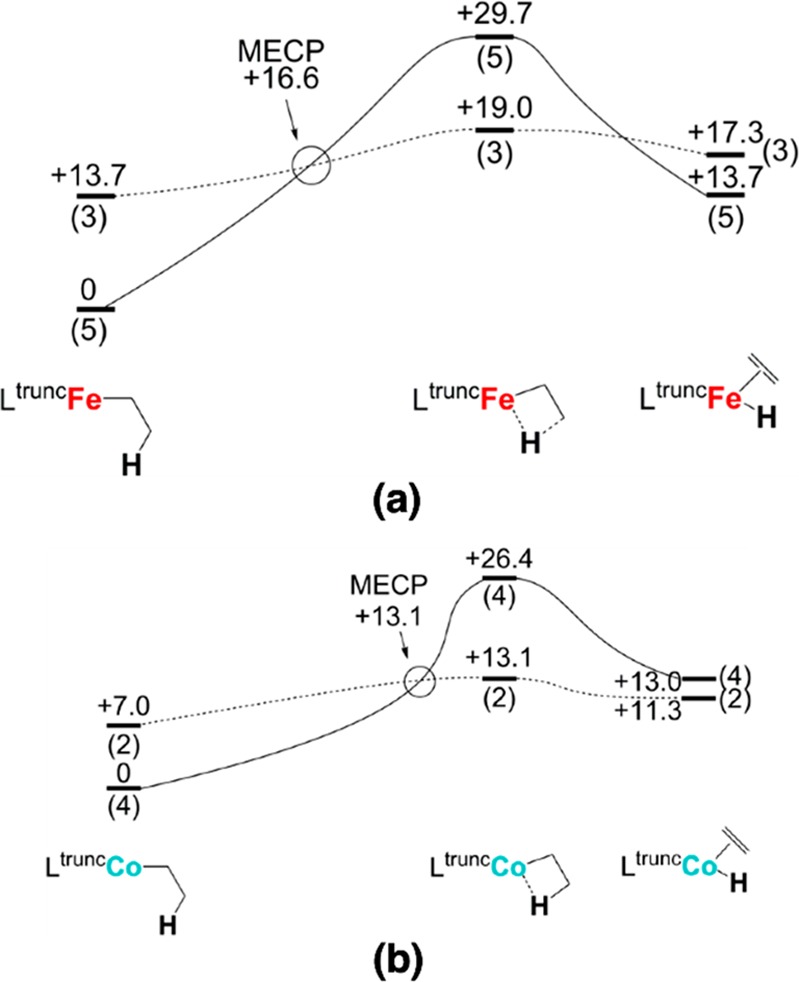
Energy profiles of the β-hydride elimination in [Et-M-L] complexes [L = (Z)-(3-iminoprop-1-en-1-yl)amide], (a) M = Fe and (b) M = Co. The free energies are given in kcal mol–1, and spin states are given in parentheses. Adapted from ref (659). Copyright 2013 American Chemical Society.
A weak ligand field often enables the spin acceleration in 3d metal complexes, and this leads to unusual reactivity in mediated coupling reactions. β-Hydride elimination in Fe(II) and Co(II) complexes with β-diketiminate ligands was shown to proceed under spin acceleration (Figure 31),659 where the quintet-triplet and quartet-doublet PES crossings were calculated at the B3LYP-D3/def2-TZVP&def2-SVP level of theory. Accounting for two-state reactivity lowered the activation barriers of the β-hydride elimination for about 1.5 and 2.0 times in [Et-Fe-L] and [Et-Co-L] complexes (L = (Z)-(3-iminoprop-1-en-1-yl)amide), respectively.
Direct C–H activation by Fe(II) complexes, however, may proceed with spin acceleration even in a strong-field ligand environment.656 The reaction was modeled at the B3LYP-D3/def2-TZVP&SDD//B3LYP-D3/def2-SVP&SDD level (with SMD continuum solvation model), and the high-level reference calculations were performed additionally with partially spin-adapted open-shell coupled-cluster CCSD(T) method and CBS extrapolation (considering energies obtained with double- and triple-ζ basis sets). Two-state reactivity in C–H activation with Fe(II) complexes was shown to take place with bidentate N-heterocyclic (dipyridine, phenanthroline) and THF weak-field ligands as well as with strong-field ligands such as 1,2-diphosphinobenzene and bis(3-methylimidazole-2-ylidene)-methane. Moreover, C–H activation with middle- and late-first row metals was modeled, and the two-state reactivity appeared to be a rather general phenomenon in C–H activation mediated by 3d metal complexes with phenanthroline ligands (Figure 32). The two-state reactivity may also be expected in the C(Ar)-H amination process with Ni(II) tris(pyrazolyl)borate complexes according to computational results reported in ref (660). Therefore, the consideration of several PES corresponding to the low-energy spin states is highly desirable in the modeling of catalytic processes with 3d metal-catalyzed coupling reactions because two-state reactivity in these systems may qualitatively affect the mechanism of the reactions.
Figure 32.

Two-state reactivity in C–H activation promoted by 3d transition metal complexes [M(substrate)(phen)(Ph)Cl] where substrate = α-benzoquinoline. The relative free energies are given in kcal mol–1 and computed at the B3LYP-D3/def2-TZVP&SDD//B3LYP-D3/def2-SVP&SDD level with SMD solvation model; phen = phenanthroline. Adapted from ref (656). Copyright 2016 American Chemical Society.
3.4.3.2. “Active Site Heterogeneity” in Homogeneous Catalysis
Experimental results suggest the simultaneity of the aforementioned even-electron Ni(0)/Ni(II) and odd-electron Ni(II)/Ni(III) catalytic processes in Suzuki, Negishi, and Kumada coupling reactions as well as in Buchwald-Hartwig amination and the corresponding differing reaction rates (i.e., TOF values corresponding to the Ni(0)/Ni(II) and Ni(II)/Ni(III) cycles). Since the reactions may proceed through both pathways irrespectively of the precatalyst used,598,624,661,662 both cycles should be accounted for in computational studies. The even-electron and odd-electron pathways are connected through the comproportionation/disproportionation equilibrium between Ni(0), Ni(I), and Ni(II) species.
The formation of Ni(I) complexes from [(COD)Ni(dppf)] was shown to proceed through the comproportionation of Ni(0) species with the Ar–X oxidative addition products (Figure 33a).625 In a joint experimental-theoretical study of Suzuki coupling of aryl sulfamates with [Cl(o-tolyl)Ni(dppf)] complex as the catalyst precursor, formation of Ni(I) species in reaction media was observed with EPR spectroscopy and modeled as a competing mechanism to the oxidative addition process (Figure 33b). Here, a dimeric Ni(0)–Ni(II)-cluster in singlet spin state is formed by the association of the [Ni(dppf)] catalyst with [Cl(o-tolyl)Ni(dppf)] precursor species. Then the Ni-containing dimer undergoes the transition to the triplet Ni(I)–Ni(I) cluster through the low-lying MECP, and the subsequent exergonic formation of the free [Cl–Ni(dppf)] and [(o-tolyl)Ni(dppf)] species occurs. Calculations were performed at the M06-L/triple-ζ//M06-L/double-ζ level with bulk solvent effects modeled with the SMD method.598
Figure 33.

Formation of Ni(I) species in cross-coupling reactions. (a) The observed experimentally Ni(I) species formation from the oxidative addition products. Adapted with permission from (625). Copyright 2017 American Chemical Society. (b) The formation of Ni(I) species via a competing mechanism to oxidative addition process in Suzuki coupling reaction, free energy values were calculated at the M06-L/triple-ζ//M06-L/double-ζ (SMD solvation) level. Adapted from ref (598). Copyright 2017 American Chemical Society.
The mechanistic picture in Ni-catalyzed coupling reactions may often be complicated by Ni(I)–Ni(I) dimeric clusters, which can introduce enhanced reactivity. Particularly, the formation of a highly oxidized and thermodynamically unfavorable Ni(III)-intermediate through the oxidative addition of an organic (pseudo)halide to Ni(I) may be circumvented, as has been shown in an experimental-theoretical study of the Kumada cross-coupling reaction. Computations at the PBE0/6-31G(d,p)&SDD level showed the feasibility of the catalytic process driven by [(μ-Cl)-Ni-NHC]2 species (NHC = N,N′-dimethylimidazol-2-ylidene). The initial formation of the [NHC-Ni(μ-Cl)(μ-Ph)Ni-NHC] (Ni(I)–Ni(I) intermediate) was followed by the oxidative addition of PhCl resulting in the formation of a [(NHC)(Ph)Ni(μ-Cl)]2 (Ni(II)–Ni(II) cluster), thus avoiding the highly oxidized Ni(III)-intermediate. The elimination of Ph-Ph molecule from [(NHC)(Ph)Ni(μ-Cl)]2 was preceded by the Ph-substituent migration (the formation of the Ph-Ni-Ph center) and led to the formation of the [(μ-Cl)-Ni-NHC]2 species. The [(NHC)(Ph)Ni(μ-Cl)]2 (Ni(II)–Ni(II) dimer) was somewhat overstabilized, and the Ph-migration proceeded with the high activation barrier of 26.2 kcal mol–1. However, the barriers did not exceed 16.1 kcal mol–1 in the other mechanistic steps of the process.663 The involvement of [(μ-I)-Ni-NHC]2 [Ni(I)–Ni(I) species] in catalysis was also proposed in the joint experimental-theoretical study of trifluoromethylselenolation of arylhalides. In accordance with the DFT computations, Ni(0) and Ni(I)–Ni(I) dimeric species had qualitatively different reactivity in the oxidative addition of PhI and PhSeCF3, with [NHC-Ni(0)] complexes being more reactive toward PhSeCF3 and Ni(I)–Ni(I) dimers being more reactive toward phenyl iodide (Figure 34).664
Figure 34.
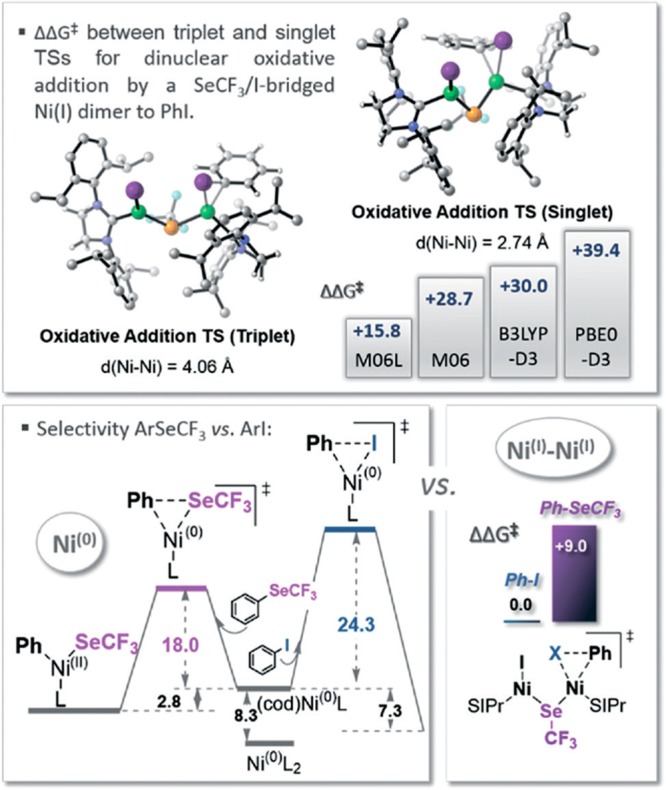
Qualitatively different reactivity of Ni(0) and Ni(I)–Ni(I)-dimeric species in the PhI and PhSeCF3 oxidative addition according to the DFT modeling. Adapted with permission from ref (664). Copyright 2017 Wiley-VCH.
The involvement of heterobimetallic particles may also be a non-negligible mechanistic step in the modeling of Ni-catalyzed coupling reactions. The Kumada cross-coupling of Grignard reagents (RMgX) with arylalkylethers (ArOR) is a feasible catalytic process if the pre-OA η2-complex of [NiPCy3] catalytic species with PhOMe is stabilized by the interaction with [(R)(Me2O)Mg(X)] organic nucleophiles (Figure 35) according to the calculations at the M06/6-311++G**&SDD//B3LYP/6-31G*&LANL2DZ level with the PCM solvation included (i-Pr2O solvent). Formation of the nickelate complex allowed avoiding the classic pathway with direct PhOMe oxidative addition to the [NiPCy3] complex that was kinetically unfavorable (with the activation barrier of 26.2 kcal mol–1) and endergonic (by 3.2 kcal mol–1) leading to the nonclassic Kumada coupling mechanism. Moreover, Pd catalysis along the similar pathway was kinetically and thermodynamically unfavorable, which was illustrated by the ineffectiveness of Pd catalysts in Kumada coupling with arylalkylethers.665
Figure 35.

Mechanism of the heterobimetallic Ni-Mg-pathway in Kumada cross-coupling as modeled at the M06/6-311++G**&SDD//B3LYP/6-31G*&LANL2DZ level with the PCM solvation included (i-Pr2O). Adapted with permission from ref (665). Copyright 2015 Wiley-VCH.
The mechanistic assistance of [PivOCs·CsCO3]− clusters was shown to be critical in benzoxazole and naphthalen-2-yl pivalate C–H/C–O-coupling catalyzed with [Ni(dcype)] species (dcype = Cy2PCH2CH2PCy2) by a combination of kinetic experiments and DFT calculations. The oxidative addition of naphthalen-2-yl pivalate proceeded through the single-site mechanism via a five-membered transition state for the case of ArOPiv, while the C–H activation stage was base-accelerated (Figure 36). The energy barrier corresponding to the direct C–H activation was 34.7 kcal mol–1, while the nonvalent H-bond- and Cs–N-interactions between the benzoxazole molecule and [PivOCs·CsCO3]− cluster stabilized the transition state in the base-assisted pathway leading to a lower barrier of 31.1 kcal mol–1. The DFT computations performed with M06-L and M06 exchange-correlation functionals, 6-31(d) and LANL2DZ basis sets, and the PCM model were consistent with the experimentally observed three-fold increase in the reaction rate and the increase from 32% to 67% reaction yield in the case of Cs2CO3 used as the base.626
Figure 36.

Mechanism of C–H/C-O-coupling catalyzed by [Ni(dcype)] and promoted by [PivOCs·CsCO3]− clusters, corroborated by DFT. Reprinted from ref (626). Copyright 2014 American Chemical Society.
Palladium catalysts used in cross-coupling, Heck, and C–H functionalization reactions may lead to so-called dynamic catalysis.666 The complexes, salts, and nanoparticles used as precatalysts in these reactions undergo chemical transformations leading to the formation of the active catalyst form. The molecular species may lose the ligand (see previous subsection), while the nanoparticles are prone to leaching of Pd from the surface (Figure 37). This leads to sigmoidal reaction kinetics and dynamic reaction mechanisms in which the catalytic activity is directly related to the accompanying formation of Pd nanoparticle followed by leaching of molecular Pd species in solution and their redeposition on the nanoparticle surface.666 This dynamic catalyst behavior is often unavoidable and leads to complications regarding catalytic efficiency, selectivity, reproducibility, and the contamination of reaction products and the environment by palladium.
Figure 37.
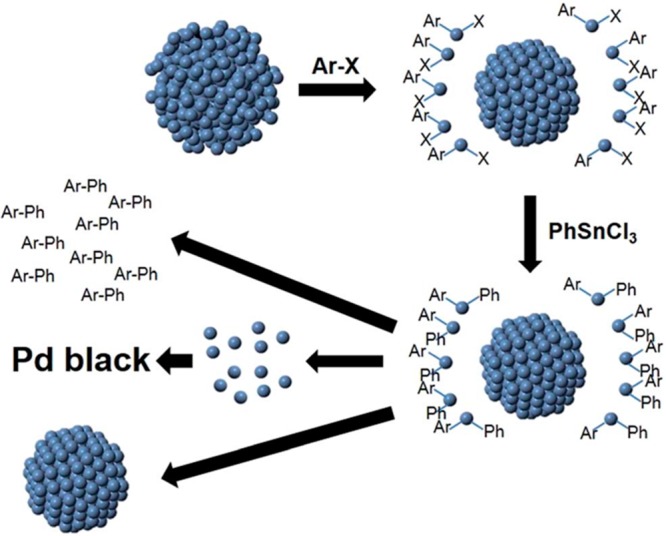
Dynamic catalyst behavior: palladium nanoparticle leaching in the Stille cross-coupling reaction. Adapted from ref (669). Copyright 2015 Royal Society of Chemistry.
The interconversions of 3d transition metal complexes, nanoparticles, and bulk aggregates under the catalytic conditions are often overlooked in theoretical studies. One may expect that the aforementioned dynamic behavior is affected by the energetics of the metal–metal and metal–ligand bonds. The first-row transition metals that have shown activity in coupling reactions (Cr to Ni) have considerably higher cohesive energies thus correspond to stronger metal–metal bonding (e.g., Fe, Co, and Ni have respectively 8.8, 12.2, and 12.5 kcal mol–1 higher cohesive energy according to experimental data) (see ref (667) for the thorough comparison of the experimental cohesive energy values as well as the benchmarking of DFT methods in modeling of transition metals in the bulk form). Because of the stronger metal–metal bonding in 3d transition metals, one expects lower feasibility of leaching and fast precipitation of M(0)-forms as colloid metal nanoparticles or the metal bulk. Indeed, ligand-free Ni catalytic systems were active in the Heck reaction and were sensitive to the solvent choice, as in the cases of noncoordinating solvents and absence of ligands, where Ni precipitated rapidly as colloid particles and no catalytic activity was observed. On the other hand, the ligand-free Ni catalytic system was active in the coordinating NMP solvent.668
Stabilization by, for example, charcoal, ligands, or surfactants is critical for heterogeneous Ni(0) catalysts. Supported metallic Ni and colloid Ni nanoparticles show activity in the Heck reaction,586 Suzuki670 and Negishi couplings,671 and Buchwald-Hartwig amination.672,673 Some detailed mechanistic insights in Ni-complexes/Ni-nanoparticles equilibrium are needed since the same activity via leaching and subsequent homogeneous catalysis was proposed for Ni/C coupling catalyst674 as in the Pd case.669,675,676 Therefore, the account of dynamic catalyst behavior, catalyst evolution analogous to the Pd case, and heterogeneous Ni catalysis is desirable in the computational studies of Ni catalysts. The heterogeneous Ullmann coupling of bromobenzene to biphenyl on Co nanoparticles proceeds below room temperature, and the feasibility of the process was clearly demonstrated by the combination of high-resolution scanning tunneling microscopy and DFT calculations.677 In the light of reports on ligand-free Fe-618,678 and Co-catalytic679 systems that are active in coupling reactions, more mechanistic computational studies considering the nanoparticle involvement in the Fe- and Co-catalytic coupling reactions are desirable as well.
3.4.3.3. Solvent-Assisted Mechanisms
Adequate accounting for solvation effects is often crucial in mechanistic studies in homogeneous catalysis. Coupling reactions are exemplary in this regard because they often involve charged (anionic and cationic) reaction intermediates in the main catalytic pathways. Their stabilization via specific interactions with solvent molecules is quite common. The electrostatic stabilization of strongly polarized and charged transition states encountered during the anionic oxidative addition mechanism may lead to substantially reduced activation barriers in the respective paths compared to alternative neutral routes.622,680 The regioselectivity of Heck arylation of acrolein acetals may be controlled by addition of anionic additives (Cl– or AcO–) that bind to the catalytic species.681 Moreover, the cationic pathway in the Heck reaction is of key importance in the case of Ni metal-complex catalysis. The neutral β-hydride elimination step in Ni complexes is problematic compared to Pd counterparts, and the cationic mechanism allows for facile β-hydride elimination, which shows lower energy barriers both computationally and experimentally.586,623,682
The importance of anionic species is well-addressed in Pd-catalyzed cross-coupling reactions. Hydroxide anions play a key role in the transmetalation step of Suzuki coupling allowing for the formation of [ArB(OH)3]− boronate anions,683 while fluoride F– additives affect the mechanism of transmetalation in Stille coupling.684 Anionic and neutral precatalyst activation mechanisms were demonstrated in Suzuki-Miyaura catalyst-transfer polymerization by the combination of kinetic experiments and computations at the PBE0/6-311+G(d,p)&LANL2DZ level of theory, where the C-PCM method used to model bulk solvent effects.685 Catalytic activity of anionic Pd-complexes in Suzuki coupling performed in polar solvents was proposed on the basis of experimental study corroborated with computational modeling results.686
Catalytic species in cross-coupling reactions are undercoordinated transition metal complexes; therefore, the explicit inclusion of solvent molecules in the transition metal coordination sphere (along with bulk solvent effects modeled with implicit solvation models as PCM, SMD, or COSMO-RS) is often non-negligible (Figure 38). A number of ligand-free 3d metal catalysts were proposed in coupling reactions as discussed above. Therefore, the addition of solvent molecules or weakly bound anionic ligands (as halide anions) becomes mandatory since they will form the first coordination sphere of the metal (see, for example, ref (618)). Moreover, water impurities in Ni-catalyzed Suzuki coupling of aryl carbamates and sulfamates led to the formation of resting states with the H2O molecule directly coordinated to the Ni center, which leads to the degradation of the activity of the catalyst according to DFT modeling.577 Therefore, the explicit consideration of possible solvent impurities may also be needed for proper understanding of the reaction mechanism.
Figure 38.

Structures of intermediates in Pd- (left) and Ni-catalyzed (right) cationic Heck reaction (top) and proton abstraction transition states (down) optimized at the M06/6-31G(d,p)&CEP-31G level of theory. The solvent molecule (DMSO) is included explicitly while the bulk solvent effects were modeled implicitly with the SMD method. Reprinted from ref (623). Copyright 2016 American Chemical Society.
In the two-level treatment of solvent effects, known as the cluster-continuum model, short-range and “outer-sphere” bulk solvent effects are considered with explicit inclusion of solvent molecules and continuum solvation model, respectively.687 The model helps to eliminate an error caused by the incompleteness of the computational models of transition metal complexes through filling coordination vacancies with solvent molecules and is useful in the computation of reaction free energies in solution.688 By means of the cluster-continuum approach, specific transition metal complex-solvent interactions are modeled explicitly and the entropic terms may be calculated more accurately. For example, the model was used to elucidate the structure of Me2Zn, MeZnCl, and ZnCl2 species and to study the thermodynamics of the corresponding Schlenk-type equilibrium in THF solvent with DFT modeling combined with IR spectroscopy and calorimetric experiments. The understanding of the organozinc reagent structure in solution allowed construction of an adequate model of the transmetalation step in the Negishi cross-coupling reaction in THF solvent.689
Different practical approaches for the correction of free energies routinely computed within the ideal gas approximation to estimate free energies in solution are considered. An empirical correction term of 1.89 kcal mol–1 was proposed to be added to every isolated species free energy which is simply equal to the RT ln(V/Vg) term value at room temperature (298 K) and V eq 1 L (Vg is the ideal gas molar volume).690,691
3.4.3.4. Radical and Photo-Redox Paths
The interaction of 3d transition metal species with organic halide molecules (R–X) may proceed through nonclassic mechanisms as opposed to the classic oxidative addition with a three-center transition state (see section 3.4.3.2). Mechanistically, these correspond to single-electron transfer (SET) from R–X molecules to transition metal species (Figure 39a) and halide atom (X•) abstraction from organic halide molecule (R–X) by transition metal complex (atom transfer, AT), leading to the formation of an organic radical (R•, Figure 39b). In both cases, the oxidation state of the transition metal center increases by 1e-unit, and the reaction can be viewed as a consecutive two-step process involving radical species. If the organic radical (R•) does not undergo side-reactions in SET or AT pathways, it binds to the transition metal so that SET, AT, and classic concerted oxidative addition result in the 2e-oxidation.
Figure 39.

(a) Heck-type reaction catalyzed by [CoCl2(dpph)] complex salt (dpph = 1,6-bis(diphenylphosphino)hexane), note single-electron transfer between R–X-molecule and [Co(dpph)]-catalytic species leading to [R–X]•– cation radical formation. Adapted from ref (692). Copyright 2006 American Chemical Society. (b) Cross-coupling of C(sp3)-chlorides with Grignard reagents catalyzed with Fe-phosphine complex salt, note Fe(II) specie formation through Cl-atom abstraction (atom transfer process). Adapted from ref (617). Copyright 2017 American Chemical Society.
The formation of a Ni(III) intermediate through SET oxidation of [(PEt3)2Ni(Me)(o-Tol)] by Ar–X species is a well-known process,629 and reductive elimination of 1,2-dimethylbenzene from the formed Ni(III) complex leads to Ni(I) that can initiate the Ni(I)/Ni(III) cross-coupling path. Concomitantly, an arylhalide anion radical is formed that should be considerably more stable toward cleavage of the Ar–X bond (compared to alkyl halides R–X).693 With the progress in understanding of organometallic reaction mechanisms, the 3d transition metal catalyzed coupling reactions with 1e-oxidations through SET and AT mechanisms became effective targets for catalyst development. A prominent example is the C(sp2)-C(sp3)-coupling reactions catalyzed by Fe(II)-chiral-bisphosphine complexes through the radical reaction mechanism.617,694 Although the nonclassic 1e-pathways often alter the mechanistic picture of the catalytic coupling reactions drastically, DFT modeling can be especially helpful to clarify the reaction mechanism as long as the mechanistic analysis thoroughly considers the participation of different spin states at various stages of the reaction and the presence of radical pathways and potential multiconfigurational effects.
The easy accessibility of 1e-oxidation chemistry with 3d transition metals offers ways to combine photoredox catalytic cycles with the conventional organometallic paths to drive the cross-coupling chemistry more efficiently. Dual Ni-photoredox systems have become quite popular in catalysis of photoassisted cross-coupling reactions.566 Since DFT modeling of open-shell molecular species is a challenging but surmountable complication, we anticipate computational or modeling-assisted studies of these dual Ni-photoredox systems to become a common practice soon.
Computational modeling of Ni-photoredox catalytic systems may help to rationalize the way photoredox processes boost Ni-catalyzed coupling reactions. The Ir–Ni catalyzed C–N coupling of (4-trifluoromethyl)phenyl bromide and pyrrolidine was studied at the M06/6-311G(d,p) level of theory with bulk solvent effects modeled with PCM implicit solvation method (Figure 40).695 The first step that was considered in the computational study was the oxidative addition of the CF3-C6H4-Br molecule to [Ni(0)L2] species (L = pyrrolidine). The addition was reported to proceed via the nonclassic SET or SNAr-type mechanism, and the often-reported ArBr oxidative addition to [Ni(I)L2] was found to be much less facile kinetically compared to the [Ni(0)L2] species. SNAr-type addition of CF3-C6H4-Br to the Ni(0) complex proceeded with the low free energy barrier of 1.6 kcal mol–1, while in the case of OA to the Ni(I) counterpart, only the classic three-center transition state corresponding to a relatively high barrier of 10.9 kcal mol–1 was found. Thus, the Ni(I)/Ni(III) catalytic cycle was found to be inoperative due to the high OA barrier.
Figure 40.

Gibbs free energy profile for the Ir–Ni-catalyzed C–N-coupling of 4-trifluoromethylphenyl bromide and pyrrolidine in N,N-dimethylacetamide (PCM) computed at the M06/6-311G(d,p) level of theory. Reprinted from ref (695). Copyright 2018 American Chemical Society.
The photoredox cycle plays a triple role. The [(CF3-C6H4)(Br)Ni(II)L2] oxidation to the Ni(III) complex via SET mechanism proceeds with the corresponding photoexcited *[Ir(III)]+ reduction to [Ir(II)]. One of the pyrrolidine ligands in the formed Ni(III) complex undergoes proton abstraction by a DABCO (1,4-diazabicyclo[2.2.2]octane) molecule, and the coupling product reductive elimination is thus facile from the highly oxidized Ni(III) species, leading to the Ni(I) complex formation. Ground-state [Ir(III)]+ is regenerated via [Ir(II)] to Ni(I) SET oxidation. Therefore, the photoredox cycle promotes the formation of Ni(III) species that are prone to reductive elimination processes and regenerates the catalytically active [Ni(0)L2] species. Moreover, as the [Ni(0)L2] complex was the active form, precatalyst activation was required for the process to start, and the activation of Ni(II) salt precatalyst proceeded via photoredox-catalyzed hydrogen atom transfer mechanism according to the reported modeling. For a more detailed description of computational electrochemistry and photoredox reactions, we refer the reader to the excellent review articles of Marenich et al.696 and Demissie et al.697
3.5. Concluding Remarks on the Model Accuracy in Homogenous Catalysis
To summarize, despite the apparent simplicity and well-defined nature of homogeneous catalysts and related catalytically active intra- or extraframework 3d transition metals of MOFs, the associated practical catalytic systems and underlying mechanisms governing their performance are enormously complex. Unlike the noble metal catalysis, 3d transition metals cannot be well-rationalized based on the single-cycle, single-site, two-electron chemistry concepts. Additionally, the generally more reactive 3d transition metals tend to open up highly branched reaction networks of chemical transformations, which effectively define the overall performance of the catalytic system. Conventionally, the mechanistic analysis and computational studies on catalytic reaction paths have been predominantly guided by the chemical insight of the researcher providing a strong and sometimes determinative bias to the reaction outcome. Given the large number of approximations made in all practical computational studies and the associated widespread computational results that can be obtained using “reliable computational protocols”, the comprehensiveness of the mechanistic analysis and accounting for all the key chemical phenomena are the most crucial factors that determine the model accuracy of the calculations and the general predictive power of their results. The minimization of such expert bias is an important target in the development of theory-based predictive models needed to progress toward the realization of the catalysis by design concept. The current state of the art in the field of automated reaction path predictions and analysis that hold promise in becoming the practical tools for reducing expert bias is discussed in the next and final section of this review.
4. Toward Bias-Free Mechanistic Models: Emerging Tools for Automated Reaction Path Analysis and Predictions
Previous sections have presented selected examples of the power of computational chemistry methods for elucidating mechanisms of catalytic reactions and formulating theoretical concepts that could contribute toward realization of the catalysis by design paradigm. As was highlighted in section 2, modern DFT698 and WFT methods699 provide the necessary computational toolbox for sufficiently and accurately evaluating the structures and energetics of sequences of intermediates and transition states within an envisioned catalytic reaction mechanism.700,701 Conventionally, the research strategies commonly employed in computational catalysis imply that the quantum chemical methods are used to analyze predefined mechanistic proposals formulated based either on previous suggestions for (often vaguely) related systems, expert opinion, or chemical intuition. The importance of expert input in mechanistic analysis of catalytic paths has been one of the focal points of section 3. In other words, conventional computational chemistry methods do not provide means to enable discovery of new catalytic paths, but their role is limited to evaluating reactions within the scope of the existing chemical knowledge. Such a situation is common for many fields of science and is often referred to as the “streetlight effect”.702−704 Dewyer and Zimmerman in their excellent recent perspective state that “for reaction mechanisms, where no hypotheses are available —and the researchers “just don’t know”—computation has not offered practical solutions to discover these unknown mechanisms”.705
Recent developments in the field are transforming the ability of computational chemistry to minimize the expert bias in mechanistic analysis and even to discover reaction paths that could not be deduced based on prior knowledge or “chemical intuition”. The basic idea behind these emerging methodologies is that with specified reactant molecules and catalyst, the program automatically determines feasible sequences of elementary reaction steps. Such approaches may give rise to practical tools for identifying unexpected reaction mechanisms at reasonable computational cost, enabling a new paradigm of research in quantum chemistry. From the computational perspective, the construction of a reaction path even for a single elementary step is a quite demanding and a nontrivial multistep procedure. Most contemporary approaches to locating reaction paths start from the approximation of the TSs and the reaction path followed by optimization of the stationary point and IRC computations. Because many of the steps involved in this procedure are highly demanding and can easily fail, substantial efforts of the research community are currently put into the development of alternative streamlined approaches for finding reaction paths directly from mechanistic hypotheses.706 Automated methods capable of formulating mechanistic hypotheses for elementary steps in combination with efficient approaches for reaction path and transition state (TS) optimization would drastically lower the amount of chemical intuition and expert bias involved in mechanistic research and may become the basis for the true predictive computational catalysis methodologies.
4.1. State-of-the-Art in the Reaction Path-Finding Strategies
Catalytic reactivity in a general sense is determined by the complex network of chemical reactions taking place simultaneously or consequently between the different (transient) components of the catalytic mixture. Each of the stages of the catalytic process, that is the catalyst activation, catalytic cycle propagation, catalyst deactivation, and nonselective conversion paths, may involve multiple elementary steps that proceed via mechanism and involve reaction intermediates that are not known a priori. Even the most advanced experimental operando techniques are not able to unravel such a high molecular-level complexity, and its computational analysis requires a much broader exploration of the chemical and configuration space to identify the minima on the PES and the pathways connecting them. Such a computational reaction discovery may be facilitated by narrowing down the reaction space by either applying predefined heuristic rules (e.g., bond breaking) to generate intermediates or by artificially pushing the reactants together in a simulation to induce chemical transformations. In principle, ab initio molecular dynamics provide the direct means to probe reaction events at the molecular level; however, the major challenge here is that even the fastest chemical reactions are considered rare events, making the adequate scanning of the reaction space by the direct atomistic AIMD simulations based on sufficiently accurate electronic structure methods challenging. The frequency of the reaction events can be greatly accelerated by applying bias potentials that push the system away from the free energy minima along a collective variable, which assumes some knowledge of the reaction coordinate or collective variable along which to apply the biasing potential.707−709 A similar reactivity enhancement can be achieved by simulating reactions at extremely high-temperature or high-pressure regimes, which effectively shift the equilibrium to products with higher entropy or lower volume, respectively.710
The determination of the reaction paths and, particularly, the transition state search with quantum chemical methods is a nontrivial task, and it is commonly associated with high computational demands. The tutorial review by Schlegel711 presents a comprehensive overview of the methodological aspects and capabilities of modern approaches for geometry optimization and transition state search. The state-of-the-art in reaction pathway finding strategies is summarized in a comprehensive review by the Zimmerman group.706 We refer the interested reader to these works for the details on the methodologies and strategies. In this review, we will limit ourselves to a brief description of the main concepts underlying these strategies and highlighting the most relevant examples of their applications to the topics relevant to the field of molecular catalysis.
The reaction path exploration strategies can be categorized in four main groups summarized in Figure 41. All these methods require explicit definition of a designated set of reactants and catalysts, but all subsequent steps are supposed to operate without external bias or with minimal possible interference from a researcher. The first class of methods, designated in Figure 41 as Concept 1, largely follows the way chemist researchers develop mechanistic proposals. The basis for this approach is a set of chemistry rules formalized by encoded elementary step types from databases or rules of chemical heuristics. These are used to describe reaction pathways between the reacting molecules defined at the start. Methods in this category commonly use activation barriers and reaction rates estimated using approximate methods and only seldom involve the explicit TS search. In the second category (Concept 2) approximate TSs are first generated from the reactants, followed by local TS optimization and IRC calculations. One of the most common methods for generation of the initial TS approximation in such methods involves exposing two molecules to artificial forces that would push them together to induce a reaction. The methods within Concept 3 start with the generation of a collection of putative elementary steps with the corresponding intermediates formed, followed by the application of double-ended methods to refine reaction paths and locate TSs. The methods within Concept 4 involve the generation of hypothetical reaction coordinates that are generated, followed by applying single-ended methods to carry out the reaction path searches along these coordinates. All these four methodological concepts share a common spirit but differ in the conceptual implementation and details that may be paramount to their degree of success. They are all designed to generate approximate reaction paths, estimate reaction barriers, and then integrate these paths into reaction networks of elementary steps. The current practical realization and implementations of these methods are still far from perfection. They all can fail at one point or another during the reaction search, and none of them can deliver an ideal balance of high accuracy and comprehensive PES analysis at a reasonable computational cost. Despite the ultimate goal to establish a bias-free reaction prediction and the promise of the nonuser interaction reaction exploration, chemical intuition in one form or another still needs to be introduced to some extent for the successful utilization of these methods. In the next sections, we will briefly discuss the available methods for automated reaction path analysis.
Figure 41.

Classification of automated reaction path exploration methods. Reprinted with permission from ref (706). Copyright 2017 Wiley-VCH.
4.2. Computational Tools to Explore Catalytic Reaction Mechanisms
Catalytic systems are commonly represented by the complex mixtures of reactants, catalyst precursors, ligands, additives, and solvent, giving rise to the formation of a wide variety of complexes that may show varied catalytic activity and behavior toward other components of the reaction mixture. The primary tasks of computational catalysis are to identify among these prereaction complexes those that contribute most to the catalytic reaction and identify mechanisms of the main catalytic cycle and competing reaction channels giving rise to unselective conversion routes or catalyst deactivation. As we have demonstrated throughout this review, the development of a comprehensive molecular-level picture of a catalytic system is a very challenging task due to the enormous complexity of the associated chemical reactions. However, it is believed that if all these tasks are accomplished, the resulting reactivity model could be used to guide the development of more active and efficient catalysts, which is the core idea of the rational catalyst design strategy.712
The strategies for automated reaction mechanism exploration tools discussed above are quite general, and they are designed to analyze general (most often organic chemistry) reactions occurring over a barrier.713 The extension of such methods to transition metal catalysis requires several auxiliary algorithms designed to deal with the specific features of transition metal catalytic reactions, mainly the particular sensitivity to the 3D geometric structure of transition metal complexes and the need for comprehensive sampling of reactions within and outside the catalytic cycles.
ZStruct2 is a reaction exploration tool introduced by the Zimmerman group. The method combinatorically samples driving coordinates (DC), which are bond-addition or bond-breaking vectors describing elementary reactions.705,714 These reactive coordinates are designed for use with the single-ended GSM that generates reaction paths, TSs, and intermediate structures for single elementary steps consistent with the DC. ZStruct2 handles intramolecular and bimolecular reactions by aligning reactants in a way consistent with the DC. The incorporation of knowledge of the transition metal center geometry and the alignment of the reactants enabled the application of ZStruct2 to perform studies on transition metal-catalyzed reactions. ZStruct2 has been successfully used to explore mechanisms of such processes as Pd-catalyzed C–H arylation of piperidine,714 FeCl3-catalyzed carbonyl-olefin metathesis,715,716 and Ni-catalyzed thiazole polymerization,717 and others.718,719 For example, in the case of piperidine arylation, ZStruct2 was able to identify all major steps of the catalytic cycles, including the roles that the multiple supporting reagents play in driving forward the reaction. In the study of thiazole polymerization, ZStruct identified an unexpected route for chain termination that prevents the controlled polymer growth.717
The Artificial Force Induced Reaction (AFIR) Method712 provides a more comprehensive and systematic approach to reaction path search, mechanistic analysis, and selectivity prediction of catalytic reactions. Starting from a given set of reactants and catalyst, AFIR searches all of the important (known, unknown, or unexpected) reaction pathways. The AFIR method not only predicts reaction mechanisms for the desired product but also explores the side paths resulting in the formation of byproducts. The AFIR methods can be successfully employed to explore reaction paths for relatively simple catalytic systems (small models, limited number of participating species, etc.), but when applied to realistic molecular systems, the full AFIR becomes prohibitively demanding and a restricted AFIR search is more appropriate. In this case, an artificial force should be added between selected fragments. Here, a restricted AFIR is more useful. The basic principles and application of AFIR in computational catalysis studies using the AFIR method has been explained in a recent comprehensive article by Morokuma and co-workers.712
The AFIR method combined with DFT calculations was used to explore the mechanism and the selectivity of the aqueous Mukaiyama aldol reaction catalyzed by an Fe-based chiral complex.712 More than 40 approximate TSs were located by AFIR and classified into 12 groups. It is worth mentioning that the AFIR method located 10 TSs relevant for the selectivity of the reaction, some of which may have been missed when following the traditional chemical intuition-guided approaches. This systematic study provides important mechanistic insights relevant for the development of Fe-based catalysts for carbon–carbon bond formation reactions. Besides, the AFIR method has also been successfully employed to analyze the reaction mechanism and identify factors controlling the stereoselectivity of the Kobayashi modification of the Mukaiyama aldol reaction, catalyzed by water-tolerant lanthanide-based Ln(OTf)3 Lewis acid catalysts in aqueous media.720−722
For relatively large molecular systems, the computational cost of AFIR searches can be reduced by using the hybrid model definitions within the ONIOM(QM:QM) or ONIOM(QM:MM) methods. Conventionally, the AFIR analysis is carried out with the higher-level model described at the DFT level with a relatively small basis set, while the lower-level part of the system is treated using semiempirical or force field methods. After approximate local minima (LM) and TSs are identified, standard more accurate computational methods (e.g., DFT with a large basis set) are used for the full molecular system to determine the true LMs and TSs and to rationalize the reaction mechanism and selectivity of the catalytic reaction.
Transition State Search using Chemical Dynamics Simulations (TSSCDS) is an automated strategy to predict the reaction mechanisms and kinetics of organometallic-catalyzed reactions. This method starts with the division of the catalytic system into smaller subsystems, which are sorted by order of increasing complexity. The TSSCDS method is then applied within each of the subsystems to locate the TSs and intermediates, which are subsequently merged into a single reaction network. Finally, this reaction network is used to calculate overall kinetics. The TSSCDS method is based on a procedure that combines accelerated direct dynamics with an efficient geometry-based postprocessing algorithm to find transition states. The method operates with the starting geometries and concentrations of the catalyst and reagents as well as the viscosity of the solvent. The geometries provide the starting configurations for locating intermediates and transition states, while concentrations and the parameters of the solvent are used as the input for the kinetic simulations.723
This method has been successfully tested on the cobalt-catalyzed hydroformylation of ethylene and provided a mechanistic outcome that verified the main pathway proposed by Heck and Breslow.724 The predicted rate law reproduced the one obtained experimentally. Importantly, the TSSCDS can reveal wasteful side reactions and predict their yields, a unique feature that can be used to optimize the reaction conditions and tune selectivity of the catalytic process. For the test hydroformylation process, alkene hydrogenation was identified as the undesirable side-path and it was found to dominate the catalytic process at very low CO pressures.723
The TSSCDS strategy can be successfully employed for studying larger catalytic systems. It uses dynamics simulations, which can be efficiently parallelized. The dynamics module contains algorithms allowing nonuniform sampling of the phase space,725 which can accelerate the TS search or guide the dynamics to mechanisms of greater interest. Finally, the method allows using specific reaction parameters in the semiempirical Hamiltonian for systems where standard parametrization is not efficient or where the procedure needs to be sped up by skipping high-energy paths.
Reiher and co-workers726 have put forward an original algorithm for finding vertices in the reaction network that makes use of conceptual electronic-structure theory to apply heuristic rules for the search of potential chemical transformations within complex reaction mechanisms. The heuristic rules guide the construction of high-energy guess-structures of supermolecules composed of the components of the reactive systems, from which the products of transformations (intermediates in the reaction networks) are derived upon structure optimization. The structures of these intermediates enter an emerging reaction network, in which elementary reactions can be identified in an automated way. In a standard procedure, a species of interest (e.g., a catalyst complex) reacts with a reactive species (e.g., a radical or a charged particle) to produce an intermediate. A collection of all intermediates is arranged in a reaction network. This chemical reaction network can be pruned by defining a certain energy cutoff that excludes consideration of high-energy intermediates that are not inaccessible under a range of reasonable physical reaction conditions and within a characteristic time scale. The heuristics-guided exploration protocol by Reiher and co-workers has been applied to the Chatt–Schrock nitrogen-fixation cycle (Figure 42).726 Its competing reaction paths were not studied in sufficient detail before. A vast number of possible elementary reactions were explored that describe protonation, proton-rearrangement, and reduction steps. The resulting network turned out to be highly complex and alternative routes that still sustain the catalytic cycle emerge.
Figure 42.

Chatt–Schrock network of catalytic nitrogen fixation. Dark-blue vertices refer to the lowest-energy intermediates of a subnetwork, and dark-red vertices refer to the corresponding highest-energy intermediates. Vertices representing Schrock intermediates are enlarged. Low-energy transition barriers between intermediates of the same subnetwork are indicated by dark-gray edges and high-energy transition barriers by light-gray edges. Internetwork connections are indicated by dashed lines. In (a–g), a selection of intermediates is shown. Element color code: gray, C; blue, N; turquoise, Mo; white, H; orange, H added to reactive sites. Reprinted from ref (726). Copyright 2015 American Chemical Society.
4.3. Comparison of Reaction Mechanism Exploration Tools
Table 1 lists the available strategies for reaction mechanism exploration. These can vary significantly in both computational demand and comprehensiveness of the description of the reaction networks that they produce. The ADDF method appears to provide the most comprehensive analysis of the chemical space, but its use is also fundamentally limited by rapid increases in cost with growing system size. On the other side of (in)completeness is the heuristics-based approach by the group of Reiher that is largely based on chemical intuition that potentially greatly limits the number of reaction pathways to be explored. Nevertheless, this method still allows identification of a great number of pathways that could not be directly envisaged with the expert knowledge only. Similar to other aspects of computational chemistry and catalysis, automated reaction path analysis faces the same problem needed for establishing a balance between accuracy and quality of the model for the investigated problem and the associated computational burden.
Table 1. Comparison of Recent Methodsa for Reaction Path Discovery and TS Optimizationb.
| method* | input required | method of change | TS finding strategy | intermediate generation strategy |
|---|---|---|---|---|
| ADDF721 | reactant(s) | anharmonic downward distortion (ADD) | anharmonic mode following then TS optimization | IRC |
| AFIR729 | reactant(s) | artificial external force | TS optimization along biased pathway | IRC |
| TSSCDS730 | reactant(s) | high-energy dynamics | optimize TS from where bond change occurs | dynamics and IRC |
| West731 | reactants, library of TS geometries | interatomic distances | reactive atom constraints followed by TS optimization | IRC |
| ZStruct732 | reactant, reactive atoms | graph rules | double-ended reaction path optimization | graph rules |
| Green733 | reactant, reactive atoms | graph rules | freezing string then local TS optimization | graph rules |
| Habershon734 | initial reactants and intermediates | reaction/graphical hamiltonian | double-ended reaction path optimization | graph rules |
| Reiher726 | reactants, reactive sites | reactive sites (Heuristics) | interpolation then local TS optimization | Heuristic rules and IRC |
| Nanoreactor735 | reactants | high p, T dynamics | double-ended reaction path optimization | MD trajectories |
| Zstruct2706 | reactants, reactive atoms | graph rules | single-ended growing string | single-ended growing string |
| MD/CD736 | reactant(s) | distance between reactive atoms | interpolation then local TS optimization | trajectories, interatomic distances |
Abbreviations: ADDF, anharmonic downward distortion following; AFIR, artificial force-induced reaction; CD, coordinate driving; MD, molecular dynamics; IRC, intrinsic reaction coordinate; TS, transition state; TSSCDS, transition state search using chemical dynamics simulation.
Adapted with permission from ref (706). Copyright 2017 Wiley-VCH.
The different approaches for automated reaction path analysis can be classified by the degree and type of human guidance required for their optimal functioning. The knowledge-based approaches such as RMG that involve decision making based on the similarities found with the reactions from predefined libraries, the performance, and depth of analysis is ultimately limited by the quality of the available reaction data.
The reaction exploration by AFIR and ZStruct strategies could also be limited by the need to impose configurational preferences on the reacting configurations. The initial implementation of ZStruct worked best for intramolecular reactions due to the requirement that reactants need to be prealigned. This requirement limited the applicability of the method for systematic reaction exploration. Furthermore, the ZStruct approach did not guarantee that pairs of intermediates were connected by a single elementary step, causing double-ended GSM to struggle in obtaining a single representative TS for a multistep pathway.
Enhanced collision and reaction acceleration strategies within TSSCDS could also be considered as a limiting factor for large systems as vibrational mode selection for reactivity analysis will inevitably be incomplete and require manual guidance due to the large number of mode combinations that may be populated. Human input is also required in AFIR strategy in the form of selecting which pairs of molecules/catalyst react as well as intramolecular fragment selection (i.e., active atom selection). Generally speaking, all available strategies, with the exception of the Nanoreactor implementation,727,728 have deficiencies when multiple reactants are involved or solvent participates in the reaction. Such methods still lack capacity for the truly bias-free mechanism exploration desired for a comprehensive analysis of multistep, multicomponent reaction paths.
For all the available methods, the efficient TS search and optimization is paramount for the overall success in the mechanism discovery. The availability of robust and efficient tools that would consistently converge reaction pathways and TSs is critical for the overall convergence of the above strategies. While methods such as the single-ended GSM and West’s TS estimator have provided some advances in this regard, there is so far no method available that would provide a failsafe TS search algorithm. A failed reaction path optimization may mean the path does not exist, it is highly unfavorable or simply that the optimizer could not reach convergence. This uncertainty is particularly troubling, as the automated approaches would disregard any failed path, even if it were the actual major reaction pathway.
The development of automated methods in reaction mechanism exploration is a very active field in computational chemistry, and with the steady progress in the field, we will move closer to uncovering the full details of chemical reactions with less and less guidance needed from the user or user’s chemical intuition. Realistic catalytic systems are multicomponent complex systems, in which myriads of potential reaction channels can in principle be found. Therefore, the practical application of the automated and comprehensive reaction network analysis tools requires establishing a balance of exploration-exploitation approaches. Currently, this is achieved either in the framework of the graph-representation of molecular systems or by predefining the reactive centers within the molecular ensembles. The former approach is particularly attractive in terms of computational efficiency but has specific limitations when applied to systems with complex electronic structures such as transition metal complexes and clusters, where the application of the concept of valence is not straightforward. The available methodologies require trimming the explored reaction networks to keep them computationally tractable at the expense of potential loss of some of the relevant pathways. Furthermore, to ensure an exhaustive exploration of the chemical space, the completeness of the set of transformation rules is required. However, for an arbitrary, unknown chemical system, this cannot be guaranteed. One will then be restricted to known or anticipated chemical transformations, which may hamper the discovery of new chemical processes.
Besides these general problems that need to be solved to progress further in this direction such as the inclusion of multicomponent reaction paths, efficient conformational screening and identification of reaction paths, there are more specific challenges related to the field of catalysis by earth-abundant 3d transition metals. This is mainly related to the failure of most of the approximate fast methods suitable for exhaustive mechanistic analysis; they may produce qualitatively incorrect results particularly when dealing with multimetallic and/or paramagnetic systems. Similarly, the less common paths involving such effects as single-electron transfer or excitation-induced reactivity may represent a particular challenge for the approximate methods used in such exploration schemes. Thus, an additional major and crucial limitation of all the current approaches for the automated reaction network analysis originates from the method accuracy of the underlying quantum chemical approaches. Because it is not known which reaction paths will emerge from the automated analysis, one cannot apply the expert knowledge to the selection of the most appropriate quantum chemical method for each specific transformation and reaction channel. The reliance on the expert bias and intuition at least at the stage of the initial selection of the computational methodology for a given system appears to be a persistent issue in modern computational chemistry and catalysis.
5. Concluding Remarks
Electronic structure calculations have become an indispensable tool in catalysis research. They are currently routinely employed to rationalize experimental observations, support mechanistic proposals, and even to guide the design of new catalytic systems. Nevertheless, the vast majority of computational studies in catalysis by transition metals still has an explanatory character and focus on describing only a small part of the actual catalyst system. The transition to truly predictive computational modeling requires the development of more complex chemical models that would allow an adequate description of the full reaction networks underlying the catalytic processes. The elimination of the expert bias in the mechanistic studies is one of the important targets, and here, the emergence of automated methods for reaction network analysis is particularly exciting and holds great promise in delivering a paradigm shift in catalysis research. Particularly important is the extension of mechanistic studies from the descriptions of catalytic cycles to other reaction paths resulting in selectivity losses and catalyst deactivation as these processes actually determine the efficiency of the catalysts and their durability.
Similarly, the adequate description of the complex chemistry of 3d transition metals necessitates the introduction of newer, more accurate, fast, and expert-bias-free methodologies suitable for dealing with the multiconfigurational effects in such catalytic systems. The development of methods to understand reactivity as defined by complicated reaction mechanisms involving multiple spin states and their interconversions in practical 3d metal-based catalysts would provide one more gear to optimize activity and selectivity. Additionally, there is a need for accurate automated reaction discovery tools; however, this necessitates fast and reasonably accurate electronic structure calculations to make them applicable to realistic catalytic systems. The development of increasingly accurate and efficient reaction discovery tools and electronic structure methods with broader applicability will aid in the development of practical, earth-abundant metal catalysts for many vital reactions.
An alternative approach that has also started becoming very popular is the application of machine learning for the fast exploration of the chemical space. Data-driven methodologies are expected to accelerate the identification of key properties that can be used as descriptors of the catalytic and contribute to computational catalyst design.737−739 Those approaches can be combined with the topics discussed in this review article (novel electronic structure theory methods, complex modeling, computation of multiple reaction pathways, and reaction network analysis) for building a comprehensive theoretical framework for computational catalysis.
Acknowledgments
The authors thank Dr. Georgy Filonenko (TU Delft) for creating the table of contents image for this manuscript. This project was in part supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 725686). K.D.V., J.K.K., and J.T. would like to acknowledge the University of Tennessee for financial support of this work (start-up grant). This material is based upon work partially supported by the National Science Foundation under Grant CHE-1800237 (K.D.V., J.K.K., and J.T.). M.V.P. acknowledges the partial support from the Government of the Russian Federation (Grant 08-08) and the Ministry of Education and Science of Russian Federation (Project 11.1706.2017/4.6).
Biographies
Konstantinos D. Vogiatzis (Athens, Greece, 1983) obtained his MSc in applied molecular spectroscopy at the University of Crete, Greece. In 2012, he completed his doctorate in theoretical chemistry at the Karlsruhe Institute of Technology, Germany, where he worked on coupled-cluster theory. In 2014, he worked as a postdoctoral research associate at the University of Minnesota. In 2016, he joined the chemistry department of the University of Tennessee-Knoxville, as an assistant professor. His research interests center on the development of computational methods based on electronic structure theory and machine learning algorithms for describing chemical systems relevant to green chemistry, with particular focus on biomimetic catalysis and gas separation processes.
Mikhail V. Polynski (Moscow, Russia, 1990) graduated from the Higher Chemical College of the Russian Academy of Sciences in 2013. In 2013–2017, he followed a Ph.D. program at Lomonosov Moscow State University and carried out research in computational chemistry and catalysis at Zelinsky Institute of Organic Chemistry, Moscow, under the guidance of Prof. Valentine Ananikov. Since Fall 2017, he assists Prof. Pidko in building and leading the theoretical chemistry group (TheoMAT) at ITMO University, St. Petersburg, Russia. His main research interests are automation of computational chemistry research, ab initio MD, and theory of catalysis.
Justin K. Kirkland (Orange Park, Florida, United States, 1992) received his B.Sc. degree in chemistry in 2014 at the University of Texas at Austin. In 2016, he joined the group of Dr. Konstantinos D. Vogiatzis at the University of Tennessee-Knoxville to begin his doctoral studies in theoretical and computational chemistry. His research interests include catalysis, small molecule activation, electronic structure theory, and machine learning in chemistry.
Jacob Townsend (Atlanta, Georgia, United States, 1992) is a chemistry Ph.D. student under the supervision of Dr. Konstantinos D. Vogiatzis at the University of Tennessee-Knoxville. His research focuses on the study of advanced materials utilizing machine learning and electronic structure methods.
Ali Hashemi (Isfahan, Iran, 1988) completed his B.Sc. in Chemical Engineering (2013) at Sharif University of Technology and his M.Sc. in Pharmaceutical Engineering (2016) at University of Tehran. Ali’s prior research was focused on molecular dynamics simulations on self-assembling polymeric controlled release drug delivery systems. Since November 2017, he has been a Ph.D. student in the Inorganic System Engineering group at Delft University of Technology, where he is working under the supervision of Prof. Pidko on reaction networks analysis in selective hydrogenation catalysis.
Chong Liu (Henan, China, 1988) received both his BSc (2010) and MSc (2013) in chemistry from Lanzhou University. He received his Ph.D. (2017) from Eindhoven University of Technology, where his research mainly focused on theory of acidity and reactivity in zeolite catalysis. Since Fall 2017, he has been a postdoc in the Inorganic System Engineering group at Delft University of Technology. His research interests lie in computational catalysis, reactivity descriptors, and C1 chemistry.
Evgeny A. Pidko (Moscow, Russia, 1982) received his Ph.D. from Eindhoven University of Technology in 2008, wherein in 2011–2017, he was an Assistant Professor of Catalysis for Sustainability. Since 2016, he has been a part-time professor of theoretical chemistry at ITMO University, St. Petersburg. Since Fall 2017, he has been an Associate Professor and head of the Inorganic Systems Engineering group at Delft University of Technology. In his research, he combines theory and experiment to study mechanisms of homogeneous and heterogeneous catalysts and guide the development of new and improved catalyst systems relevant to sustainable chemistry and energy technologies.
The authors declare no competing financial interest.
References
- Chu S.; Majumdar A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. 10.1038/nature11475. [DOI] [PubMed] [Google Scholar]
- Kerr R. A.; Service R. F. What Can Replace Cheap Oil--and When?. Science 2005, 309, 101–101. 10.1126/science.309.5731.101. [DOI] [PubMed] [Google Scholar]
- Hunt A. J.; Farmer T. J. In Sustainable Catalysis: With Non-endangered Metals, Part 1; North M., Ed.; The Royal Society of Chemistry, 2016. [Google Scholar]
- Chirik P.; Morris R. Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res. 2015, 48, 2495–2495. 10.1021/acs.accounts.5b00385. [DOI] [PubMed] [Google Scholar]
- Schafer L. L.; Mountford P.; Piers W. E. Earth abundant element compounds in homogeneous catalysis. Dalton Trans. 2015, 44, 12027–12028. 10.1039/C5DT90105G. [DOI] [PubMed] [Google Scholar]
- Tolman C. A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- Busch M.; Wodrich M. D.; Corminboeuf C. Linear scaling relationships and volcano plots in homogeneous catalysis - revisiting the Suzuki reaction. Chem. Sci. 2015, 6, 6754–6761. 10.1039/C5SC02910D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medford A. J.; Vojvodic A.; Hummelshøj J. S.; Voss J.; Abild-Pedersen F.; Studt F.; Bligaard T.; Nilsson A.; Nørskov J. K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. 10.1016/j.jcat.2014.12.033. [DOI] [Google Scholar]
- Cheng G.-J.; Zhang X.; Chung L. W.; Xu L.; Wu Y.-D. Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions. J. Am. Chem. Soc. 2015, 137, 1706–1725. 10.1021/ja5112749. [DOI] [PubMed] [Google Scholar]
- Sperger T.; Sanhueza I. A.; Schoenebeck F. Computation and Experiment: A Powerful Combination to Understand and Predict Reactivities. Acc. Chem. Res. 2016, 49, 1311–1319. 10.1021/acs.accounts.6b00068. [DOI] [PubMed] [Google Scholar]
- Sperger T.; Sanhueza I. A.; Kalvet I.; Schoenebeck F. Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights. Chem. Rev. 2015, 115, 9532–9586. 10.1021/acs.chemrev.5b00163. [DOI] [PubMed] [Google Scholar]
- Spivey J. J.; Krishna K. S.; Kumar C. S. S. R.; Dooley K. M.; Flake J. C.; Haber L. H.; Xu Y.; Janik M. J.; Sinnott S. B.; Cheng Y.-T.; et al. Synthesis, Characterization, and Computation of Catalysts at the Center for Atomic-Level Catalyst Design. J. Phys. Chem. C 2014, 118, 20043–20069. 10.1021/jp502556u. [DOI] [Google Scholar]
- Van Speybroeck V.; Hemelsoet K.; Joos L.; Waroquier M.; Bell R. G.; Catlow C. R. A. Advances in theory and their application within the field of zeolite chemistry. Chem. Soc. Rev. 2015, 44, 7044–7111. 10.1039/C5CS00029G. [DOI] [PubMed] [Google Scholar]
- Grindy S.; Meredig B.; Kirklin S.; Saal J. E.; Wolverton C. Approaching chemical accuracy with density functional calculations: Diatomic energy corrections. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 87, 075150. 10.1103/PhysRevB.87.075150. [DOI] [Google Scholar]
- Piccini G.; Alessio M.; Sauer J. Ab Initio Calculation of Rate Constants for Molecule–Surface Reactions with Chemical Accuracy. Angew. Chem., Int. Ed. 2016, 55, 5235–5237. 10.1002/anie.201601534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falivene L.; Kozlov S. M.; Cavallo L. Constructing Bridges between Computational Tools in Heterogeneous and Homogeneous Catalysis. ACS Catal. 2018, 8, 5637–5656. 10.1021/acscatal.8b00042. [DOI] [Google Scholar]
- Greeley J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 605–635. 10.1146/annurev-chembioeng-080615-034413. [DOI] [PubMed] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlögl R. Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2015, 54, 3465–3520. 10.1002/anie.201410738. [DOI] [PubMed] [Google Scholar]
- van Santen R. A.; Sengar A.; Steur E. The challenge of catalyst prediction. Faraday Discuss. 2018, 208, 35–52. 10.1039/C7FD00208D. [DOI] [PubMed] [Google Scholar]
- Grajciar L.; Heard C. J.; Bondarenko A. A.; Polynski M. V.; Meeprasert J.; Pidko E. A.; Nachtigall P. Towards operando computational modeling in heterogeneous catalysis. Chem. Soc. Rev. 2018, 10.1039/C8CS00398J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananikov V. P. E.Understanding Organometallic Reaction Mechanisms and Catalysis; Wiley, 2014. [Google Scholar]
- Stirling A.; Nair N. N.; Lledos A.; Ujaque G. Challenges in modelling homogeneous catalysis: new answers from ab initio molecular dynamics to the controversy over the Wacker process. Chem. Soc. Rev. 2014, 43, 4940–4952. 10.1039/C3CS60469A. [DOI] [PubMed] [Google Scholar]
- Torrent M.; Solà M.; Frenking G. Theoretical Studies of Some Transition-Metal-Mediated Reactions of Industrial and Synthetic Importance. Chem. Rev. 2000, 100, 439–494. 10.1021/cr980452i. [DOI] [PubMed] [Google Scholar]
- Koch W.; Holthausen M. C.. A Chemist’s Guide to Density Functional Theory; Wiley: Weinheim, Germany, 2001. [Google Scholar]
- Cramer C. J.Essentials of Computational Chemistry: Theories and Models, 2nd ed.; Wiley: Chichester, UK, 2004. [Google Scholar]
- Lewars E. G.Computational Chemistry, 3rd ed.; Springer: Berlin, Germany, 2016. [Google Scholar]
- Jensen F.Introduction to Computational Chemistry, 3rd ed.; Wiley: Chichester, UK, 2017. [Google Scholar]
- Sholl D. S.; Steckel J. A.. Density Functional Theory: A Practical Introduction; Wiley & Sons: NJ, 2009. [Google Scholar]
- Cohen A. J.; Mori-Sanchez P.; Yang W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. 10.1021/cr200107z. [DOI] [PubMed] [Google Scholar]
- Burke K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. 10.1063/1.4704546. [DOI] [PubMed] [Google Scholar]
- Jones R. O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. 10.1103/RevModPhys.87.897. [DOI] [Google Scholar]
- Pribram-Jones A.; Gross D. A.; Burke K. DFT: A Theory Full of Holes?. Annu. Rev. Phys. Chem. 2015, 66, 283–304. 10.1146/annurev-physchem-040214-121420. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. 10.1063/1.4869598. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; Li S. L.; Truhlar D. G. Perspective: Kohn-Sham density functional theory descending a staircase. J. Chem. Phys. 2016, 145, 130901. 10.1063/1.4963168. [DOI] [PubMed] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Tao J.; Staroverov V. N.; Scuseria G. E.; Csonka G. I. Prescription for the design and selection of density functional approximations: more constraint satisfaction with fewer fits. J. Chem. Phys. 2005, 123, 062201. 10.1063/1.1904565. [DOI] [PubMed] [Google Scholar]
- Tew D. P.; Klopper W.; Helgaker T. Electron correlation: the many-body problem at the heart of chemistry. J. Comput. Chem. 2007, 28, 1307–1320. 10.1002/jcc.20581. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and 12 other functionals. Theor. Chem. Acc. 2008, 119, 525–525. 10.1007/s00214-007-0401-8. [DOI] [Google Scholar]
- Becke A. D. Density - functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Furche F. Molecular tests of the random phase approximation to the exchange-correlation energy functional. Phys. Rev. B: Condens. Matter Mater. Phys. 2001, 64, 195120. 10.1103/PhysRevB.64.195120. [DOI] [Google Scholar]
- Fuchs M.; Niquet Y. M.; Gonze X.; Burke K. Describing static correlation in bond dissociation by Kohn-Sham density functional theory. J. Chem. Phys. 2005, 122, 094116. 10.1063/1.1858371. [DOI] [PubMed] [Google Scholar]
- Furche F. Developing the random phase approximation into a practical post-Kohn-Sham correlation model. J. Chem. Phys. 2008, 129, 114105. 10.1063/1.2977789. [DOI] [PubMed] [Google Scholar]
- Janesko B. G.; Henderson T. M.; Scuseria G. E. Long-range-corrected hybrid density functionals including random phase approximation correlation: application to noncovalent interactions. J. Chem. Phys. 2009, 131, 034110. 10.1063/1.3176514. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. Exact Kohn-Sham scheme based on perturbation theory. Phys. Rev. A: At., Mol., Opt. Phys. 1994, 50, 196–204. 10.1103/PhysRevA.50.196. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. Doubly Hybrid Meta DFT: New Multi-Coefficient Correlation and Density Functional Methods for Thermochemistry and Thermochemical Kinetics. J. Phys. Chem. A 2004, 108, 4786–4791. 10.1021/jp049253v. [DOI] [Google Scholar]
- Cruz F. G.; Lam K.-C.; Burke K. Exchange–Correlation Energy Density from Virial Theorem. J. Phys. Chem. A 1998, 102, 4911–4917. 10.1021/jp980950v. [DOI] [Google Scholar]
- Jaramillo J.; Scuseria G. E.; Ernzerhof M. Local hybrid functionals. J. Chem. Phys. 2003, 118, 1068–1073. 10.1063/1.1528936. [DOI] [Google Scholar]
- Bahmann H.; Rodenberg A.; Arbuznikov A. V.; Kaupp M. A thermochemically competitive local hybrid functional without gradient corrections. J. Chem. Phys. 2007, 126, 011103. 10.1063/1.2429058. [DOI] [PubMed] [Google Scholar]
- Savin A.; Flad H. J. Density functionals for the Yukawa electron-electron interaction. Int. J. Quantum Chem. 1995, 56, 327–332. 10.1002/qua.560560417. [DOI] [Google Scholar]
- Leininger T.; Stoll H.; Werner H.-J.; Savin A. Combining long-range configuration interaction with short-range density functionals. Chem. Phys. Lett. 1997, 275, 151–160. 10.1016/S0009-2614(97)00758-6. [DOI] [Google Scholar]
- Toulouse J.; Colonna F.; Savin A. Long-range–short-range separation of the electron-electron interaction in density-functional theory. Phys. Rev. A: At., Mol., Opt. Phys. 2004, 70, 062505. 10.1103/PhysRevA.70.062505. [DOI] [Google Scholar]
- Wu Q.; Yang W. Empirical correction to density functional theory for van der Waals interactions. J. Chem. Phys. 2002, 116, 515–524. 10.1063/1.1424928. [DOI] [Google Scholar]
- Grimme S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Goerigk L. In Non-Covalent Interactions in Quantum Chemistry and Physics; Otero de la Roza A., DiLabio G. A., Eds.; Elsevier, 2017. [Google Scholar]
- Johnson E. R.; Becke A. D. A post-Hartree-Fock model of intermolecular interactions: inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. 10.1063/1.2190220. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Medvedev M. G.; Bushmarinov I. S.; Sun J.; Perdew J. P.; Lyssenko K. A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. 10.1126/science.aah5975. [DOI] [PubMed] [Google Scholar]
- Brorsen K. R.; Yang Y.; Pak M. V.; Hammes-Schiffer S. Is the Accuracy of Density Functional Theory for Atomization Energies and Densities in Bonding Regions Correlated?. J. Phys. Chem. Lett. 2017, 8, 2076–2081. 10.1021/acs.jpclett.7b00774. [DOI] [PubMed] [Google Scholar]
- Kepp K. P. Comment on “Density functional theory is straying from the path toward the exact functional. Science 2017, 356, 496. 10.1126/science.aam9364. [DOI] [PubMed] [Google Scholar]
- Medvedev M. G.; Bushmarinov I. S.; Sun J.; Perdew J. P.; Lyssenko K. A. Response to Comment on “Density functional theory is straying from the path toward the exact functional. Science 2017, 356, 496. 10.1126/science.aam9550. [DOI] [PubMed] [Google Scholar]
- Korth M. Density Functional Theory: Not Quite the Right Answer for the Right Reason Yet. Angew. Chem., Int. Ed. 2017, 56, 5396–5398. 10.1002/anie.201701894. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Mardirossian N.; Head-Gordon M. omegaB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110. 10.1063/1.4952647. [DOI] [PubMed] [Google Scholar]
- Vydrov O. A.; Van Voorhis T. Nonlocal van der Waals density functional: the simpler the better. J. Chem. Phys. 2010, 133, 244103. 10.1063/1.3521275. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; He X.; Li S. L.; Truhlar D. G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. 10.1039/C6SC00705H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moltved K. A.; Kepp K. P. Chemical Bond Energies of 3d Transition Metals Studied by Density Functional Theory. J. Chem. Theory Comput. 2018, 14, 3479–3492. 10.1021/acs.jctc.8b00143. [DOI] [PubMed] [Google Scholar]
- Freysoldt C.; Grabowski B.; Hickel T.; Neugebauer J.; Kresse G.; Janotti A.; Van de Walle C. G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. 10.1103/RevModPhys.86.253. [DOI] [Google Scholar]
- Kumar G.; Lau S. L. J.; Krcha M. D.; Janik M. J. Correlation of Methane Activation and Oxide Catalyst Reducibility and Its Implications for Oxidative Coupling. ACS Catal. 2016, 6, 1812–1821. 10.1021/acscatal.5b02657. [DOI] [Google Scholar]
- Liechtenstein A. I.; Anisimov V. I.; Zaanen J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 52, R5467–R5470. 10.1103/PhysRevB.52.R5467. [DOI] [PubMed] [Google Scholar]
- Anisimov V. I.; Zaanen J.; Andersen O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B: Condens. Matter Mater. Phys. 1991, 44, 943–954. 10.1103/PhysRevB.44.943. [DOI] [PubMed] [Google Scholar]
- Himmetoglu B.; Floris A.; de Gironcoli S.; Cococcioni M. Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems. Int. J. Quantum Chem. 2014, 114, 14–49. 10.1002/qua.24521. [DOI] [Google Scholar]
- Himmetoglu B.; Marchenko A.; Dabo I.; Cococcioni M. Role of electronic localization in the phosphorescence of iridium sensitizing dyes. J. Chem. Phys. 2012, 137, 154309. 10.1063/1.4757286. [DOI] [PubMed] [Google Scholar]
- Verma P.; Truhlar D. G. Does DFT+U mimic hybrid density functionals?. Theor. Chem. Acc. 2016, 135, 182. 10.1007/s00214-016-1927-4. [DOI] [Google Scholar]
- Beridze G.; Kowalski P. M. Benchmarking the DFT+U Method for Thermochemical Calculations of Uranium Molecular Compounds and Solids. J. Phys. Chem. A 2014, 118, 11797–11810. 10.1021/jp5101126. [DOI] [PubMed] [Google Scholar]
- Varga Z.; Verma P.; Truhlar D. G. Hyper Open-Shell States: The Lowest Excited Spin States of O Atom, Fe2+ Ion, and FeF2. J. Am. Chem. Soc. 2017, 139, 12569–12578. 10.1021/jacs.7b06107. [DOI] [PubMed] [Google Scholar]
- Verma P.; Varga Z.; Truhlar D. G. Hyper Open-Shell Excited Spin States of Transition-Metal Compounds: FeF2, FeF2···Ethane, and FeF2···Ethylene. J. Phys. Chem. A 2018, 122, 2563–2579. 10.1021/acs.jpca.7b12652. [DOI] [PubMed] [Google Scholar]
- De Loth P.; Cassoux P.; Daudey J. P.; Malrieu J. P. Ab initio direct calculation of the singlet-triplet separation in cupric acetate hydrate dimer. J. Am. Chem. Soc. 1981, 103, 4007–4016. 10.1021/ja00404a007. [DOI] [Google Scholar]
- Moreira I.; Illas F. A unified view of the theoretical description of magnetic coupling in molecular chemistry and solid state physics. Phys. Chem. Chem. Phys. 2006, 8, 1645–1659. 10.1039/b515732c. [DOI] [PubMed] [Google Scholar]
- Vogiatzis K. D.; Klopper W.; Mavrandonakis A.; Fink K. Magnetic properties of paddlewheels and trinuclear clusters with exposed metal sites. ChemPhysChem 2011, 12, 3307–3319. 10.1002/cphc.201100559. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A.; Krewald V.; Orio M.; Neese F. Theoretical magnetochemistry of dinuclear manganese complexes: broken symmetry density functional theory investigation on the influence of bridging motifs on structure and magnetism. Dalton Trans 2010, 39, 4959–4967. 10.1039/c001286f. [DOI] [PubMed] [Google Scholar]
- Seeger R.; Pople J. A. Self-consistent molecular orbital methods. XVIII. Constraints and stability in Hartree–Fock theory. J. Chem. Phys. 1977, 66, 3045–3050. 10.1063/1.434318. [DOI] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Stability analysis for solutions of the closed shell Kohn–Sham equation. J. Chem. Phys. 1996, 104, 9047–9052. 10.1063/1.471637. [DOI] [Google Scholar]
- Szalay P. G.; Muller T.; Gidofalvi G.; Lischka H.; Shepard R. Multiconfiguration self-consistent field and multireference configuration interaction methods and applications. Chem. Rev. 2012, 112, 108–181. 10.1021/cr200137a. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P. Å.; Veryazov V.; Widmark P. O.. Multiconfigurational Quantum Chemistry; Wiley, 2016. [Google Scholar]
- de Jong W. A.; Bylaska E.; Govind N.; Janssen C. L.; Kowalski K.; Muller T.; Nielsen I. M.; van Dam H. J.; Veryazov V.; Lindh R. Utilizing high performance computing for chemistry: parallel computational chemistry. Phys. Chem. Chem. Phys. 2010, 12, 6896–6920. 10.1039/c002859b. [DOI] [PubMed] [Google Scholar]
- Vogiatzis K. D.; Ma D.; Olsen J.; Gagliardi L.; de Jong W. A. Pushing configuration-interaction to the limit: Towards massively parallel MCSCF calculations. J. Chem. Phys. 2017, 147, 184111. 10.1063/1.4989858. [DOI] [PubMed] [Google Scholar]
- Olsen J.; Roos B. O.; Jørgensen P.; Jensen H. J. A. Determinant based configuration interaction algorithms for complete and restricted configuration interaction spaces. J. Chem. Phys. 1988, 89, 2185–2192. 10.1063/1.455063. [DOI] [Google Scholar]
- Vogiatzis K. D.; Li Manni G.; Stoneburner S. J.; Ma D.; Gagliardi L. Systematic Expansion of Active Spaces beyond the CASSCF Limit: A GASSCF/SplitGAS Benchmark Study. J. Chem. Theory Comput. 2015, 11, 3010–3021. 10.1021/acs.jctc.5b00191. [DOI] [PubMed] [Google Scholar]
- Ma D.; Li Manni G.; Gagliardi L. The generalized active space concept in multiconfigurational self-consistent field methods. J. Chem. Phys. 2011, 135, 044128. 10.1063/1.3611401. [DOI] [PubMed] [Google Scholar]
- Chan G. K.; Sharma S. The density matrix renormalization group in quantum chemistry. Annu. Rev. Phys. Chem. 2011, 62, 465–481. 10.1146/annurev-physchem-032210-103338. [DOI] [PubMed] [Google Scholar]
- Schollwöck U. The density-matrix renormalization group. Rev. Mod. Phys. 2005, 77, 259–315. 10.1103/RevModPhys.77.259. [DOI] [Google Scholar]
- Knecht S.; Hedegard E. D.; Keller S.; Kovyrshin A.; Ma Y.; Muolo A.; Stein C. J.; Reiher M. New Approaches for ab initio Calculations of Molecules with Strong Electron Correlation. Chimia 2016, 70, 244–251. 10.2533/chimia.2016.244. [DOI] [PubMed] [Google Scholar]
- Booth G. H.; Thom A. J.; Alavi A. Fermion Monte Carlo without fixed nodes: a game of life, death, and annihilation in Slater determinant space. J. Chem. Phys. 2009, 131, 054106. 10.1063/1.3193710. [DOI] [PubMed] [Google Scholar]
- Buenker R. J.; Peyerimhoff S. D. Individualized configuration selection in CI calculations with subsequent energy extrapolation. Theor. Chim. Acta 1974, 35, 33–58. 10.1007/BF02394557. [DOI] [Google Scholar]
- Huron B.; Malrieu J. P.; Rancurel P. Iterative perturbation calculations of ground and excited state energies from multiconfigurational zeroth-order wavefunctions. J. Chem. Phys. 1973, 58, 5745–5759. 10.1063/1.1679199. [DOI] [Google Scholar]
- Smith J. E. T.; Mussard B.; Holmes A. A.; Sharma S. Cheap and Near Exact CASSCF with Large Active Spaces. J. Chem. Theory Comput. 2017, 13, 5468–5478. 10.1021/acs.jctc.7b00900. [DOI] [PubMed] [Google Scholar]
- Schriber J. B.; Evangelista F. A. Communication: An adaptive configuration interaction approach for strongly correlated electrons with tunable accuracy. J. Chem. Phys. 2016, 144, 161106. 10.1063/1.4948308. [DOI] [PubMed] [Google Scholar]
- Nakatani N.; Guo S. Density matrix renormalization group (DMRG) method as a common tool for large active-space CASSCF/CASPT2 calculations. J. Chem. Phys. 2017, 146, 094102. 10.1063/1.4976644. [DOI] [Google Scholar]
- Li Manni G.; Smart S. D.; Alavi A. Combining the Complete Active Space Self-Consistent Field Method and the Full Configuration Interaction Quantum Monte Carlo within a Super-CI Framework, with Application to Challenging Metal-Porphyrins. J. Chem. Theory Comput. 2016, 12, 1245–1258. 10.1021/acs.jctc.5b01190. [DOI] [PubMed] [Google Scholar]
- Roca-Sanjuán D.; Aquilante F.; Lindh R. Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. WIREs Comput. Mol. Sci. 2012, 2, 585–603. 10.1002/wcms.97. [DOI] [Google Scholar]
- Lyakh D. I.; Musial M.; Lotrich V. F.; Bartlett R. J. Multireference nature of chemistry: the coupled-cluster view. Chem. Rev. 2012, 112, 182–243. 10.1021/cr2001417. [DOI] [PubMed] [Google Scholar]
- Miechlich B.; Stoll H.; Savin A. A correlation-energy density functional for multideterminantal wavefunctions. Mol. Phys. 1997, 91, 527–536. 10.1080/002689797171418. [DOI] [Google Scholar]
- Pollet R.; Savin A.; Leininger T.; Stoll H. Combining multideterminantal wave functions with density functionals to handle near-degeneracy in atoms and molecules. J. Chem. Phys. 2002, 116, 1250–1258. 10.1063/1.1430739. [DOI] [Google Scholar]
- Sharkas K.; Savin A.; Jensen H. J.; Toulouse J. A multiconfigurational hybrid density-functional theory. J. Chem. Phys. 2012, 137, 044104. 10.1063/1.4733672. [DOI] [PubMed] [Google Scholar]
- Gagliardi L.; Truhlar D. G.; Li Manni G.; Carlson R. K.; Hoyer C. E.; Bao J. L. Multiconfiguration Pair-Density Functional Theory: A New Way To Treat Strongly Correlated Systems. Acc. Chem. Res. 2017, 50, 66–73. 10.1021/acs.accounts.6b00471. [DOI] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P. Å.; Roos B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. 10.1063/1.462209. [DOI] [Google Scholar]
- Angeli C.; Cimiraglia R.; Evangelisti S.; Leininger T.; Malrieu J. P. Introduction ofn-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. 10.1063/1.1361246. [DOI] [Google Scholar]
- Guo Y.; Sivalingam K.; Valeev E. F.; Neese F. SparseMaps--A systematic infrastructure for reduced-scaling electronic structure methods. III. Linear-scaling multireference domain-based pair natural orbital N-electron valence perturbation theory. J. Chem. Phys. 2016, 144, 094111. 10.1063/1.4942769. [DOI] [PubMed] [Google Scholar]
- Veryazov V.; Malmqvist P. Å.; Roos B. O. How to select active space for multiconfigurational quantum chemistry?. Int. J. Quantum Chem. 2011, 111, 3329–3338. 10.1002/qua.23068. [DOI] [Google Scholar]
- Jensen H. J. A.; Jørgensen P.; Ågren H.; Olsen J. Second-order Møller–Plesset perturbation theory as a configuration and orbital generator in multiconfiguration self-consistent field calculations. J. Chem. Phys. 1988, 88, 3834–3839. 10.1063/1.453884. [DOI] [Google Scholar]
- Pulay P.; Hamilton T. P. UHF natural orbitals for defining and starting MC-SCF calculations. J. Chem. Phys. 1988, 88, 4926–4933. 10.1063/1.454704. [DOI] [Google Scholar]
- Sayfutyarova E. R.; Sun Q.; Chan G. K.; Knizia G. Automated Construction of Molecular Active Spaces from Atomic Valence Orbitals. J. Chem. Theory Comput. 2017, 13, 4063–4078. 10.1021/acs.jctc.7b00128. [DOI] [PubMed] [Google Scholar]
- Stein C. J.; Reiher M. Automated Selection of Active Orbital Spaces. J. Chem. Theory Comput. 2016, 12, 1760–1771. 10.1021/acs.jctc.6b00156. [DOI] [PubMed] [Google Scholar]
- Raghavachari K.; Trucks G. W.; Pople J. A.; Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. 10.1016/S0009-2614(89)87395-6. [DOI] [Google Scholar]
- Dutta A.; Sherrill C. D. Full configuration interaction potential energy curves for breaking bonds to hydrogen: An assessment of single-reference correlation methods. J. Chem. Phys. 2003, 118, 1610–1619. 10.1063/1.1531658. [DOI] [Google Scholar]
- Lee T. J.; Taylor P. R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. 10.1002/qua.560360824. [DOI] [Google Scholar]
- Nielsen I. M. B.; Janssen C. L. Double-substitution-based diagnostics for coupled-cluster and Møller–Plesset perturbation theory. Chem. Phys. Lett. 1999, 310, 568–576. 10.1016/S0009-2614(99)00770-8. [DOI] [Google Scholar]
- Jiang W.; DeYonker N. J.; Wilson A. K. Multireference Character for 3d Transition-Metal-Containing Molecules. J. Chem. Theory Comput. 2012, 8, 460–468. 10.1021/ct2006852. [DOI] [PubMed] [Google Scholar]
- Schultz N. E.; Zhao Y.; Truhlar D. G. Density Functionals for Inorganometallic and Organometallic Chemistry. J. Phys. Chem. A 2005, 109, 11127–11143. 10.1021/jp0539223. [DOI] [PubMed] [Google Scholar]
- Yu H.; Truhlar D. G. Components of the Bond Energy in Polar Diatomic Molecules, Radicals, and Ions Formed by Group-1 and Group-2 Metal Atoms. J. Chem. Theory Comput. 2015, 11, 2968–2983. 10.1021/acs.jctc.5b00083. [DOI] [PubMed] [Google Scholar]
- Hattig C.; Klopper W.; Kohn A.; Tew D. P. Explicitly correlated electrons in molecules. Chem. Rev. 2012, 112, 4–74. 10.1021/cr200168z. [DOI] [PubMed] [Google Scholar]
- Kong L.; Bischoff F. A.; Valeev E. F. Explicitly correlated R12/F12 methods for electronic structure. Chem. Rev. 2012, 112, 75–107. 10.1021/cr200204r. [DOI] [PubMed] [Google Scholar]
- Pulay P. Localizability of dynamic electron correlation. Chem. Phys. Lett. 1983, 100, 151–154. 10.1016/0009-2614(83)80703-9. [DOI] [Google Scholar]
- Kjaergaard T.; Baudin P.; Bykov D.; Kristensen K.; Jørgensen P. The divide-expand-consolidate coupled cluster scheme. WIREs Comput. Mol. Sci. 2017, 7, e1319. 10.1002/wcms.1319. [DOI] [Google Scholar]
- Liakos D. G.; Neese F. Interplay of Correlation and Relativistic Effects in Correlated Calculations on Transition-Metal Complexes: The (Cu2O2)2+ Core Revisited. J. Chem. Theory Comput. 2011, 7, 1511–1523. 10.1021/ct1006949. [DOI] [PubMed] [Google Scholar]
- Mondal B.; Neese F.; Ye S. Control in the Rate-Determining Step Provides a Promising Strategy To Develop New Catalysts for CO2 Hydrogenation: A Local Pair Natural Orbital Coupled Cluster Theory Study. Inorg. Chem. 2015, 54, 7192–7198. 10.1021/acs.inorgchem.5b00469. [DOI] [PubMed] [Google Scholar]
- Minenkov Y.; Chermak E.; Cavallo L. Accuracy of DLPNO-CCSD(T) method for noncovalent bond dissociation enthalpies from coinage metal cation complexes. J. Chem. Theory Comput. 2015, 11, 4664–4676. 10.1021/acs.jctc.5b00584. [DOI] [PubMed] [Google Scholar]
- Da Silva J. C.; Pennifold R. C.; Harvey J. N.; Rocha W. R. A radical rebound mechanism for the methane oxidation reaction promoted by the dicopper center of a pMMO enzyme: a computational perspective. Dalton Trans 2016, 45, 2492–2504. 10.1039/C5DT02638E. [DOI] [PubMed] [Google Scholar]
- Minenkov Y.; Chermak E.; Cavallo L. Troubles in the Systematic Prediction of Transition Metal Thermochemistry with Contemporary Out-of-the-Box Methods. J. Chem. Theory Comput. 2016, 12, 1542–1560. 10.1021/acs.jctc.5b01163. [DOI] [PubMed] [Google Scholar]
- Gani T. Z. H.; Kulik H. J. Unifying Exchange Sensitivity in Transition-Metal Spin-State Ordering and Catalysis through Bond Valence Metrics. J. Chem. Theory Comput. 2017, 13, 5443–5457. 10.1021/acs.jctc.7b00848. [DOI] [PubMed] [Google Scholar]
- Dereli B.; Ortuno M. A.; Cramer C. J. Accurate Ionization Energies for Mononuclear Copper Complexes Remain a Challenge for Density Functional Theory. ChemPhysChem 2018, 19, 959–966. 10.1002/cphc.201701334. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Włoch M.; Piecuch P.; Puzzarini C.; Gagliardi L. Theoretical Models on the Cu2O2 Torture Track: Mechanistic Implications for Oxytyrosinase and Small-Molecule Analogues. J. Phys. Chem. A 2006, 110, 1991–2004. 10.1021/jp056791e. [DOI] [PubMed] [Google Scholar]
- Kozlowski P. M.; Kumar M.; Piecuch P.; Li W.; Bauman N. P.; Hansen J. A.; Lodowski P.; Jaworska M. The Cobalt–Methyl Bond Dissociation in Methylcobalamin: New Benchmark Analysis Based on Density Functional Theory and Completely Renormalized Coupled-Cluster Calculations. J. Chem. Theory Comput. 2012, 8, 1870–1894. 10.1021/ct300170y. [DOI] [PubMed] [Google Scholar]
- Ochsenfeld C.; Kussmann J.; Lambrecht D. S. In Reviews in Computational Chemistry; Lipkowitz K. B., Cundari T. R., Eds.; Wiley, 2007. [Google Scholar]
- Collins M. A.; Bettens R. P. Energy-Based Molecular Fragmentation Methods. Chem. Rev. 2015, 115, 5607–5642. 10.1021/cr500455b. [DOI] [PubMed] [Google Scholar]
- Fedorov D. G.; Nagata T.; Kitaura K. Exploring chemistry with the fragment molecular orbital method. Phys. Chem. Chem. Phys. 2012, 14, 7562–7577. 10.1039/c2cp23784a. [DOI] [PubMed] [Google Scholar]
- Raghavachari K.; Saha A. Accurate Composite and Fragment-Based Quantum Chemical Models for Large Molecules. Chem. Rev. 2015, 115, 5643–5677. 10.1021/cr500606e. [DOI] [PubMed] [Google Scholar]
- Richard R. M.; Lao K. U.; Herbert J. M. Aiming for benchmark accuracy with the many-body expansion. Acc. Chem. Res. 2014, 47, 2828–2836. 10.1021/ar500119q. [DOI] [PubMed] [Google Scholar]
- Fragmentation: Toward Accurate Calculations on Complex Molecular Systems; Gordon M. S., Ed.; Wiley, 2017. [Google Scholar]
- Tsuchiya T.; Shrestha K.; Jakubikova E. Orbital Analysis and Excited-State Calculations in an Energy-Based Fragmentation Method. J. Chem. Theory Comput. 2013, 9, 3350–3363. 10.1021/ct400025a. [DOI] [PubMed] [Google Scholar]
- Stoll H. The correlation energy of crystalline silicon. Chem. Phys. Lett. 1992, 191, 548–552. 10.1016/0009-2614(92)85587-Z. [DOI] [Google Scholar]
- Friedrich J.; Hanrath M.; Dolg M. Fully automated implementation of the incremental scheme: application to CCSD energies for hydrocarbons and transition metal compounds. J. Chem. Phys. 2007, 126, 154110. 10.1063/1.2721538. [DOI] [PubMed] [Google Scholar]
- Vogiatzis K. D.; Klopper W.; Friedrich J. Non-covalent Interactions of CO2 with Functional Groups of Metal-Organic Frameworks from a CCSD(T) Scheme Applicable to Large Systems. J. Chem. Theory Comput. 2015, 11, 1574–1584. 10.1021/ct5011888. [DOI] [PubMed] [Google Scholar]
- Hillier I. H. Chemical reactivity studied by hybrid QM/MM methods. J. Mol. Struct.: THEOCHEM 1999, 463, 45–52. 10.1016/S0166-1280(98)00391-1. [DOI] [Google Scholar]
- Gao J.; Truhlar D. G. Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem. 2002, 53, 467–505. 10.1146/annurev.physchem.53.091301.150114. [DOI] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM methods for biomolecular systems. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- Lin H.; Truhlar D. G. QM/MM: what have we learned, where are we, and where do we go from here?. Theor. Chem. Acc. 2007, 117, 185–199. 10.1007/s00214-006-0143-z. [DOI] [Google Scholar]
- Bo C.; Maseras F. QM/MM methods in inorganic chemistry. Dalton Trans 2008, 37 (22), 2911–2919. 10.1039/b718076d. [DOI] [PubMed] [Google Scholar]
- Chung L. W.; Sameera W. M.; Ramozzi R.; Page A. J.; Hatanaka M.; Petrova G. P.; Harris T. V.; Li X.; Ke Z.; Liu F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. 10.1021/cr5004419. [DOI] [PubMed] [Google Scholar]
- Gordon M. S.; Fedorov D. G.; Pruitt S. R.; Slipchenko L. V. Fragmentation methods: a route to accurate calculations on large systems. Chem. Rev. 2012, 112, 632–672. 10.1021/cr200093j. [DOI] [PubMed] [Google Scholar]
- Wang B.; Truhlar D. G. Including Charge Penetration Effects in Molecular Modeling. J. Chem. Theory Comput. 2010, 6, 3330–3342. 10.1021/ct1003862. [DOI] [PubMed] [Google Scholar]
- Severo Pereira Gomes A.; Jacob C. R. Quantum-chemical embedding methods for treating local electronic excitations in complex chemical systems. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. 2012, 108, 222. 10.1039/c2pc90007f. [DOI] [Google Scholar]
- Jacob C. R.; Neugebauer J. Subsystem density-functional theory. WIREs Comput. Mol. Sci. 2014, 4, 325–362. 10.1002/wcms.1175. [DOI] [Google Scholar]
- Libisch F.; Huang C.; Carter E. A. Embedded Correlated Wavefunction Schemes: Theory and Applications. Acc. Chem. Res. 2014, 47, 2768–2775. 10.1021/ar500086h. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Chan G. K. Quantum Embedding Theories. Acc. Chem. Res. 2016, 49, 2705–2712. 10.1021/acs.accounts.6b00356. [DOI] [PubMed] [Google Scholar]
- Manby F. R.; Stella M.; Goodpaster J. D.; Miller T. F. 3rd. A Simple, Exact Density-Functional-Theory Embedding Scheme. J. Chem. Theory Comput. 2012, 8, 2564–2568. 10.1021/ct300544e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hégely B.; Nagy P. R.; Ferenczy G. G.; Kállay M. Exact density functional and wave function embedding schemes based on orbital localization. J. Chem. Phys. 2016, 145, 064107. 10.1063/1.4960177. [DOI] [Google Scholar]
- Wesołowski T. A.; Warshel A. Frozen density functional approach for ab initio calculations of solvated molecules. J. Phys. Chem. 1993, 97, 8050–8053. 10.1021/j100132a040. [DOI] [Google Scholar]
- Fux S.; Kiewisch K.; Jacob C. R.; Neugebauer J.; Reiher M. Analysis of electron density distributions from subsystem density functional theory applied to coordination bonds. Chem. Phys. Lett. 2008, 461, 353–359. 10.1016/j.cplett.2008.07.038. [DOI] [Google Scholar]
- Wesołowski T. A. Embedding a multideterminantal wave function in an orbital-free environment. Phys. Rev. A: At., Mol., Opt. Phys. 2008, 77, 012504. 10.1103/PhysRevA.77.012504. [DOI] [Google Scholar]
- Huang P.; Carter E. A. Advances in correlated electronic structure methods for solids, surfaces, and nanostructures. Annu. Rev. Phys. Chem. 2008, 59, 261–290. 10.1146/annurev.physchem.59.032607.093528. [DOI] [PubMed] [Google Scholar]
- Kluner T.; Govind N.; Wang Y. A.; Carter E. A. Prediction of electronic excited states of adsorbates on metal surfaces from first principles. Phys. Rev. Lett. 2001, 86, 5954–5957. 10.1103/PhysRevLett.86.5954. [DOI] [PubMed] [Google Scholar]
- Klüner T.; Govind N.; Wang Y. A.; Carter E. A. Periodic density functional embedding theory for complete active space self-consistent field and configuration interaction calculations: Ground and excited states. J. Chem. Phys. 2002, 116, 42. 10.1063/1.1420748. [DOI] [Google Scholar]
- Huang P.; Carter E. A. Self-consistent embedding theory for locally correlated configuration interaction wave functions in condensed matter. J. Chem. Phys. 2006, 125, 084102. 10.1063/1.2336428. [DOI] [PubMed] [Google Scholar]
- Khait Y. G.; Hoffmann M. R. Embedding theory for excited states. J. Chem. Phys. 2010, 133, 044107. 10.1063/1.3460594. [DOI] [PubMed] [Google Scholar]
- Hofener S.; Gomes A. S. P.; Visscher L. Molecular properties via a subsystem density functional theory formulation: a common framework for electronic embedding. J. Chem. Phys. 2012, 136, 044104. 10.1063/1.3675845. [DOI] [PubMed] [Google Scholar]
- Huang C.; Carter E. A. Potential-functional embedding theory for molecules and materials. J. Chem. Phys. 2011, 135, 194104. 10.1063/1.3659293. [DOI] [PubMed] [Google Scholar]
- Roncero O.; de Lara-Castells M. P.; Villarreal P.; Flores F.; Ortega J.; Paniagua M.; Aguado A. An inversion technique for the calculation of embedding potentials. J. Chem. Phys. 2008, 129, 184104. 10.1063/1.3007987. [DOI] [PubMed] [Google Scholar]
- Roncero O.; Zanchet A.; Villarreal P.; Aguado A. A density-division embedding potential inversion technique. J. Chem. Phys. 2009, 131, 234110. 10.1063/1.3274823. [DOI] [PubMed] [Google Scholar]
- Goodpaster J. D.; Barnes T. A.; Miller T. F. 3rd. Embedded density functional theory for covalently bonded and strongly interacting subsystems. J. Chem. Phys. 2011, 134, 164108. 10.1063/1.3582913. [DOI] [PubMed] [Google Scholar]
- Goodpaster J. D.; Barnes T. A.; Manby F. R.; Miller T. F. 3rd. Density functional theory embedding for correlated wavefunctions: Improved methods for open-shell systems and transition metal complexes. J. Chem. Phys. 2012, 137, 224113. 10.1063/1.4770226. [DOI] [PubMed] [Google Scholar]
- Huo P.; Uyeda C.; Goodpaster J. D.; Peters J. C.; Miller T. F. 3rd. Breaking the Correlation between Energy Costs and Kinetic Barriers in Hydrogen Evolution via a Cobalt Pyridine-Diimine-Dioxime Catalyst. ACS Catal. 2016, 6, 6114–6123. 10.1021/acscatal.6b01387. [DOI] [Google Scholar]
- Coughtrie D. J.; Giereth R.; Kats D.; Werner H. J.; Köhn A. Embedded Multireference Coupled Cluster Theory. J. Chem. Theory Comput. 2018, 14, 693–709. 10.1021/acs.jctc.7b01144. [DOI] [PubMed] [Google Scholar]
- Banerjee A.; Simons J. The coupled-cluster method with a multiconfiguration reference state. Int. J. Quantum Chem. 1981, 19, 207–216. 10.1002/qua.560190203. [DOI] [Google Scholar]
- Evangelista F. A.; Gauss J. An orbital-invariant internally contracted multireference coupled cluster approach. J. Chem. Phys. 2011, 134, 114102. 10.1063/1.3559149. [DOI] [PubMed] [Google Scholar]
- Hanauer M.; Kohn A. Communication: Restoring full size extensivity in internally contracted multireference coupled cluster theory. J. Chem. Phys. 2012, 137, 131103. 10.1063/1.4757728. [DOI] [PubMed] [Google Scholar]
- Menezes F.; Kats D.; Werner H. J. Local complete active space second-order perturbation theory using pair natural orbitals (PNO-CASPT2). J. Chem. Phys. 2016, 145, 124115. 10.1063/1.4963019. [DOI] [PubMed] [Google Scholar]
- Garino C.; Salassa L. The photochemistry of transition metal complexes using density functional theory. Philos. Trans. R. Soc., A 2013, 371, 20120134. 10.1098/rsta.2012.0134. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Light K. M.; Liu L. V.; Srnec M.; Wong S. D. Geometric and electronic structure contributions to function in non-heme iron enzymes. Acc. Chem. Res. 2013, 46, 2725–2739. 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srnec M.; Wong S. D.; Matthews M. L.; Krebs C.; Bollinger J. M. Jr.; Solomon E. I. Electronic Structure of the Ferryl Intermediate in the alpha-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. J. Am. Chem. Soc. 2016, 138, 5110–5122. 10.1021/jacs.6b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Draksharapu A.; Harvey E.; Rasheed W.; Que L.; Browne W. R. Direct photochemical activation of non-heme Fe(IV)=O complexes. Chem. Commun. 2017, 53, 12357–12360. 10.1039/C7CC07452B. [DOI] [PubMed] [Google Scholar]
- Kupper C.; Mondal B.; Serrano-Plana J.; Klawitter I.; Neese F.; Costas M.; Ye S.; Meyer F. Nonclassical Single-State Reactivity of an Oxo-Iron(IV) Complex Confined to Triplet Pathways. J. Am. Chem. Soc. 2017, 139, 8939–8949. 10.1021/jacs.7b03255. [DOI] [PubMed] [Google Scholar]
- Werner H. J.; Meyer W. A quadratically convergent MCSCF method for the simultaneous optimization of several states. J. Chem. Phys. 1981, 74, 5794–5801. 10.1063/1.440892. [DOI] [Google Scholar]
- Finley J.; Malmqvist P.-Å.; Roos B. O.; Serrano-Andrés L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. 10.1016/S0009-2614(98)00252-8. [DOI] [Google Scholar]
- Grimme S.; Waletzke M. A combination of Kohn–Sham density functional theory and multi-reference configuration interaction methods. J. Chem. Phys. 1999, 111, 5645–5655. 10.1063/1.479866. [DOI] [Google Scholar]
- Escudero D.; Thiel W. Assessing the density functional theory-based multireference configuration interaction (DFT/MRCI) method for transition metal complexes. J. Chem. Phys. 2014, 140, 194105. 10.1063/1.4875810. [DOI] [PubMed] [Google Scholar]
- Bartlett R. J.; Musiał M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. 10.1103/RevModPhys.79.291. [DOI] [Google Scholar]
- Christiansen O.; Koch H.; Jørgensen P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. 10.1016/0009-2614(95)00841-Q. [DOI] [Google Scholar]
- Cederbaum L. S.; Hohlneicher G.; Peyerimhoff S. Calculation of the vertical ionization potentials of formaldehyde by means of perturbation theory. Chem. Phys. Lett. 1971, 11, 421–424. 10.1016/0009-2614(71)80375-5. [DOI] [Google Scholar]
- Dreuw A.; Wormit M. The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states. WIREs Comput. Mol. Sci. 2015, 5, 82–95. 10.1002/wcms.1206. [DOI] [Google Scholar]
- Oddershede J. Polarization Propagator Calculations. Adv. Quantum Chem. 1978, 11, 275–352. 10.1016/S0065-3276(08)60240-3. [DOI] [Google Scholar]
- Bak K. L.; Koch H.; Oddershede J.; Christiansen O.; Sauer S. P. A. Atomic integral driven second order polarization propagator calculations of the excitation spectra of naphthalene and anthracene. J. Chem. Phys. 2000, 112, 4173–4185. 10.1063/1.480963. [DOI] [Google Scholar]
- Casida M. E.; Huix-Rotllant M. Progress in time-dependent density-functional theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. 10.1146/annurev-physchem-032511-143803. [DOI] [PubMed] [Google Scholar]
- Marques M. A.; Gross E. K. Time-dependent density functional theory. Annu. Rev. Phys. Chem. 2004, 55, 427–455. 10.1146/annurev.physchem.55.091602.094449. [DOI] [PubMed] [Google Scholar]
- Ipatov A.; Cordova F.; Doriol L. J.; Casida M. E. Excited-state spin-contamination in time-dependent density-functional theory for molecules with open-shell ground states. J. Mol. Struct.: THEOCHEM 2009, 914, 60–73. 10.1016/j.theochem.2009.07.036. [DOI] [Google Scholar]
- Daniel C. Electronic spectroscopy and photoreactivity in transition metal complexes. Coord. Chem. Rev. 2003, 238–239, 143–166. 10.1016/S0010-8545(02)00295-3. [DOI] [Google Scholar]
- Neese F.; Petrenko T.; Ganyushin D.; Olbrich G. Advanced aspects of ab initio theoretical optical spectroscopy of transition metal complexes: Multiplets, spin-orbit coupling and resonance Raman intensities. Coord. Chem. Rev. 2007, 251, 288–327. 10.1016/j.ccr.2006.05.019. [DOI] [Google Scholar]
- Cramer C. J.; Truhlar D. G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. 10.1039/b907148b. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Jacquemin D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. 10.1039/C2CS35394F. [DOI] [PubMed] [Google Scholar]
- Milichko V. A.; Khramenkova E. V.; Dzyuba V. P.; Pidko E. A. Response to Comment “On the Existence of Excitonic Signatures in the Optical Response of Metal–Organic Frameworks. Adv. Mater. 2017, 29, 1705261. 10.1002/adma.201705261. [DOI] [PubMed] [Google Scholar]
- Blase X.; Duchemin I.; Jacquemin D. The Bethe-Salpeter equation in chemistry: relations with TD-DFT, applications and challenges. Chem. Soc. Rev. 2018, 47, 1022–1043. 10.1039/C7CS00049A. [DOI] [PubMed] [Google Scholar]
- Korbel S.; Boulanger P.; Duchemin I.; Blase X.; Marques M. A.; Botti S. Benchmark Many-Body GW and Bethe-Salpeter Calculations for Small Transition Metal Molecules. J. Chem. Theory Comput. 2014, 10, 3934–3943. 10.1021/ct5003658. [DOI] [PubMed] [Google Scholar]
- Marx D.; Hutter J.. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press, 2009. [Google Scholar]
- Kühne T. D. Second generation Car-Parrinello molecular dynamics. WIREs Comput. Mol. Sci. 2014, 4, 391–406. 10.1002/wcms.1176. [DOI] [Google Scholar]
- Tse J. S. Ab initio molecular dynamics with density functional theory. Annu. Rev. Phys. Chem. 2002, 53, 249–290. 10.1146/annurev.physchem.53.090401.105737. [DOI] [PubMed] [Google Scholar]
- Tuckerman M. E. Ab initio molecular dynamics: basic concepts, current trends and novel applications. J. Phys.: Condens. Matter 2002, 14, R1297. 10.1088/0953-8984/14/50/202. [DOI] [Google Scholar]
- Rovira C. The description of electronic processes inside proteins from Car-Parrinello molecular dynamics: chemical transformations. WIREs Comput. Mol. Sci. 2013, 3, 393–407. 10.1002/wcms.1153. [DOI] [Google Scholar]
- Carloni P.; Rothlisberger U.; Parrinello M. The Role and Perspective of Ab Initio Molecular Dynamics in the Study of Biological Systems. Acc. Chem. Res. 2002, 35, 455–464. 10.1021/ar010018u. [DOI] [PubMed] [Google Scholar]
- Vidossich P.; Magistrato A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules 2014, 4, 616–645. 10.3390/biom4030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni P.; Bloechl P. E.; Parrinello M. Electronic structure of the Cu, Zn superoxide dismutase active site and its interactions with the substrate. J. Phys. Chem. 1995, 99, 1338–1348. 10.1021/j100004a039. [DOI] [Google Scholar]
- Alfonso-Prieto M.; Biarnes X.; Vidossich P.; Rovira C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. 10.1021/ja9018572. [DOI] [PubMed] [Google Scholar]
- Alfonso-Prieto M.; Borovik A.; Carpena X.; Murshudov G.; Melik-Adamyan W.; Fita I.; Rovira C.; Loewen P. C. The structures and electronic configuration of compound I intermediates of Helicobacter pylori and Penicillium vitale catalases determined by X-ray crystallography and QM/MM density functional theory calculations. J. Am. Chem. Soc. 2007, 129, 4193–4205. 10.1021/ja063660y. [DOI] [PubMed] [Google Scholar]
- Alfonso-Prieto M.; Oberhofer H.; Klein M. L.; Rovira C.; Blumberger J. Proton transfer drives protein radical formation in Helicobacter pylori catalase but not in Penicillium vitale catalase. J. Am. Chem. Soc. 2011, 133, 4285–4298. 10.1021/ja1110706. [DOI] [PubMed] [Google Scholar]
- Campomanes P.; Rothlisberger U.; Alfonso-Prieto M.; Rovira C. The Molecular Mechanism of the Catalase-like Activity in Horseradish Peroxidase. J. Am. Chem. Soc. 2015, 137, 11170–11178. 10.1021/jacs.5b06796. [DOI] [PubMed] [Google Scholar]
- Lian P.; Li J.; Wang D. Q.; Wei D. Q. Car-Parrinello molecular dynamics/molecular mechanics (CPMD/MM) simulation study of coupling and uncoupling mechanisms of Cytochrome P450cam. J. Phys. Chem. B 2013, 117, 7849–7856. 10.1021/jp312107r. [DOI] [PubMed] [Google Scholar]
- Zipoli F.; Car R.; Cohen M. H.; Selloni A. Hydrogen production by the naked active site of the di-iron hydrogenases in water. J. Phys. Chem. B 2009, 113, 13096–13106. 10.1021/jp9059328. [DOI] [PubMed] [Google Scholar]
- Dal Peraro M.; Llarrull L. I.; Rothlisberger U.; Vila A. J.; Carloni P. Water-assisted reaction mechanism of monozinc beta-lactamases. J. Am. Chem. Soc. 2004, 126, 12661–12668. 10.1021/ja048071b. [DOI] [PubMed] [Google Scholar]
- Dal Peraro M.; Vila A. J.; Carloni P.; Klein M. L. Role of zinc content on the catalytic efficiency of B1 metallo beta-lactamases. J. Am. Chem. Soc. 2007, 129, 2808–2816. 10.1021/ja0657556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A.; Rodriguez E. E.; Valensin D.; D’Amelio N.; Ippoliti E.; Obal G.; Duran R.; Magistrato A.; Pritsch O.; Zweckstetter M.; et al. Bioinorganic chemistry of Parkinson’s disease: structural determinants for the copper-mediated amyloid formation of alpha-synuclein. Inorg. Chem. 2010, 49, 10668–10679. 10.1021/ic1016752. [DOI] [PubMed] [Google Scholar]
- Díaz N.; Suárez D.; Merz K. M. Molecular Dynamics Simulations of the Mononuclear Zinc-β-lactamase from Bacilluscereus Complexed with Benzylpenicillin and a Quantum Chemical Study of the Reaction Mechanism. J. Am. Chem. Soc. 2001, 123, 9867–9879. 10.1021/ja0113246. [DOI] [PubMed] [Google Scholar]
- Furlan S.; La Penna G. The mechanism of hydrogen uptake in [NiFe] hydrogenase: first-principles molecular dynamics investigation of a model compound. JBIC, J. Biol. Inorg. Chem. 2012, 17, 149–164. 10.1007/s00775-011-0838-z. [DOI] [PubMed] [Google Scholar]
- Sit P. H.; Car R.; Cohen M. H.; Selloni A. Oxygen tolerance of an in silico-designed bioinspired hydrogen-evolving catalyst in water. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 2017–2022. 10.1073/pnas.1215149110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Sit P. H. Density Functional Theory and Car-Parrinello Molecular Dynamics Study of the Hydrogen-Producing Mechanism of the Co(dmgBF2)2 and Co(dmgH)2 Cobaloxime Complexes in Acetonitrile-Water Solvent. J. Phys. Chem. A 2017, 121, 3515–3525. 10.1021/acs.jpca.7b00163. [DOI] [PubMed] [Google Scholar]
- Pavlova A.; Rösler E.; Meijer E. J. Mechanistic Aspects of Using Formate as a Hydrogen Donor in Aqueous Transfer Hydrogenation. ACS Catal. 2016, 6, 5350–5358. 10.1021/acscatal.6b00323. [DOI] [Google Scholar]
- Scalambra F.; Holzmann N.; Bernasconi L.; Imberti S.; Romerosa A. Water Participation in Catalysis: An Atomistic Approach to Solvent Effects in the Catalytic Isomerization of Allylic Alcohols. ACS Catal. 2018, 8, 3812–3819. 10.1021/acscatal.8b00199. [DOI] [Google Scholar]
- Alhambra C.; Corchado J. C.; Sánchez M. L.; Gao J.; Truhlar D. G. Quantum Dynamics of Hydride Transfer in Enzyme Catalysis. J. Am. Chem. Soc. 2000, 122, 8197–8203. 10.1021/ja001476l. [DOI] [Google Scholar]
- Polyak I.; Allan C. S. M.; Worth G. A. A complete description of tunnelling using direct quantum dynamics simulation: Salicylaldimine proton transfer. J. Chem. Phys. 2015, 143, 084121. 10.1063/1.4929478. [DOI] [PubMed] [Google Scholar]
- Tully J. C. Perspective: Nonadiabatic dynamics theory. J. Chem. Phys. 2012, 137, 22A301. 10.1063/1.4757762. [DOI] [PubMed] [Google Scholar]
- Tapavicza E.; Bellchambers G. D.; Vincent J. C.; Furche F. Ab initio non-adiabatic molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 18336–18348. 10.1039/c3cp51514a. [DOI] [PubMed] [Google Scholar]
- Fang H.; Jing H.; Ge H.; Brothers P. J.; Fu X.; Ye S. The Mechanism of E–H (E = N, O) Bond Activation by a Germanium Corrole Complex: A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2015, 137, 7122–7127. 10.1021/jacs.5b01121. [DOI] [PubMed] [Google Scholar]
- Klähn M.; Garland M. V. On the Mechanism of the Catalytic Binuclear Elimination Reaction in Hydroformylation Systems. ACS Catal. 2015, 5, 2301–2316. 10.1021/cs5019925. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Orozco M.; Luque F. J. Theoretical Methods for the Description of the Solvent Effect in Biomolecular Systems. Chem. Rev. 2000, 100, 4187–4226. 10.1021/cr990052a. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Truhlar D. G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev. 1999, 99, 2161–2200. 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- Klamt A. The COSMO and COSMO-RS solvation models. WIREs Comput. Mol. Sci. 2018, 8, e1338. 10.1002/wcms.1338. [DOI] [Google Scholar]
- Mennucci B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. 10.1002/wcms.1086. [DOI] [Google Scholar]
- Cramer C. J.; Truhlar D. G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. 10.1021/ar800019z. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Mohrig J. R.; Truhlar D. G. Free-Energy Surfaces for Liquid-Phase Reactions and Their Use To Study the Border Between Concerted and Nonconcerted α,β-Elimination Reactions of Esters and Thioesters. J. Am. Chem. Soc. 2010, 132, 11071–11082. 10.1021/ja101104q. [DOI] [PubMed] [Google Scholar]
- Klippenstein S. J.; Pande V. S.; Truhlar D. G. Chemical Kinetics and Mechanisms of Complex Systems: A Perspective on Recent Theoretical Advances. J. Am. Chem. Soc. 2014, 136, 528–546. 10.1021/ja408723a. [DOI] [PubMed] [Google Scholar]
- Reuter K. In Operando Research in Heterogeneous Catalysis; Frenken J.; Groot I., Eds.; Springer International Publishing: Cham, 2017. [Google Scholar]
- Kosinov N.; Liu C.; Hensen E. J. M.; Pidko E. A. Engineering of Transition Metal Catalysts Confined in Zeolites. Chem. Mater. 2018, 30, 3177–3198. 10.1021/acs.chemmater.8b01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidko E. A., Hensen E. J. M. In Zeolites and Zeolite-Like Materials; Kustov L. M., Ed.; Elsevier: Amsterdam, 2016. [Google Scholar]
- Filonenko G. A.; Smykowski D.; Szyja B. M.; Li G.; Szczygiel J.; Hensen E. J. M.; Pidko E. A. Catalytic Hydrogenation of CO2 to Formates by a Lutidine-Derived Ru-CNC Pincer Complex: Theoretical Insight into the Unrealized Potential. ACS Catal. 2015, 5, 1145–1154. 10.1021/cs501990c. [DOI] [Google Scholar]
- Liu C.; van Putten R.; Kulyaev P. O.; Filonenko G. A.; Pidko E. A. Computational insights into the catalytic role of the base promoters in ester hydrogenation with homogeneous non-pincer-based Mn-P,N catalyst. J. Catal. 2018, 363, 136–143. 10.1016/j.jcat.2018.04.018. [DOI] [Google Scholar]
- Klamt A. COSMO-RSC: Second-Order Quasi-Chemical Theory Recovering Local Surface Correlation Effects. J. Phys. Chem. A 2016, 120, 2049–2056. 10.1021/acs.jpca.6b00757. [DOI] [PubMed] [Google Scholar]
- Schröder D.; Shaik S.; Schwarz H. Two-State Reactivity as a New Concept in Organometallic Chemistry. Acc. Chem. Res. 2000, 33, 139–145. 10.1021/ar990028j. [DOI] [PubMed] [Google Scholar]
- Pidko E. A. Toward the Balance between the Reductionist and Systems Approaches in Computational Catalysis: Model versus Method Accuracy for the Description of Catalytic Systems. ACS Catal. 2017, 7, 4230–4234. 10.1021/acscatal.7b00290. [DOI] [Google Scholar]
- Mahyuddin M. H.; Staykov A.; Shiota Y.; Miyanishi M.; Yoshizawa K. Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged AEI, CHA, AFX, and MFI Zeolites. ACS Catal. 2017, 7, 3741–3751. 10.1021/acscatal.7b00588. [DOI] [Google Scholar]
- Göltl F.; Michel C.; Andrikopoulos P. C.; Love A. M.; Hafner J.; Hermans I.; Sautet P. Computationally Exploring Confinement Effects in the Methane-to-Methanol Conversion Over Iron-Oxo Centers in Zeolites. ACS Catal. 2016, 6, 8404–8409. 10.1021/acscatal.6b02640. [DOI] [Google Scholar]
- Maurice R.; Verma P.; Zadrozny J. M.; Luo S.; Borycz J.; Long J. R.; Truhlar D. G.; Gagliardi L. Single-ion magnetic anisotropy and isotropic magnetic couplings in the metal-organic framework Fe2(dobdc). Inorg. Chem. 2013, 52, 9379–9389. 10.1021/ic400953e. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Big Data Meets Quantum Chemistry Approximations: The Delta-Machine Learning Approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. 10.1021/acs.jctc.5b00099. [DOI] [PubMed] [Google Scholar]
- Supady A.; Blum V.; Baldauf C. First-Principles Molecular Structure Search with a Genetic Algorithm. J. Chem. Inf. Model. 2015, 55, 2338–2348. 10.1021/acs.jcim.5b00243. [DOI] [PubMed] [Google Scholar]
- Gomez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernandez-Lobato J. M.; Sanchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadukova M.; Grudinin S. Knodle: A Support Vector Machines-Based Automatic Perception of Organic Molecules from 3D Coordinates. J. Chem. Inf. Model. 2016, 56, 1410–1419. 10.1021/acs.jcim.5b00512. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D. Conformation Generation: The State of the Art. J. Chem. Inf. Model. 2017, 57, 1747–1756. 10.1021/acs.jcim.7b00221. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T. C.; Winkler D. A. Discovery and Optimization of Materials Using Evolutionary Approaches. Chem. Rev. 2016, 116, 6107–6132. 10.1021/acs.chemrev.5b00691. [DOI] [PubMed] [Google Scholar]
- Oliynyk A. O.; Mar A. Discovery of Intermetallic Compounds from Traditional to Machine-Learning Approaches. Acc. Chem. Res. 2018, 51, 59–68. 10.1021/acs.accounts.7b00490. [DOI] [PubMed] [Google Scholar]
- Witman M.; Ling S.; Boyd P.; Barthel S.; Haranczyk M.; Slater B.; Smit B. Cutting Materials in Half: A Graph Theory Approach for Generating Crystal Surfaces and Its Prediction of 2D Zeolites. ACS Cent. Sci. 2018, 4, 235–245. 10.1021/acscentsci.7b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Bombarelli R.; Aguilera-Iparraguirre J.; Hirzel T. D.; Duvenaud D.; Maclaurin D.; Blood-Forsythe M. A.; Chae H. S.; Einzinger M.; Ha D. G.; Wu T.; et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 2016, 15, 1120–1127. 10.1038/nmat4717. [DOI] [PubMed] [Google Scholar]
- Crippen G. M. Note rapid calculation of coordinates from distance matrices. J. Comput. Phys. 1978, 26, 449–452. 10.1016/0021-9991(78)90081-5. [DOI] [Google Scholar]
- Crippen G. M., Havel T. F.. Distance Geometry and Molecular Conformation; Research Studies Press, 1988. [Google Scholar]
- Sadowski J.; Gasteiger J. From atoms and bonds to three-dimensional atomic coordinates: automatic model builders. Chem. Rev. 1993, 93, 2567–2581. 10.1021/cr00023a012. [DOI] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Izrailev S.; Agrafiotis D. K. Conformational Sampling by Self-Organization. J. Chem. Inf. Model. 2003, 43, 1186–1191. 10.1021/ci0340557. [DOI] [PubMed] [Google Scholar]
- Vainio M. J.; Johnson M. S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 2007, 47, 2462–2474. 10.1021/ci6005646. [DOI] [PubMed] [Google Scholar]
- Riniker S.; Landrum G. A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. 10.1021/acs.jcim.5b00654. [DOI] [PubMed] [Google Scholar]
- Miteva M. A.; Guyon F.; Tuffery P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. 10.1093/nar/gkq325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y.; Heyndrickx W.; Occhipinti G.; Jensen V. R.; Alsberg B. K. An evolutionary algorithm for de novo optimization of functional transition metal compounds. J. Am. Chem. Soc. 2012, 134, 8885–8895. 10.1021/ja300865u. [DOI] [PubMed] [Google Scholar]
- Foscato M.; Occhipinti G.; Venkatraman V.; Alsberg B. K.; Jensen V. R. Automated Design of Realistic Organometallic Molecules from Fragments. J. Chem. Inf. Model. 2014, 54, 767–780. 10.1021/ci4007497. [DOI] [PubMed] [Google Scholar]
- Foscato M.; Venkatraman V.; Occhipinti G.; Alsberg B. K.; Jensen V. R. Automated building of organometallic complexes from 3D fragments. J. Chem. Inf. Model. 2014, 54, 1919–1931. 10.1021/ci5003153. [DOI] [PubMed] [Google Scholar]
- Janet J. P.; Gani T. Z. H.; Steeves A. H.; Ioannidis E. I.; Kulik H. J. Leveraging Cheminformatics Strategies for Inorganic Discovery: Application to Redox Potential Design. Ind. Eng. Chem. Res. 2017, 56, 4898–4910. 10.1021/acs.iecr.7b00808. [DOI] [Google Scholar]
- Janet J. P.; Chan L.; Kulik H. J. Accelerating Chemical Discovery with Machine Learning: Simulated Evolution of Spin Crossover Complexes with an Artificial Neural Network. J. Phys. Chem. Lett. 2018, 9, 1064–1071. 10.1021/acs.jpclett.8b00170. [DOI] [PubMed] [Google Scholar]
- Doney A. C.; Rooks B. J.; Lu T.; Wheeler S. E. Design of Organocatalysts for Asymmetric Propargylations through Computational Screening. ACS Catal. 2016, 6, 7948–7955. 10.1021/acscatal.6b02366. [DOI] [Google Scholar]
- Wheeler S. E.; Seguin T. J.; Guan Y.; Doney A. C. Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Design. Acc. Chem. Res. 2016, 49, 1061–1069. 10.1021/acs.accounts.6b00096. [DOI] [PubMed] [Google Scholar]
- Wagner A.; Himmel H. J. aRMSD: A Comprehensive Tool for Structural Analysis. J. Chem. Inf. Model. 2017, 57, 428–438. 10.1021/acs.jcim.6b00516. [DOI] [PubMed] [Google Scholar]
- SMILES - A Simplified Chemical Language; Daylight Chemical Information Systems Inc. [Google Scholar]
- Heller S.; McNaught A.; Stein S.; Tchekhovskoi D.; Pletnev I. InChI - the worldwide chemical structure identifier standard. J. Cheminf. 2013, 5, 7. 10.1186/1758-2946-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M. Accurate specification of molecular structures: the case for zero-order bonds and explicit hydrogen counting. J. Chem. Inf. Model. 2011, 51, 3149–3157. 10.1021/ci200488k. [DOI] [PubMed] [Google Scholar]
- Andronico A.; Randall A.; Benz R. W.; Baldi P. Data-driven high-throughput prediction of the 3-D structure of small molecules: review and progress. J. Chem. Inf. Model. 2011, 51, 760–776. 10.1021/ci100223t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski P.; Baldi P. Small-molecule 3D structure prediction using open crystallography data. J. Chem. Inf. Model. 2013, 53, 3127–3130. 10.1021/ci4005282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzel A.; Kästner J. Gaussian process regression for geometry optimization. J. Chem. Phys. 2018, 148, 094114. 10.1063/1.5017103. [DOI] [Google Scholar]
- Milstein D. Metal–ligand cooperation by aromatization–dearomatization as a tool in single bond activation. Philos. Trans. R. Soc., A 2015, 373, 20140189. 10.1098/rsta.2014.0189. [DOI] [PubMed] [Google Scholar]
- Khusnutdinova J. R.; Milstein D. Metal–Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]
- Wodrich M. D.; Hu X. Natural inspirations for metal–ligand cooperative catalysis. Nat. Rev. Chem. 2017, 2, 0099. 10.1038/s41570-017-0099. [DOI] [Google Scholar]
- van der Vlugt J. I. Cooperative Catalysis with First-Row Late Transition Metals. Eur. J. Inorg. Chem. 2012, 2012, 363–375. 10.1002/ejic.201100752. [DOI] [Google Scholar]
- Karunananda M. K.; Mankad N. P. Cooperative Strategies for Catalytic Hydrogenation of Unsaturated Hydrocarbons. ACS Catal. 2017, 7, 6110–6119. 10.1021/acscatal.7b02203. [DOI] [Google Scholar]
- Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Catalytic (de)hydrogenation promoted by non-precious metals - Co, Fe and Mn: recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. 10.1039/C7CS00334J. [DOI] [PubMed] [Google Scholar]
- Pritchard J.; Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 2015, 44, 3808–3833. 10.1039/C5CS00038F. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Ikariya T. Catalytic Reductive Transformations of Carboxylic and Carbonic Acid Derivatives Using Molecular Hydrogen. ACS Catal. 2012, 2, 1718–1741. 10.1021/cs300341g. [DOI] [Google Scholar]
- Wang D.; Astruc D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. 10.1021/acs.chemrev.5b00203. [DOI] [PubMed] [Google Scholar]
- Zell T.; Langer R. From Ruthenium to Iron and Manganese-A Mechanistic View on Challenges and Design Principles of Base-Metal Hydrogenation Catalysts. ChemCatChem 2018, 10, 1930–1940. 10.1002/cctc.201701722. [DOI] [Google Scholar]
- Kallmeier F.; Kempe R. Manganese Complexes for (De)Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem., Int. Ed. 2018, 57, 46–60. 10.1002/anie.201709010. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Gordon J. C. The mechanism of enantioselective ketone reduction with Noyori and Noyori-Ikariya bifunctional catalysts. Dalton Trans 2016, 45, 6756–6781. 10.1039/C6DT00476H. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Scott B. L.; Gordon J. C. Why Does Alkylation of the N-H Functionality within M/NH Bifunctional Noyori-Type Catalysts Lead to Turnover?. J. Am. Chem. Soc. 2017, 139, 1245–1260. 10.1021/jacs.6b11666. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Ikariya T. Quantum chemical calculations with the inclusion of nonspecific and specific solvation: asymmetric transfer hydrogenation with bifunctional ruthenium catalysts. J. Am. Chem. Soc. 2013, 135, 2604–2619. 10.1021/ja3097674. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Henson N. J.; Martin R. L.; Gordon J. C. Unravelling the mechanism of the asymmetric hydrogenation of acetophenone by [RuX2(diphosphine)(1,2-diamine)] catalysts. J. Am. Chem. Soc. 2014, 136, 3505–3521. 10.1021/ja411374j. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Gordon J. C. Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms. ACS Catal. 2017, 7, 6635–6655. 10.1021/acscatal.7b01791. [DOI] [Google Scholar]
- van Putten R.; Uslamin E. A.; Garbe M.; Liu C.; Gonzalez-de-Castro A.; Lutz M.; Junge K.; Hensen E. J. M.; Beller M.; Lefort L.; et al. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem., Int. Ed. 2017, 56, 7531–7534. 10.1002/anie.201701365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; van Putten R.; Filonenko G. A.; Kulyaev P. O.; Pidko E. A. Computational Insights into the Catalytic Role of the Base Promoters In Ester Hydrogenation with Homogeneous Non-Pincer-Based Mn-P,N Catalyst. J. Catal. 2018, 363, 136–143. 10.1016/j.jcat.2018.04.018. [DOI] [Google Scholar]
- Elangovan S.; Topf C.; Fischer S.; Jiao H.; Spannenberg A.; Baumann W.; Ludwig R.; Junge K.; Beller M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. 10.1021/jacs.6b03709. [DOI] [PubMed] [Google Scholar]
- Nguyen D. H.; Trivelli X.; Capet F.; Paul J.-F.; Dumeignil F.; Gauvin R. M. Manganese Pincer Complexes for the Base-Free, Acceptorless Dehydrogenative Coupling of Alcohols to Esters: Development, Scope, and Understanding. ACS Catal. 2017, 7, 2022–2032. 10.1021/acscatal.6b03554. [DOI] [Google Scholar]
- Gediga M.; Feil C. M.; Schlindwein S. H.; Bender J.; Nieger M.; Gudat D. N-Heterocyclic Phosphenium Complex of Manganese: Synthesis and Catalytic Activity in Ammonia Borane Dehydrogenation. Chem. - Eur. J. 2017, 23, 11560–11569. 10.1002/chem.201701442. [DOI] [PubMed] [Google Scholar]
- Mastalir M.; Glatz M.; Gorgas N.; Stoger B.; Pittenauer E.; Allmaier G.; Veiros L. F.; Kirchner K. Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride Mn(I) and Fe(II) PNP Pincer Complexes. Chem. - Eur. J. 2016, 22, 12316–12320. 10.1002/chem.201603148. [DOI] [PubMed] [Google Scholar]
- Gorgas N.; Stoger B.; Veiros L. F.; Pittenauer E.; Allmaier G.; Kirchner K. Efficient Hydrogenation of Ketones and Aldehydes Catalyzed by Well-Defined Iron(II) PNP Pincer Complexes: Evidence for an Insertion Mechanism. Organometallics 2014, 33, 6905–6914. 10.1021/om5009814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S.; Lagaditis P. O.; Förster M.; Bielinski E. A.; Hazari N.; Holthausen M. C.; Jones W. D.; Schneider S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. 10.1021/cs5009656. [DOI] [Google Scholar]
- Bielinski E. A.; Förster M.; Zhang Y.; Bernskoetter W. H.; Hazari N.; Holthausen M. C. Base-Free Methanol Dehydrogenation Using a Pincer-Supported Iron Compound and Lewis Acid Co-catalyst. ACS Catal. 2015, 5, 2404–2415. 10.1021/acscatal.5b00137. [DOI] [Google Scholar]
- Sawatlon B.; Surawatanawong P. Mechanisms for dehydrogenation and hydrogenation of N-heterocycles using PNP-pincer-supported iron catalysts: a density functional study. Dalton Trans 2016, 45, 14965–14978. 10.1039/C6DT02431A. [DOI] [PubMed] [Google Scholar]
- Xu R.; Chakraborty S.; Bellows S. M.; Yuan H.; Cundari T. R.; Jones W. D. Iron-Catalyzed Homogeneous Hydrogenation of Alkenes under Mild Conditions by a Stepwise, Bifunctional Mechanism. ACS Catal. 2016, 6, 2127–2135. 10.1021/acscatal.5b02674. [DOI] [Google Scholar]
- Zhang Y.; Zhang Y.; Qi Z.-H.; Gao Y.; Liu W.; Wang Y. Ammonia-borane dehydrogenation catalyzed by Iron pincer complexes: A concerted metal-ligand cooperation mechanism. Int. J. Hydrogen Energy 2016, 41, 17208–17215. 10.1016/j.ijhydene.2016.07.209. [DOI] [Google Scholar]
- Li H.; Hall M. B. Computational Mechanistic Studies on Reactions of Transition Metal Complexes with Noninnocent Pincer Ligands: Aromatization–Dearomatization or Not. ACS Catal. 2015, 5, 1895–1913. 10.1021/cs501875z. [DOI] [Google Scholar]
- Berry J. F. Metal–metal multiple bonded intermediates in catalysis. J. Chem. Sci. 2015, 127, 209–214. 10.1007/s12039-015-0773-6. [DOI] [Google Scholar]
- Stephan D. W. Early-late heterobimetallics. Coord. Chem. Rev. 1989, 95, 41–107. 10.1016/0010-8545(89)80002-5. [DOI] [Google Scholar]
- Wheatley N.; Kalck P. Structure and Reactivity of Early–Late Heterobimetallic Complexes. Chem. Rev. 1999, 99, 3379–3420. 10.1021/cr980325m. [DOI] [PubMed] [Google Scholar]
- Gade L. H. Highly Polar Metal–Metal Bonds in “Early–Late” Heterodimetallic Complexes. Angew. Chem., Int. Ed. 2000, 39, 2658–2678. . [DOI] [PubMed] [Google Scholar]
- Bullock R. M.; Casey C. P. Heterobimetallic compounds linked by heterodifunctional ligands. Acc. Chem. Res. 1987, 20, 167–173. 10.1021/ar00137a002. [DOI] [Google Scholar]
- van der Vlugt J. I. Cooperative Catalysis with First-Row Late Transition Metals. Eur. J. Inorg. Chem. 2012, 2012, 363–375. 10.1002/ejic.201100752. [DOI] [Google Scholar]
- Buchwalter P.; Rose J.; Braunstein P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. 10.1021/cr500208k. [DOI] [PubMed] [Google Scholar]
- Powers I. G.; Uyeda C. Metal–Metal Bonds in Catalysis. ACS Catal. 2017, 7, 936–958. 10.1021/acscatal.6b02692. [DOI] [Google Scholar]
- Volbeda A.; Charon M. H.; Piras C.; Hatchikian E. C.; Frey M.; Fontecilla-Camps J. C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- Kim J.; Rees D. C. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677–1682. 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Einsle O.; Howard J. B.; Rees D. C. Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620–1623. 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippel D.; Rohde M.; Netzer J.; Trncik C.; Gies J.; Grunau K.; Djurdjevic I.; Decamps L.; Andrade S. L. A.; Einsle O. A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–1489. 10.1126/science.aar2765. [DOI] [PubMed] [Google Scholar]
- Kuntzleman T.; Yocum C. F. Reduction-induced inhibition and Mn(II) release from the photosystem II oxygen-evolving complex by hydroquinone or NH2OH are consistent with a Mn(III)/Mn(III)/Mn(IV)/Mn(IV) oxidation state for the dark-adapted enzyme. Biochemistry 2005, 44, 2129–2142. 10.1021/bi048460i. [DOI] [PubMed] [Google Scholar]
- Cotton F. A. Discovering and understanding multiple metal-to-metal bonds. Acc. Chem. Res. 1978, 11, 225–232. 10.1021/ar50126a001. [DOI] [Google Scholar]
- Cotton F. A. Strong homonuclear metal-metal bonds. Acc. Chem. Res. 1969, 2, 240–247. 10.1021/ar50020a003. [DOI] [Google Scholar]
- Cotton F. A. Metal-Metal Bonding in [Re2X8]2-Ions and Other Metal Atom Clusters. Inorg. Chem. 1965, 4, 334–336. 10.1021/ic50025a016. [DOI] [Google Scholar]
- Cotton F. A. Centenary Lecture. Quadruple bonds and other multiple metal to metal bonds. Chem. Soc. Rev. 1975, 4, 27–53. 10.1039/cs9750400027. [DOI] [Google Scholar]
- Fontecilla-Camps J. C.; Amara P.; Cavazza C.; Nicolet Y.; Volbeda A. Structure-function relationships of anaerobic gas-processing metalloenzymes. Nature 2009, 460, 814–822. 10.1038/nature08299. [DOI] [PubMed] [Google Scholar]
- Que L. Jr.; Tolman W. B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- Engelmann X.; Monte-Perez I.; Ray K. Oxidation Reactions with Bioinspired Mononuclear Non-Heme Metal-Oxo Complexes. Angew. Chem., Int. Ed. 2016, 55, 7632–7649. 10.1002/anie.201600507. [DOI] [PubMed] [Google Scholar]
- Snyder B. E.; Vanelderen P.; Bols M. L.; Hallaert S. D.; Bottger L. H.; Ungur L.; Pierloot K.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317–321. 10.1038/nature19059. [DOI] [PubMed] [Google Scholar]
- Snyder B. E. R.; Bottger L. H.; Bols M. L.; Yan J. J.; Rhoda H. M.; Jacobs A. B.; Hu M. Y.; Zhao J.; Alp E. E.; Hedman B.; et al. Structural characterization of a non-heme iron active site in zeolites that hydroxylates methane. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4565–4570. 10.1073/pnas.1721717115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelmann A. R.; Stiebritz M. T.; Reiher M. Activation Barriers of Oxygen Transformation at the Active Site of [FeFe] Hydrogenases. Inorg. Chem. 2014, 53, 11890–11902. 10.1021/ic501049z. [DOI] [PubMed] [Google Scholar]
- Vignais P. M.; Billoud B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 2007, 107, 4206–4272. 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Fontecilla-Camps J. C.; Volbeda A.; Cavazza C.; Nicolet Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 2007, 107, 4273–4303. 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- Schilter D.; Camara J. M.; Huynh M. T.; Hammes-Schiffer S.; Rauchfuss T. B. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem. Rev. 2016, 116, 8693–8749. 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G.; Ryde U.; Jensen H. J. A.; Hedegard E. D. Exploration of H2 binding to the [NiFe]-hydrogenase active site with multiconfigurational density functional theory. Phys. Chem. Chem. Phys. 2018, 20, 794–801. 10.1039/C7CP06767D. [DOI] [PubMed] [Google Scholar]
- McCullagh M.; Voth G. A. Unraveling the role of the protein environment for [FeFe]-hydrogenase: a new application of coarse-graining. J. Phys. Chem. B 2013, 117, 4062–4071. 10.1021/jp402441s. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E.; Tye J. W.; Hall M. B. Computational studies of [NiFe] and [FeFe] hydrogenases. Chem. Rev. 2007, 107, 4414–4435. 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- Ryde U.; Greco C.; De Gioia L. Quantum refinement of [FeFe] hydrogenase indicates a dithiomethylamine ligand. J. Am. Chem. Soc. 2010, 132, 4512–4513. 10.1021/ja909194f. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; de Lacey A. L.; Vernède X.; Fernandez V. M.; Hatchikian E. C.; Fontecilla-Camps J. C. Crystallographic and FTIR Spectroscopic Evidence of Changes in Fe Coordination Upon Reduction of the Active Site of the Fe-Only Hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 2001, 123, 1596–1601. 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- Greco C.; Bruschi M.; De Gioia L.; Ryde U. A QM/MM investigation of the activation and catalytic mechanism of Fe-only hydrogenases. Inorg. Chem. 2007, 46, 5911–5921. 10.1021/ic062320a. [DOI] [PubMed] [Google Scholar]
- Greco C.; Silakov A.; Bruschi M.; Ryde U.; De Gioia L.; Lubitz W. Magnetic Properties of [FeFe]-Hydrogenases: A Theoretical Investigation Based on Extended QM and QM/MM Models of the H-Cluster and Its Surroundings. Eur. J. Inorg. Chem. 2011, 2011, 1043–1049. 10.1002/ejic.201001058. [DOI] [Google Scholar]
- Thomas C. M.; Darensbourg M. Y.; Hall M. B. Computational definition of a mixed valent Fe(II)Fe(I) model of the [FeFe]hydrogenase active site resting state. J. Inorg. Biochem. 2007, 101, 1752–1757. 10.1016/j.jinorgbio.2007.06.037. [DOI] [PubMed] [Google Scholar]
- Chang C. H. Computational chemical analysis of [FeFe] hydrogenase H-cluster analogues to discern catalytically relevant features of the natural diatomic ligand configuration. J. Phys. Chem. A 2011, 115, 8691–8704. 10.1021/jp112296d. [DOI] [PubMed] [Google Scholar]
- Long H.; King P. W.; Chang C. H. Proton transport in Clostridium pasteurianum [FeFe] hydrogenase I: a computational study. J. Phys. Chem. B 2014, 118, 890–900. 10.1021/jp408621r. [DOI] [PubMed] [Google Scholar]
- Finkelmann A. R.; Stiebritz M. T.; Reiher M. Electric-field effects on the [FeFe]-hydrogenase active site. Chem. Commun. 2013, 49, 8099–8101. 10.1039/c3cc44112a. [DOI] [PubMed] [Google Scholar]
- Pelmenschikov V.; Birrell J. A.; Pham C. C.; Mishra N.; Wang H.; Sommer C.; Reijerse E.; Richers C. P.; Tamasaku K.; Yoda Y.; et al. Reaction Coordinate Leading to H2 Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc. 2017, 139, 16894–16902. 10.1021/jacs.7b09751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt U. W.; Voth G. A. Multistate Empirical Valence Bond Model for Proton Transport in Water. J. Phys. Chem. B 1998, 102, 5547–5551. 10.1021/jp9818131. [DOI] [Google Scholar]
- Wu Y.; Chen H.; Wang F.; Paesani F.; Voth G. A. An improved multistate empirical valence bond model for aqueous proton solvation and transport. J. Phys. Chem. B 2008, 112, 467–482. 10.1021/jp076658h. [DOI] [PubMed] [Google Scholar]
- Sode O.; Voth G. A. Electron transfer activation of a second water channel for proton transport in [FeFe]-hydrogenase. J. Chem. Phys. 2014, 141, 22D527. 10.1063/1.4902236. [DOI] [PubMed] [Google Scholar]
- Liu C.; Liu T.; Hall M. B. Influence of the density functional and basis set on the relative stabilities of oxygenated isomers of diiron models for the active site of [FeFe]-hydrogenase. J. Chem. Theory Comput. 2015, 11, 205–214. 10.1021/ct500594z. [DOI] [PubMed] [Google Scholar]
- Dong G.; Ryde U. Protonation states of intermediates in the reaction mechanism of [NiFe] hydrogenase studied by computational methods. JBIC, J. Biol. Inorg. Chem. 2016, 21, 383–394. 10.1007/s00775-016-1348-9. [DOI] [PubMed] [Google Scholar]
- Krämer T.; Kampa M.; Lubitz W.; van Gastel M. v.; Neese F. Theoretical Spectroscopy of the NiII Intermediate States in the Catalytic Cycle and the Activation of [NiFe] Hydrogenases. ChemBioChem 2013, 14, 1898–1905. 10.1002/cbic.201300104. [DOI] [PubMed] [Google Scholar]
- Kampa M.; Pandelia M. E.; Lubitz W.; van Gastel M.; Neese F. A metal-metal bond in the light-induced state of [NiFe] hydrogenases with relevance to hydrogen evolution. J. Am. Chem. Soc. 2013, 135, 3915–3925. 10.1021/ja3115899. [DOI] [PubMed] [Google Scholar]
- Breglia R.; Greco C.; Fantucci P.; De Gioia L.; Bruschi M. Theoretical investigation of aerobic and anaerobic oxidative inactivation of the [NiFe]-hydrogenase active site. Phys. Chem. Chem. Phys. 2018, 20, 1693–1706. 10.1039/C7CP06228A. [DOI] [PubMed] [Google Scholar]
- Delcey M. G.; Pierloot K.; Phung Q. M.; Vancoillie S.; Lindh R.; Ryde U. Accurate calculations of geometries and singlet-triplet energy differences for active-site models of [NiFe] hydrogenase. Phys. Chem. Chem. Phys. 2014, 16, 7927–7938. 10.1039/C4CP00253A. [DOI] [PubMed] [Google Scholar]
- Dong G.; Phung Q. M.; Hallaert S. D.; Pierloot K.; Ryde U. H2 binding to the active site of [NiFe] hydrogenase studied by multiconfigurational and coupled-cluster methods. Phys. Chem. Chem. Phys. 2017, 19, 10590–10601. 10.1039/C7CP01331K. [DOI] [PubMed] [Google Scholar]
- Huynh M. T.; Wang W.; Rauchfuss T. B.; Hammes-Schiffer S. Computational investigation of [FeFe]-hydrogenase models: characterization of singly and doubly protonated intermediates and mechanistic insights. Inorg. Chem. 2014, 53, 10301–10311. 10.1021/ic5013523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll M. E.; Barton B. E.; Rauchfuss T. B.; Carroll P. J. Synthetic models for the active site of the [FeFe]-hydrogenase: catalytic proton reduction and the structure of the doubly protonated intermediate. J. Am. Chem. Soc. 2012, 134, 18843–18852. 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogo S.; Ichikawa K.; Kishima T.; Matsumoto T.; Nakai H.; Kusaka K.; Ohhara T. A functional [NiFe]hydrogenase mimic that catalyzes electron and hydride transfer from H2. Science 2013, 339, 682–684. 10.1126/science.1231345. [DOI] [PubMed] [Google Scholar]
- Kochem A.; Bill E.; Neese F.; van Gastel M. Mossbauer and computational investigation of a functional [NiFe] hydrogenase model complex. Chem. Commun. 2015, 51, 2099–2102. 10.1039/C4CC09035G. [DOI] [PubMed] [Google Scholar]
- Chen P.; DeBeer George S.; Cabrito I.; Antholine W. E.; Moura J. J. G.; Moura I.; Hedman B.; Hodgson K. O.; Solomon E. I. Electronic Structure Description of the μ4-Sulfide Bridged Tetranuclear CuZCenter in N2O Reductase. J. Am. Chem. Soc. 2002, 124, 744–745. 10.1021/ja0169623. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L. Copper active sites in biology. Chem. Rev. 2014, 114, 3659–3853. 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner D. E.; Kjaergaard C. H.; Solomon E. I. Mechanism of the Reduction of the Native Intermediate in the Multicopper Oxidases: Insights into Rapid Intramolecular Electron Transfer in Turnover. J. Am. Chem. Soc. 2014, 136, 17788–17801. 10.1021/ja509150j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.; Li N.; Kwak Y.; Srnec M.; Bell C. B.; Liu L. V.; Wong S. D.; Yoda Y.; Kitao S.; Seto M.; et al. Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-heme Iron Enzyme AurF. J. Am. Chem. Soc. 2017, 139, 7062–7070. 10.1021/jacs.7b02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. F.; Du L. Theoretical Study of the Oxidation of Methane to Methanol by the [CuIICuII(μ-O)2CuIII(7-N-Etppz)]1+ Complex. Inorg. Chem. 2018, 57, 3261–3271. 10.1021/acs.inorgchem.8b00054. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. Nitrogenase MoFe-protein at 1.16 A resolution: a central ligand in the FeMo-cofactor. Science 2002, 297, 1696–1700. 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 2011, 334, 940. 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Schlesier J.; Burger M.; Sippel D.; Zhang L.; Andrade S. L. A.; Rees D. C.; Einsle O. Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. 10.1038/ncomms10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunenberg J. The Interstitial Carbon of the Nitrogenase FeMo Cofactor is Far Better Stabilized than Previously Assumed. Angew. Chem., Int. Ed. 2017, 56, 7288–7291. 10.1002/anie.201701790. [DOI] [PubMed] [Google Scholar]
- Scott A. D.; Pelmenschikov V.; Guo Y.; Yan L.; Wang H.; George S. J.; Dapper C. H.; Newton W. E.; Yoda Y.; Tanaka Y.; et al. Structural characterization of CO-inhibited Mo-nitrogenase by combined application of nuclear resonance vibrational spectroscopy, extended X-ray absorption fine structure, and density functional theory: new insights into the effects of CO binding and the role of the interstitial atom. J. Am. Chem. Soc. 2014, 136, 15942–15954. 10.1021/ja505720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M. Model Calculations Suggest that the Central Carbon in the FeMo-Cofactor of Nitrogenase Becomes Protonated in the Process of Nitrogen Fixation. J. Am. Chem. Soc. 2016, 138, 10485–10495. 10.1021/jacs.6b03846. [DOI] [PubMed] [Google Scholar]
- Blomberg M. R.; Borowski T.; Himo F.; Liao R. Z.; Siegbahn P. E. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]
- Siedschlag R. B.; Bernales V.; Vogiatzis K. D.; Planas N.; Clouston L. J.; Bill E.; Gagliardi L.; Lu C. C. Catalytic silylation of dinitrogen with a dicobalt complex. J. Am. Chem. Soc. 2015, 137, 4638–4641. 10.1021/jacs.5b01445. [DOI] [PubMed] [Google Scholar]
- Clouston L. J.; Bernales V.; Carlson R. K.; Gagliardi L.; Lu C. C. Bimetallic cobalt-dinitrogen complexes: impact of the supporting metal on N2 activation. Inorg. Chem. 2015, 54, 9263–9270. 10.1021/acs.inorgchem.5b00983. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Jerome S. V.; Cramer C. J.; Truhlar D. G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. 10.1021/ct200866d. [DOI] [PubMed] [Google Scholar]
- Carlson R. K.; Odoh S. O.; Tereniak S. J.; Lu C. C.; Gagliardi L. Can Multiconfigurational Self-Consistent Field Theory and Density Functional Theory Correctly Predict the Ground State of Metal-Metal-Bonded Complexes?. J. Chem. Theory Comput. 2015, 11, 4093–4101. 10.1021/acs.jctc.5b00412. [DOI] [PubMed] [Google Scholar]
- Eisenhart R. J.; Carlson R. K.; Clouston L. J.; Young V. G. Jr.; Chen Y. S.; Bill E.; Gagliardi L.; Lu C. C. Influence of Copper Oxidation State on the Bonding and Electronic Structure of Cobalt-Copper Complexes. Inorg. Chem. 2015, 54, 11330–11338. 10.1021/acs.inorgchem.5b01950. [DOI] [PubMed] [Google Scholar]
- Clouston L. J.; Bernales V.; Cammarota R. C.; Carlson R. K.; Bill E.; Gagliardi L.; Lu C. C. Heterobimetallic Complexes That Bond Vanadium to Iron, Cobalt and Nickel. Inorg. Chem. 2015, 54, 11669–11679. 10.1021/acs.inorgchem.5b01631. [DOI] [PubMed] [Google Scholar]
- Wu B.; Gramigna K. M.; Bezpalko M. W.; Foxman B. M.; Thomas C. M. Heterobimetallic Ti/Co Complexes That Promote Catalytic N-N Bond Cleavage. Inorg. Chem. 2015, 54, 10909–10917. 10.1021/acs.inorgchem.5b01962. [DOI] [PubMed] [Google Scholar]
- Krogman J. P.; Thomas C. M. Metal-metal multiple bonding in C3-symmetric bimetallic complexes of the first row transition metals. Chem. Commun. 2014, 50, 5115–5127. 10.1039/c3cc47537a. [DOI] [PubMed] [Google Scholar]
- Kuppuswamy S.; Bezpalko M. W.; Powers T. M.; Wilding M. J. T.; Brozek C. K.; Foxman B. M.; Thomas C. M. A series of C3-symmetric heterobimetallic Cr–M (M = Fe, Co and Cu) complexes. Chem. Sci. 2014, 5, 1617–1626. 10.1039/c3sc52943f. [DOI] [Google Scholar]
- Wu B.; Wilding M. J.; Kuppuswamy S.; Bezpalko M. W.; Foxman B. M.; Thomas C. M. Exploring Trends in Metal-Metal Bonding, Spectroscopic Properties, and Conformational Flexibility in a Series of Heterobimetallic Ti/M and V/M Complexes (M = Fe, Co, Ni, and Cu). Inorg. Chem. 2016, 55, 12137–12148. 10.1021/acs.inorgchem.6b01543. [DOI] [PubMed] [Google Scholar]
- Bellows S. M.; Arnet N. A.; Gurubasavaraj P. M.; Brennessel W. W.; Bill E.; Cundari T. R.; Holland P. L. The Mechanism of N-N Double Bond Cleavage by an Iron(II) Hydride Complex. J. Am. Chem. Soc. 2016, 138, 12112–12123. 10.1021/jacs.6b04654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosha D. E.; Mercado B. Q.; Lukat-Rodgers G.; Rodgers K. R.; Holland P. L. Enhancement of C-H Oxidizing Ability in Co-O2 Complexes through an Isolated Heterobimetallic Oxo Intermediate. Angew. Chem., Int. Ed. 2017, 56, 3211–3215. 10.1002/anie.201612010. [DOI] [PubMed] [Google Scholar]
- Kwon D.-H.; Proctor M.; Mendoza S.; Uyeda C.; Ess D. H. Catalytic Dinuclear Nickel Spin Crossover Mechanism and Selectivity for Alkyne Cyclotrimerization. ACS Catal. 2017, 7, 4796–4804. 10.1021/acscatal.7b00978. [DOI] [Google Scholar]
- Walker W. K.; Kay B. M.; Michaelis S. A.; Anderson D. L.; Smith S. J.; Ess D. H.; Michaelis D. J. Origin of fast catalysis in allylic amination reactions catalyzed by Pd-Ti heterobimetallic complexes. J. Am. Chem. Soc. 2015, 137, 7371–7378. 10.1021/jacs.5b02428. [DOI] [PubMed] [Google Scholar]
- Carlsen R. W.; Ess D. H. Allylic amination reactivity of Ni, Pd, and Pt heterobimetallic and monometallic complexes. Dalton. Trans. 2016, 45, 9835–9840. 10.1039/C6DT00256K. [DOI] [PubMed] [Google Scholar]
- Pal S.; Uyeda C. Evaluating the Effect of Catalyst Nuclearity in Ni-Catalyzed Alkyne Cyclotrimerizations. J. Am. Chem. Soc. 2015, 137, 8042–8045. 10.1021/jacs.5b04990. [DOI] [PubMed] [Google Scholar]
- Kilpatrick A. F.; Green J. C.; Cloke F. G. The Reductive Activation of CO2 Across a Ti = Ti Double Bond: Synthetic, Structural, and Mechanistic Studies. Organometallics 2015, 34, 4816–4829. 10.1021/acs.organomet.5b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Motta A.; Mouat A. R.; Delferro M.; Marks T. J. Very large cooperative effects in heterobimetallic titanium-chromium catalysts for ethylene polymerization/copolymerization. J. Am. Chem. Soc. 2014, 136, 10460–10469. 10.1021/ja5046742. [DOI] [PubMed] [Google Scholar]
- Brudvig G. W. Water oxidation chemistry of photosystem II. Philos. Trans. R. Soc., B 2008, 363, 1211–1219. 10.1098/rstb.2007.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore J. D.; Crabtree R. H.; Brudvig G. W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- Najafpour M. M.; Isaloo M. A.; Eaton-Rye J. J.; Tomo T.; Nishihara H.; Satoh K.; Carpentier R.; Shen J. R.; Allakhverdiev S. I. Water exchange in manganese-based water-oxidizing catalysts in photosynthetic systems: from the water-oxidizing complex in photosystem II to nano-sized manganese oxides. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 1395–1410. 10.1016/j.bbabio.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Najafpour M. M.; Renger G.; Holynska M.; Moghaddam A. N.; Aro E. M.; Carpentier R.; Nishihara H.; Eaton-Rye J. J.; Shen J. R.; Allakhverdiev S. I. Manganese Compounds as Water-Oxidizing Catalysts: From the Natural Water-Oxidizing Complex to Nanosized Manganese Oxide Structures. Chem. Rev. 2016, 116, 2886–2936. 10.1021/acs.chemrev.5b00340. [DOI] [PubMed] [Google Scholar]
- Cox N.; Pantazis D. A.; Neese F.; Lubitz W. Biological water oxidation. Acc. Chem. Res. 2013, 46, 1588–1596. 10.1021/ar3003249. [DOI] [PubMed] [Google Scholar]
- Terrett R.; Petrie S.; Stranger R.; Pace R. J. What computational chemistry and magnetic resonance reveal concerning the oxygen evolving centre in Photosystem II. J. Inorg. Biochem. 2016, 162, 178–189. 10.1016/j.jinorgbio.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Cho A. Energy’s tricky tradeoffs. Science 2010, 329, 786–587. 10.1126/science.329.5993.786. [DOI] [PubMed] [Google Scholar]
- Jia J.; Seitz L. C.; Benck J. D.; Huo Y.; Chen Y.; Ng J. W.; Bilir T.; Harris J. S.; Jaramillo T. F. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30. Nat. Commun. 2016, 7, 13237. 10.1038/ncomms13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitrogen Fixation; Nishibayashi Y., Ed.; Springer International Publishing, 2017; Vol. 60. [Google Scholar]
- Gersten S. W.; Samuels G. J.; Meyer T. J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. 10.1021/ja00378a053. [DOI] [Google Scholar]
- Moyer B. A.; Meyer T. J. Oxobis(2,2’-bipyridine)pyridineruthenium(IV) ion, [(bpy)2(py)Ru:O]2+. J. Am. Chem. Soc. 1978, 100, 3601–3603. 10.1021/ja00479a054. [DOI] [Google Scholar]
- Gagliardi C. J.; Vannucci A. K.; Concepcion J. J.; Chen Z.; Meyer T. J. The role of proton coupled electron transfer in water oxidation. Energy Environ. Sci. 2012, 5, 7704–7717. 10.1039/c2ee03311a. [DOI] [Google Scholar]
- Mavros M. G.; Tsuchimochi T.; Kowalczyk T.; McIsaac A.; Wang L. P.; Voorhis T. V. What can density functional theory tell us about artificial catalytic water splitting?. Inorg. Chem. 2014, 53, 6386–6397. 10.1021/ic5002557. [DOI] [PubMed] [Google Scholar]
- Gimbert-Surinach C.; Moonshiram D.; Francas L.; Planas N.; Bernales V.; Bozoglian F.; Guda A.; Mognon L.; Lopez I.; Hoque M. A.; et al. Structural and Spectroscopic Characterization of Reaction Intermediates Involved in a Dinuclear Co-Hbpp Water Oxidation Catalyst. J. Am. Chem. Soc. 2016, 138, 15291–15294. 10.1021/jacs.6b08532. [DOI] [PubMed] [Google Scholar]
- Brodsky C. N.; Hadt R. G.; Hayes D.; Reinhart B. J.; Li N.; Chen L. X.; Nocera D. G. In situ characterization of cofacial Co(IV) centers in Co4O4 cubane: Modeling the high-valent active site in oxygen-evolving catalysts. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3855–3860. 10.1073/pnas.1701816114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanan M. W.; Nocera D. G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- Chakrabarty R.; Bora S. J.; Das B. K. Synthesis, structure, spectral and electrochemical properties, and catalytic use of cobalt(III)-oxo cubane clusters. Inorg. Chem. 2007, 46, 9450–9462. 10.1021/ic7011759. [DOI] [PubMed] [Google Scholar]
- Kanady J. S.; Mendoza-Cortes J. L.; Tsui E. Y.; Nielsen R. J.; Goddard W. A. 3rd; Agapie T. Oxygen atom transfer and oxidative water incorporation in cuboidal Mn3MO(n) complexes based on synthetic, isotopic labeling, and computational studies. J. Am. Chem. Soc. 2013, 135, 1073–1082. 10.1021/ja310022p. [DOI] [PubMed] [Google Scholar]
- Liao R.-Z.; Siegbahn P. E. M. Quantum Chemical Modeling of Homogeneous Water Oxidation Catalysis. ChemSusChem 2017, 10, 4236–4263. 10.1002/cssc.201701374. [DOI] [PubMed] [Google Scholar]
- Sala X.; Maji S.; Bofill R.; García-Antón J.; Escriche L.; Llobet A. Molecular Water Oxidation Mechanisms Followed by Transition Metals: State of the Art. Acc. Chem. Res. 2014, 47, 504–516. 10.1021/ar400169p. [DOI] [PubMed] [Google Scholar]
- Li J.; Güttinger R.; Moré R.; Song F.; Wan W.; Patzke G. R. Frontiers of water oxidation: the quest for true catalysts. Chem. Soc. Rev. 2017, 46, 6124–6147. 10.1039/C7CS00306D. [DOI] [PubMed] [Google Scholar]
- Meyer T. J.; Sheridan M. V.; Sherman B. D. Mechanisms of molecular water oxidation in solution and on oxide surfaces. Chem. Soc. Rev. 2017, 46, 6148–6169. 10.1039/C7CS00465F. [DOI] [PubMed] [Google Scholar]
- Smestad G. P.; Steinfeld A. Review: Photochemical and Thermochemical Production of Solar Fuels from H2O and CO2 Using Metal Oxide Catalysts. Ind. Eng. Chem. Res. 2012, 51, 11828–11840. 10.1021/ie3007962. [DOI] [Google Scholar]
- Tuller H. L. Solar to fuels conversion technologies: a perspective. Mater. Renew. Sustain. Energy 2017, 6, 3. 10.1007/s40243-017-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya J. H.; Seitz L. C.; Chakthranont P.; Vojvodic A.; Jaramillo T. F.; Nørskov J. K. Materials for solar fuels and chemicals. Nat. Mater. 2017, 16, 70. 10.1038/nmat4778. [DOI] [PubMed] [Google Scholar]
- Li Y.; Chan S. H.; Sun Q. Heterogeneous catalytic conversion of CO2: a comprehensive theoretical review. Nanoscale 2015, 7, 8663–8683. 10.1039/C5NR00092K. [DOI] [PubMed] [Google Scholar]
- Artz J.; Müller T. E.; Thenert K.; Kleinekorte J.; Meys R.; Sternberg A.; Bardow A.; Leitner W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. 10.1021/acs.chemrev.7b00435. [DOI] [PubMed] [Google Scholar]
- Roy S.; Cherevotan A.; Peter S. C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. 10.1021/acsenergylett.8b00740. [DOI] [Google Scholar]
- Nielsen D. U.; Hu X.-M.; Daasbjerg K.; Skrydstrup T. Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals. Nat. Catal. 2018, 1, 244–254. 10.1038/s41929-018-0051-3. [DOI] [Google Scholar]
- Wang W.-H.; Himeda Y.; Muckerman J. T.; Manbeck G. F.; Fujita E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. 10.1021/acs.chemrev.5b00197. [DOI] [PubMed] [Google Scholar]
- Álvarez A.; Bansode A.; Urakawa A.; Bavykina A. V.; Wezendonk T. A.; Makkee M.; Gascon J.; Kapteijn F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. 10.1021/acs.chemrev.6b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt N. A., Guan H. In Pincer Compounds; Morales-Morales D., Ed.; Elsevier, 2018. [Google Scholar]
- Tatin A.; Comminges C.; Kokoh B.; Costentin C.; Robert M.; Savéant J.-M. Efficient electrolyzer for CO2 splitting in neutral water using earth-abundant materials. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5526–5529. 10.1073/pnas.1604628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco F.; Pinto M. F.; Royo B.; Lloret-Fillol J. A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem., Int. Ed. 2018, 57, 4603–4606. 10.1002/anie.201800705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H.; Schmidt L. C.; Bonin J.; Robert M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74. 10.1038/nature23016. [DOI] [PubMed] [Google Scholar]
- Sinopoli A.; La Porte N. T.; Martinez J. F.; Wasielewski M. R.; Sohail M. Manganese carbonyl complexes for CO2 reduction. Coord. Chem. Rev. 2018, 365, 60–74. 10.1016/j.ccr.2018.03.011. [DOI] [Google Scholar]
- Stanbury M.; Compain J.-D.; Chardon-Noblat S. Electro and photoreduction of CO2 driven by manganese-carbonyl molecular catalysts. Coord. Chem. Rev. 2018, 361, 120–137. 10.1016/j.ccr.2018.01.014. [DOI] [Google Scholar]
- Bonin J.; Maurin A.; Robert M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances. Coord. Chem. Rev. 2017, 334, 184–198. 10.1016/j.ccr.2016.09.005. [DOI] [Google Scholar]
- Grice K. A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. 10.1016/j.ccr.2017.01.007. [DOI] [Google Scholar]
- Wang J.-W.; Liu W.-J.; Zhong D.-C.; Lu T.-B. Nickel complexes as molecular catalysts for water splitting and CO2 reduction. Coord. Chem. Rev. 2017, 10.1016/j.ccr.2017.12.009. [DOI] [Google Scholar]
- Qiao J.; Liu Y.; Hong F.; Zhang J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. 10.1039/C3CS60323G. [DOI] [PubMed] [Google Scholar]
- Francke R.; Schille B.; Roemelt M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. 10.1021/acs.chemrev.7b00459. [DOI] [PubMed] [Google Scholar]
- Kornienko N. Enhancing Catalysis through Substitute-Driven Redox Tuning. Joule 2018, 2, 207–209. 10.1016/j.joule.2018.01.005. [DOI] [Google Scholar]
- Cao L.-M.; Huang H.-H.; Wang J.-W.; Zhong D.-C.; Lu T.-B. The synergistic catalysis effect within a dinuclear nickel complex for efficient and selective electrocatalytic reduction of CO2 to CO. Green Chem. 2018, 20, 798–803. 10.1039/C7GC03451B. [DOI] [Google Scholar]
- Ouyang T.; Huang H.-H.; Wang J.-W.; Zhong D.-C.; Lu T.-B. A Dinuclear Cobalt Cryptate as a Homogeneous Photocatalyst for Highly Selective and Efficient Visible-Light Driven CO2 Reduction to CO in CH3CN/H2O Solution. Angew. Chem., Int. Ed. 2017, 56, 738–743. 10.1002/anie.201610607. [DOI] [PubMed] [Google Scholar]
- Angamuthu R.; Byers P.; Lutz M.; Spek A. L.; Bouwman E. Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex. Science 2010, 327, 313–315. 10.1126/science.1177981. [DOI] [PubMed] [Google Scholar]
- Pokharel U. R.; Fronczek F. R.; Maverick A. W. Reduction of carbon dioxide to oxalate by a binuclear copper complex. Nat. Commun. 2014, 5, 5883. 10.1038/ncomms6883. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Sivalingam K.; Neese F.; Chan G. K. Low-energy spectrum of iron-sulfur clusters directly from many-particle quantum mechanics. Nat. Chem. 2014, 6, 927–933. 10.1038/nchem.2041. [DOI] [PubMed] [Google Scholar]
- Phung Q. M.; Wouters S.; Pierloot K. Cumulant Approximated Second-Order Perturbation Theory Based on the Density Matrix Renormalization Group for Transition Metal Complexes: A Benchmark Study. J. Chem. Theory Comput. 2016, 12, 4352–4361. 10.1021/acs.jctc.6b00714. [DOI] [PubMed] [Google Scholar]
- Pettinari C.; Marchetti F.; Mosca N.; Tosi G.; Drozdov A. Application of metal – organic frameworks. Polym. Int. 2017, 66, 731–744. 10.1002/pi.5315. [DOI] [Google Scholar]
- Odoh S. O.; Cramer C. J.; Truhlar D. G.; Gagliardi L. Quantum-Chemical Characterization of the Properties and Reactivities of Metal-Organic Frameworks. Chem. Rev. 2015, 115, 6051–6111. 10.1021/cr500551h. [DOI] [PubMed] [Google Scholar]
- Bernales V.; Ortuño M. A.; Truhlar D. G.; Cramer C. J.; Gagliardi L. Computational Design of Functionalized Metal–Organic Framework Nodes for Catalysis. ACS Cent. Sci. 2018, 4, 5–19. 10.1021/acscentsci.7b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Farha O. K.; Roberts J.; Scheidt K. A.; Nguyen S. T.; Hupp J. T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. 10.1039/b807080f. [DOI] [PubMed] [Google Scholar]
- Ma L.; Abney C.; Lin W. Enantioselective catalysis with homochiral metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1248–1256. 10.1039/b807083k. [DOI] [PubMed] [Google Scholar]
- Lillerud K. P.; Olsbye U.; Tilset M. Designing Heterogeneous Catalysts by Incorporating Enzyme-Like Functionalities into MOFs. Top. Catal. 2010, 53, 859–868. 10.1007/s11244-010-9518-4. [DOI] [Google Scholar]
- Lin W. Asymmetric Catalysis with Chiral Porous Metal–Organic Frameworks. Top. Catal. 2010, 53, 869–875. 10.1007/s11244-010-9519-3. [DOI] [Google Scholar]
- Corma A.; Garcia H.; Llabres i Xamena F. X. Engineering metal organic frameworks for heterogeneous catalysis. Chem. Rev. 2010, 110, 4606–4655. 10.1021/cr9003924. [DOI] [PubMed] [Google Scholar]
- Gu Z.-Y.; Park J.; Raiff A.; Wei Z.; Zhou H.-C. Metal-Organic Frameworks as Biomimetic Catalysts. ChemCatChem 2014, 6, 67–75. 10.1002/cctc.201300493. [DOI] [Google Scholar]
- Wu C. D.; Zhao M. Incorporation of Molecular Catalysts in Metal-Organic Frameworks for Highly Efficient Heterogeneous Catalysis. Adv. Mater. 2017, 29, 1605446. 10.1002/adma.201605446. [DOI] [PubMed] [Google Scholar]
- Rogge S. M. J.; Bavykina A.; Hajek J.; Garcia H.; Olivos-Suarez A. I.; Sepulveda-Escribano A.; Vimont A.; Clet G.; Bazin P.; Kapteijn F.; et al. Metal-organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. 10.1039/C7CS00033B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z.; Bueken B.; De Vos D. E.; Fischer R. A. Defect-Engineered Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2015, 54, 7234–7254. 10.1002/anie.201411540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getman R. B.; Bae Y. S.; Wilmer C. E.; Snurr R. Q. Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal-organic frameworks. Chem. Rev. 2012, 112, 703–723. 10.1021/cr200217c. [DOI] [PubMed] [Google Scholar]
- Choomwattana S.; Maihom T.; Khongpracha P.; Probst M.; Limtrakul J. Structures and Mechanisms of the Carbonyl-ene Reaction between MOF-11 Encapsulated Formaldehyde and Propylene: An ONIOM Study. J. Phys. Chem. C 2008, 112, 10855–10861. 10.1021/jp8021437. [DOI] [Google Scholar]
- Doitomi K.; Hirao H. Hybrid computational approaches for deriving quantum mechanical insights into metal–organic frameworks. Tetrahedron Lett. 2017, 58, 2309–2317. 10.1016/j.tetlet.2017.04.088. [DOI] [Google Scholar]
- Doitomi K.; Xu K.; Hirao H. The mechanism of an asymmetric ring-opening reaction of epoxide with amine catalyzed by a metal-organic framework: insights from combined quantum mechanics and molecular mechanics calculations. Dalton. Trans. 2017, 46, 3470–3481. 10.1039/C6DT04745A. [DOI] [PubMed] [Google Scholar]
- Deshmukh M. M.; Ohba M.; Kitagawa S.; Sakaki S. Absorption of CO2 and CS2 into the Hofmann-type porous coordination polymer: electrostatic versus dispersion interactions. J. Am. Chem. Soc. 2013, 135, 4840–4849. 10.1021/ja400537f. [DOI] [PubMed] [Google Scholar]
- Bernales V.; League A. B.; Li Z.; Schweitzer N. M.; Peters A. W.; Carlson R. K.; Hupp J. T.; Cramer C. J.; Farha O. K.; Gagliardi L. Computationally Guided Discovery of a Catalytic Cobalt-Decorated Metal–Organic Framework for Ethylene Dimerization. J. Phys. Chem. C 2016, 120, 23576–23583. 10.1021/acs.jpcc.6b07362. [DOI] [Google Scholar]
- Lee K.; Murray É. D.; Kong L.; Lundqvist B. I.; Langreth D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- Verma P.; Xu X.; Truhlar D. G. Adsorption on Fe-MOF-74 for C1–C3 Hydrocarbon Separation. J. Phys. Chem. C 2013, 117, 12648–12660. 10.1021/jp402884h. [DOI] [Google Scholar]
- Verma P.; Vogiatzis K. D.; Planas N.; Borycz J.; Xiao D. J.; Long J. R.; Gagliardi L.; Truhlar D. G. Mechanism of Oxidation of Ethane to Ethanol at Iron(IV)-Oxo Sites in Magnesium-Diluted Fe2(dobdc). J. Am. Chem. Soc. 2015, 137, 5770–5781. 10.1021/jacs.5b00382. [DOI] [PubMed] [Google Scholar]
- Que L. Jr. The road to non-heme oxoferryls and beyond. Acc. Chem. Res. 2007, 40, 493–500. 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- Hohenberger J.; Ray K.; Meyer K. The biology and chemistry of high-valent iron-oxo and iron-nitrido complexes. Nat. Commun. 2012, 3, 720. 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
- Snyder B. E. R.; Vanelderen P.; Bols M. L.; Hallaert S. D.; Böttger L. H.; Ungur L.; Pierloot K.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317. 10.1038/nature19059. [DOI] [PubMed] [Google Scholar]
- Grundner S.; Markovits M. A. C.; Li G.; Tromp M.; Pidko E. A.; Hensen E. J. M.; Jentys A.; Sanchez-Sanchez M.; Lercher J. A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. 10.1038/ncomms8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchina A.; Rivallan M.; Berlier G.; Lamberti C.; Ricchiardi G. Structure and nuclearity of active sites in Fe-zeolites: comparison with iron sites in enzymes and homogeneous catalysts. Phys. Chem. Chem. Phys. 2007, 9, 3483–3499. 10.1039/b703445h. [DOI] [PubMed] [Google Scholar]
- Sumida K.; Horike S.; Kaye S. S.; Herm Z. R.; Queen W. L.; Brown C. M.; Grandjean F.; Long G. J.; Dailly A.; Long J. R. Hydrogen storage and carbon dioxide capture in an iron-based sodalite-type metal–organic framework (Fe-BTT) discovered via high-throughput methods. Chem. Sci. 2010, 1, 184–191. 10.1039/c0sc00179a. [DOI] [Google Scholar]
- Bloch E. D.; Murray L. J.; Queen W. L.; Chavan S.; Maximoff S. N.; Bigi J. P.; Krishna R.; Peterson V. K.; Grandjean F.; Long G. J.; et al. Selective Binding of O2 over N2 in a Redox–Active Metal–Organic Framework with Open Iron(II) Coordination Sites. J. Am. Chem. Soc. 2011, 133, 14814–14822. 10.1021/ja205976v. [DOI] [PubMed] [Google Scholar]
- Ma S.; Yuan D.; Chang J.-S.; Zhou H.-C. Investigation of Gas Adsorption Performances and H2 Affinities of Porous Metal-Organic Frameworks with Different Entatic Metal Centers. Inorg. Chem. 2009, 48, 5398–5402. 10.1021/ic900475q. [DOI] [PubMed] [Google Scholar]
- Xiao D. J.; Bloch E. D.; Mason J. A.; Queen W. L.; Hudson M. R.; Planas N.; Borycz J.; Dzubak A. L.; Verma P.; Lee K.; et al. Oxidation of ethane to ethanol by N2O in a metal-organic framework with coordinatively unsaturated iron(II) sites. Nat. Chem. 2014, 6, 590–595. 10.1038/nchem.1956. [DOI] [PubMed] [Google Scholar]
- Franchini C.; Kovacik R.; Marsman M.; Murthy S. S.; He J.; Ederer C.; Kresse G. Maximally localized Wannier functions in LaMnO3 within PBE + U, hybrid functionals and partially self-consistent GW: an efficient route to construct ab initio tight-binding parameters for eg perovskites. J. Phys.: Condens. Matter 2012, 24, 235602. 10.1088/0953-8984/24/23/235602. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Kumar D.; Que L. Jr.; Shaik S. Two-state reactivity in alkane hydroxylation by non-heme iron-oxo complexes. J. Am. Chem. Soc. 2006, 128, 8590–8606. 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- Ye S.; Neese F. Nonheme oxo-iron(IV) intermediates form an oxyl radical upon approaching the C-H bond activation transition state. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1228–1233. 10.1073/pnas.1008411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England J.; Martinho M.; Farquhar E. R.; Frisch J. R.; Bominaar E. L.; Munck E.; Que L. Jr. A synthetic high-spin oxoiron(IV) complex: generation, spectroscopic characterization, and reactivity. Angew. Chem., Int. Ed. 2009, 48, 3622–3626. 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England J.; Guo Y.; Farquhar E. R.; Young V. G. Jr.; Münck E.; Que L. Jr. The crystal structure of a high-spin oxoiron(IV) complex and characterization of its self-decay pathway. J. Am. Chem. Soc. 2010, 132, 8635–8644. 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy D. C.; Gupta R.; Stone K. L.; Greaves J.; Ziller J. W.; Hendrich M. P.; Borovik A. S. Formation, structure, and EPR detection of a high spin Fe(IV)-oxo species derived from either an Fe(III)-oxo or Fe(III)-OH complex. J. Am. Chem. Soc. 2010, 132, 12188–12190. 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srnec M.; Wong S. D.; England J.; Que L. Jr.; Solomon E. I. pi-Frontier molecular orbitals in S = 2 ferryl species and elucidation of their contributions to reactivity. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14326–14331. 10.1073/pnas.1212693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usharani D.; Janardanan D.; Li C.; Shaik S. A theory for bioinorganic chemical reactivity of oxometal complexes and analogous oxidants: the exchange and orbital-selection rules. Acc. Chem. Res. 2013, 46, 471–482. 10.1021/ar300204y. [DOI] [PubMed] [Google Scholar]
- Kirkland J. K.; Khan S. N.; Casale B.; Miliordos E.; Vogiatzis K. D. Ligand Field Effects on the Ground and Excited States of the Reactive FeO2+ Species. Phys. Chem. Chem. Phys. 2018, 10.1039/C8CP05372C. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Ng W. K. H.; Moeljadi A. M. P.; Bureekaew S. Multiscale Model for a Metal–Organic Framework: High-Spin Rebound Mechanism in the Reaction of the Oxoiron(IV) Species of Fe-MOF-74. ACS Catal. 2015, 5, 3287–3291. 10.1021/acscatal.5b00475. [DOI] [Google Scholar]
- Pestovsky O.; Stoian S.; Bominaar E. L.; Shan X.; Munck E.; Que L. Jr.; Bakac A. Aqueous Fe(IV)=O: spectroscopic identification and oxo-group exchange. Angew. Chem., Int. Ed. 2005, 44, 6871–6874. 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- Chung Y. G.; Camp J.; Haranczyk M.; Sikora B. J.; Bury W.; Krungleviciute V.; Yildirim T.; Farha O. K.; Sholl D. S.; Snurr R. Q. Computation-Ready, Experimental Metal–Organic Frameworks: A Tool To Enable High-Throughput Screening of Nanoporous Crystals. Chem. Mater. 2014, 26, 6185–6192. 10.1021/cm502594j. [DOI] [Google Scholar]
- Vogiatzis K. D.; Haldoupis E.; Xiao D. J.; Long J. R.; Siepmann J. I.; Gagliardi L. Accelerated Computational Analysis of Metal–Organic Frameworks for Oxidation Catalysis. J. Phys. Chem. C 2016, 120, 18707–18712. 10.1021/acs.jpcc.6b07115. [DOI] [Google Scholar]
- Liao P.; Getman R. B.; Snurr R. Q. Optimizing Open Iron Sites in Metal-Organic Frameworks for Ethane Oxidation: A First-Principles Study. ACS Appl. Mater. Interfaces 2017, 9, 33484–33492. 10.1021/acsami.7b02195. [DOI] [PubMed] [Google Scholar]
- Osadchii D. Y.; Olivos-Suarez A. I.; Szécsényi Á.; Li G.; Nasalevich M. A.; Dugulan I. A.; Crespo P. S.; Hensen E. J. M.; Veber S. L.; Fedin M. V.; et al. Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. 10.1021/acscatal.8b00505. [DOI] [Google Scholar]
- Szécsényi Á.; Li G.; Gascon J.; Pidko E. A. Unraveling reaction networks behind the catalytic oxidation of methane with H2O2 over a mixed-metal MIL-53(Al,Fe) MOF catalyst. Chem. Sci. 2018, 9, 6765–6773. 10.1039/C8SC02376J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtch N. C.; Jasuja H.; Walton K. S. Water Stability and Adsorption in Metal–Organic Frameworks. Chem. Rev. 2014, 114, 10575–10612. 10.1021/cr5002589. [DOI] [PubMed] [Google Scholar]
- McHugh L. N.; McPherson M. J.; McCormick L. J.; Morris S. A.; Wheatley P. S.; Teat S. J.; McKay D.; Dawson D. M.; Sansome C. E. F.; Ashbrook S. E.; et al. Hydrolytic stability in hemilabile metal–organic frameworks. Nat. Chem. 2018, 10, 1096. 10.1038/s41557-018-0104-x. [DOI] [PubMed] [Google Scholar]
- Liu J.; Benin A. I.; Furtado A. M. B.; Jakubczak P.; Willis R. R.; LeVan M. D. Stability Effects on CO2 Adsorption for the DOBDC Series of Metal–Organic Frameworks. Langmuir 2011, 27, 11451–11456. 10.1021/la201774x. [DOI] [PubMed] [Google Scholar]
- Zuluaga S.; Fuentes-Fernandez E. M. A.; Tan K.; Xu F.; Li J.; Chabal Y. J.; Thonhauser T. Understanding and controlling water stability of MOF-74. J. Mater. Chem. A 2016, 4, 5176–5183. 10.1039/C5TA10416E. [DOI] [Google Scholar]
- Kuwahara Y.; Yoshimura Y.; Yamashita H. Liquid-phase oxidation of alkylaromatics to aromatic ketones with molecular oxygen over a Mn-based metal–organic framework. Dalton Trans 2017, 46, 8415–8421. 10.1039/C7DT01351E. [DOI] [PubMed] [Google Scholar]
- Wu D.; Yan W.; Xu H.; Zhang E.; Li Q. Defect engineering of Mn-based MOFs with rod-shaped building units by organic linker fragmentation. Inorg. Chim. Acta 2017, 460, 93–98. 10.1016/j.ica.2016.07.022. [DOI] [Google Scholar]
- Khramenkova E. V.; Polynski M. V.; Vinogradov A. V.; Pidko E. A. Degradation paths of manganese-based MOF materials in a model oxidative environment: a computational study. Phys. Chem. Chem. Phys. 2018, 20, 20785–20795. 10.1039/C8CP00397A. [DOI] [PubMed] [Google Scholar]
- Ikuno T.; Zheng J.; Vjunov A.; Sanchez-Sanchez M.; Ortuño M. A.; Pahls D. R.; Fulton J. L.; Camaioni D. M.; Li Z.; Ray D.; et al. Methane Oxidation to Methanol Catalyzed by Cu-Oxo Clusters Stabilized in NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2017, 139, 10294–10301. 10.1021/jacs.7b02936. [DOI] [PubMed] [Google Scholar]
- Pahls D. R.; Ortuno M. A.; Winegar P. H.; Cramer C. J.; Gagliardi L. Computational Screening of Bimetal-Functionalized Zr6O8 MOF Nodes for Methane C-H Bond Activation. Inorg. Chem. 2017, 56, 8739–8743. 10.1021/acs.inorgchem.7b01334. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peters A. W.; Platero-Prats A. E.; Liu J.; Kung C.-W.; Noh H.; DeStefano M. R.; Schweitzer N. M.; Chapman K. W.; Hupp J. T.; et al. Fine-Tuning the Activity of Metal–Organic Framework-Supported Cobalt Catalysts for the Oxidative Dehydrogenation of Propane. J. Am. Chem. Soc. 2017, 139, 15251–15258. 10.1021/jacs.7b09365. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peters A. W.; Bernales V.; Ortuño M. A.; Schweitzer N. M.; DeStefano M. R.; Gallington L. C.; Platero-Prats A. E.; Chapman K. W.; Cramer C. J.; et al. Metal–Organic Framework Supported Cobalt Catalysts for the Oxidative Dehydrogenation of Propane at Low Temperature. ACS Cent. Sci. 2017, 3, 31–38. 10.1021/acscentsci.6b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortuño M. A.; Bernales V.; Gagliardi L.; Cramer C. J. Computational Study of First-Row Transition Metals Supported on MOF NU-1000 for Catalytic Acceptorless Alcohol Dehydrogenation. J. Phys. Chem. C 2016, 120, 24697–24705. 10.1021/acs.jpcc.6b06381. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Ye J. Y.; Liu C. J. Cu3(BTC)2: CO oxidation over MOF based catalysts. Chem. Commun. 2011, 47, 2167–2169. 10.1039/c0cc04944a. [DOI] [PubMed] [Google Scholar]
- Ketrat S.; Maihom T.; Wannakao S.; Probst M.; Nokbin S.; Limtrakul J. Coordinatively Unsaturated Metal-Organic Frameworks M3(btc)2 (M = Cr, Fe, Co, Ni, Cu, and Zn) Catalyzing the Oxidation of CO by N2O: Insight from DFT Calculations. Inorg. Chem. 2017, 56, 14005–14012. 10.1021/acs.inorgchem.7b02143. [DOI] [PubMed] [Google Scholar]
- Noei H.; Amirjalayer S.; Müller M.; Zhang X.; Schmid R.; Muhler M.; Fischer R. A.; Wang Y. Low-Temperature CO Oxidation over Cu-Based Metal-Organic Frameworks Monitored by using FTIR Spectroscopy. ChemCatChem 2012, 4, 709–709. 10.1002/cctc.201290016. [DOI] [Google Scholar]
- Zou R. Q.; Sakurai H.; Xu Q. Preparation, adsorption properties, and catalytic activity of 3D porous metal-organic frameworks composed of cubic building blocks and alkali-metal ions. Angew. Chem., Int. Ed. 2006, 45, 2542–2546. 10.1002/anie.200503923. [DOI] [PubMed] [Google Scholar]
- Zou R. Q.; Sakurai H.; Han S.; Zhong R. Q.; Xu Q. Probing the Lewis acid sites and CO catalytic oxidation activity of the porous metal-organic polymer [Cu(5-methylisophthalate)]. J. Am. Chem. Soc. 2007, 129, 8402–8403. 10.1021/ja071662s. [DOI] [PubMed] [Google Scholar]
- Noodleman L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. 10.1063/1.440939. [DOI] [Google Scholar]
- Heveling J.; van der Beek A.; de Pender M. Oligomerization of ethene over nickel-exchanged zeolite y into a diesel-range product. Appl. Catal. 1988, 42, 325–336. 10.1016/0166-9834(88)80011-3. [DOI] [Google Scholar]
- Brogaard R. Y.; Olsbye U. Ethene Oligomerization in Ni-Containing Zeolites: Theoretical Discrimination of Reaction Mechanisms. ACS Catal. 2016, 6, 1205–1214. 10.1021/acscatal.5b01957. [DOI] [Google Scholar]
- Mlinar A. N.; Keitz B. K.; Gygi D.; Bloch E. D.; Long J. R.; Bell A. T. Selective Propene Oligomerization with Nickel(II)-Based Metal–Organic Frameworks. ACS Catal. 2014, 4, 717–721. 10.1021/cs401189a. [DOI] [Google Scholar]
- Li Z.; Schweitzer N. M.; League A. B.; Bernales V.; Peters A. W.; Getsoian A. B.; Wang T. C.; Miller J. T.; Vjunov A.; Fulton J. L.; et al. Sintering-Resistant Single-Site Nickel Catalyst Supported by Metal-Organic Framework. J. Am. Chem. Soc. 2016, 138, 1977–1982. 10.1021/jacs.5b12515. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Liu N.; Lei Z.; Chen B. Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts. Chem. Rev. 2016, 116, 3658–3721. 10.1021/acs.chemrev.5b00474. [DOI] [PubMed] [Google Scholar]
- Wang P.; Zhao H.; Sun H.; Yu H.; Chen S.; Quan X. Porous metal–organic framework MIL-100(Fe) as an efficient catalyst for the selective catalytic reduction of NOx with NH3. RSC Adv. 2014, 4, 48912–48919. 10.1039/C4RA07028C. [DOI] [Google Scholar]
- Zhang M.; Wang W.; Chen Y. Theoretical investigation of selective catalytic reduction of NO on MIL-100-Fe. Phys. Chem. Chem. Phys. 2018, 20, 2211–2219. 10.1039/C7CP06756A. [DOI] [PubMed] [Google Scholar]
- Brüggemann T. C.; Keil F. J. Theoretical Investigation of the Mechanism of the Oxidation of Nitrogen Oxide on Iron-Form Zeolites in the Presence of Water. J. Phys. Chem. C 2011, 115, 2114–2133. 10.1021/jp1096458. [DOI] [Google Scholar]
- McEwen J. S.; Anggara T.; Schneider W. F.; Kispersky V. F.; Miller J. T.; Delgass W. N.; Ribeiro F. H. Integrated operando X-ray absorption and DFT characterization of Cu–SSZ-13 exchange sites during the selective catalytic reduction of NOx with NH3. Catal. Today 2012, 184, 129–144. 10.1016/j.cattod.2011.11.037. [DOI] [Google Scholar]
- Metkar P. S.; Salazar N.; Muncrief R.; Balakotaiah V.; Harold M. P. Selective catalytic reduction of NO with NH3 on iron zeolite monolithic catalysts: Steady-state and transient kinetics. Appl. Catal., B 2011, 104, 110–126. 10.1016/j.apcatb.2011.02.022. [DOI] [Google Scholar]
- Gao F.; Wang Y.; Kollár M.; Washton N. M.; Szanyi J.; Peden C. H. F. A comparative kinetics study between Cu/SSZ-13 and Fe/SSZ-13 SCR catalysts. Catal. Today 2015, 258, 347–358. 10.1016/j.cattod.2015.01.025. [DOI] [Google Scholar]
- Kornienko N.; Zhao Y.; Kley C. S.; Zhu C.; Kim D.; Lin S.; Chang C. J.; Yaghi O. M.; Yang P. Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. 10.1021/jacs.5b08212. [DOI] [PubMed] [Google Scholar]
- Maihom T.; Wannakao S.; Boekfa B.; Limtrakul J. Production of Formic Acid via Hydrogenation of CO2 over a Copper-Alkoxide-Functionalized MOF: A Mechanistic Study. J. Phys. Chem. C 2013, 117, 17650–17658. 10.1021/jp405178p. [DOI] [Google Scholar]
- Stoneburner S. J.; Livermore V.; McGreal M. E.; Yu D.; Vogiatzis K. D.; Snurr R. Q.; Gagliardi L. Catechol-Ligated Transition Metals: A Quantum Chemical Study on a Promising System for Gas Separation. J. Phys. Chem. C 2017, 121, 10463–10469. 10.1021/acs.jpcc.7b02685. [DOI] [Google Scholar]
- Webber T. E.; Liu W.-G.; Desai S. P.; Lu C. C.; Truhlar D. G.; Penn R. L. Role of a Modulator in the Synthesis of Phase-Pure NU-1000. ACS Appl. Mater. Interfaces 2017, 9, 39342–39346. 10.1021/acsami.7b11348. [DOI] [PubMed] [Google Scholar]
- Sholl D. S.; Lively R. P. Defects in Metal-Organic Frameworks: Challenge or Opportunity?. J. Phys. Chem. Lett. 2015, 6, 3437–3444. 10.1021/acs.jpclett.5b01135. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Muller U.; Yaghi O. M. ″Heterogeneity within order″ in metal-organic frameworks. Angew. Chem., Int. Ed. 2015, 54, 3417–3430. 10.1002/anie.201410252. [DOI] [PubMed] [Google Scholar]
- Ren J.; Ledwaba M.; Musyoka N. M.; Langmi H. W.; Mathe M.; Liao S.; Pang W. Structural defects in metal–organic frameworks (MOFs): Formation, detection and control towards practices of interests. Coord. Chem. Rev. 2017, 349, 169–197. 10.1016/j.ccr.2017.08.017. [DOI] [Google Scholar]
- Valenzano L.; Civalleri B.; Chavan S.; Bordiga S.; Nilsen M. H.; Jakobsen S.; Lillerud K. P.; Lamberti C. Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory. Chem. Mater. 2011, 23, 1700–1718. 10.1021/cm1022882. [DOI] [Google Scholar]
- Platero-Prats A. E.; Mavrandonakis A.; Gallington L. C.; Liu Y.; Hupp J. T.; Farha O. K.; Cramer C. J.; Chapman K. W. Structural Transitions of the Metal-Oxide Nodes within Metal-Organic Frameworks: On the Local Structures of NU-1000 and UiO-66. J. Am. Chem. Soc. 2016, 138, 4178–4185. 10.1021/jacs.6b00069. [DOI] [PubMed] [Google Scholar]
- Mondloch J. E.; Katz M. J.; Isley W. C. III; Ghosh P.; Liao P.; Bury W.; Wagner G. W.; Hall M. G.; DeCoste J. B.; Peterson G. W.; et al. Destruction of chemical warfare agents using metal-organic frameworks. Nat. Mater. 2015, 14, 512–516. 10.1038/nmat4238. [DOI] [PubMed] [Google Scholar]
- Yang D.; Ortuno M. A.; Bernales V.; Cramer C. J.; Gagliardi L.; Gates B. C. Structure and Dynamics of Zr6O8 Metal-Organic Framework Node Surfaces Probed with Ethanol Dehydration as a Catalytic Test Reaction. J. Am. Chem. Soc. 2018, 140, 3751–3759. 10.1021/jacs.7b13330. [DOI] [PubMed] [Google Scholar]
- Planas N.; Mondloch J. E.; Tussupbayev S.; Borycz J.; Gagliardi L.; Hupp J. T.; Farha O. K.; Cramer C. J. Defining the Proton Topology of the Zr6-Based Metal-Organic Framework NU-1000. J. Phys. Chem. Lett. 2014, 5, 3716–3723. 10.1021/jz501899j. [DOI] [PubMed] [Google Scholar]
- Trickett C. A.; Gagnon K. J.; Lee S.; Gandara F.; Burgi H. B.; Yaghi O. M. Definitive molecular level characterization of defects in UiO-66 crystals. Angew. Chem., Int. Ed. 2015, 54, 11162–11167. 10.1002/anie.201505461. [DOI] [PubMed] [Google Scholar]
- Llabrés i Xamena F. X.; Cirujano F. G.; Corma A. An unexpected bifunctional acid base catalysis in IRMOF-3 for Knoevenagel condensation reactions. Microporous Mesoporous Mater. 2012, 157, 112–117. 10.1016/j.micromeso.2011.12.058. [DOI] [Google Scholar]
- Vermoortele F.; Vandichel M.; Van de Voorde B.; Ameloot R.; Waroquier M.; Van Speybroeck V.; De Vos D. E. Electronic Effects of Linker Substitution on Lewis Acid Catalysis with Metal–Organic Frameworks. Angew. Chem., Int. Ed. 2012, 51, 4887–4890. 10.1002/anie.201108565. [DOI] [PubMed] [Google Scholar]
- Vermoortele F.; Bueken B.; Le Bars G.; Van de Voorde B.; Vandichel M.; Houthoofd K.; Vimont A.; Daturi M.; Waroquier M.; Van Speybroeck V.; et al. Synthesis Modulation as a Tool To Increase the Catalytic Activity of Metal–Organic Frameworks: The Unique Case of UiO-66(Zr). J. Am. Chem. Soc. 2013, 135, 11465–11468. 10.1021/ja405078u. [DOI] [PubMed] [Google Scholar]
- Valvekens P.; Jonckheere D.; De Baerdemaeker T.; Kubarev A. V.; Vandichel M.; Hemelsoet K.; Waroquier M.; Van Speybroeck V.; Smolders E.; Depla D.; et al. Base catalytic activity of alkaline earth MOFs: a (micro)spectroscopic study of active site formation by the controlled transformation of structural anions. Chem. Sci. 2014, 5, 4517–4524. 10.1039/C4SC01731E. [DOI] [Google Scholar]
- Fang Z.; Dürholt J. P.; Kauer M.; Zhang W.; Lochenie C.; Jee B.; Albada B.; Metzler-Nolte N.; Poppl A.; Weber B.; et al. Structural complexity in metal-organic frameworks: simultaneous modification of open metal sites and hierarchical porosity by systematic doping with defective linkers. J. Am. Chem. Soc. 2014, 136, 9627–9636. 10.1021/ja503218j. [DOI] [PubMed] [Google Scholar]
- Bureekaew S.; Amirjalayer S.; Tafipolsky M.; Spickermann C.; Roy T. K.; Schmid R. MOF-FF - A flexible first-principles derived force field for metal-organic frameworks. Phys. Status Solidi B 2013, 250, 1128–1141. 10.1002/pssb.201248460. [DOI] [Google Scholar]
- Chen K.; Wu C.-D. Designed fabrication of biomimetic metal–organic frameworks for catalytic applications. Coord. Chem. Rev. 2018, 10.1016/j.ccr.2018.01.016. [DOI] [Google Scholar]
- Fujii H. Electronic structure and reactivity of high-valent oxo iron porphyrins. Coord. Chem. Rev. 2002, 226, 51–60. 10.1016/S0010-8545(01)00441-6. [DOI] [Google Scholar]
- Wurster B.; Grumelli D.; Hotger D.; Gutzler R.; Kern K. Driving the Oxygen Evolution Reaction by Nonlinear Cooperativity in Bimetallic Coordination Catalysts. J. Am. Chem. Soc. 2016, 138, 3623–3626. 10.1021/jacs.5b10484. [DOI] [PubMed] [Google Scholar]
- Mandal B.; Chung J. S.; Kang S. G. Theoretical Insight into M1TPyP–M2 (M1, M2 = Fe, Co) MOFs: Correlation between Electronic Structure and Catalytic Activity Extending to Potentiality in Capturing Flue Gases. J. Phys. Chem. C 2018, 122, 9899–9908. 10.1021/acs.jpcc.8b00080. [DOI] [Google Scholar]
- Roy S.; George C. B.; Ratner M. A. Catalysis by a Zinc-Porphyrin-Based Metal–Organic Framework: From Theory to Computational Design. J. Phys. Chem. C 2012, 116, 23494–23502. 10.1021/jp308496g. [DOI] [Google Scholar]
- Shultz A. M.; Farha O. K.; Hupp J. T.; Nguyen S. T. A catalytically active, permanently microporous MOF with metalloporphyrin struts. J. Am. Chem. Soc. 2009, 131, 4204–4205. 10.1021/ja900203f. [DOI] [PubMed] [Google Scholar]
- Magano J.; Dunetz J. R. Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]
- Torborg C.; Beller M. Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries. Adv. Synth. Catal. 2009, 351, 3027–3043. 10.1002/adsc.200900587. [DOI] [Google Scholar]
- Roughley S. D.; Jordan A. M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Rouhi A. M. Fine Chemicals. Chem. Eng. News 2004, 82, 49–58. 10.1021/cen-v082n036.p049. [DOI] [Google Scholar]
- Ragan J. A.; Raggon J. W.; Hill P. D.; Jones B. P.; McDermott R. E.; Munchhof M. J.; Marx M. A.; Casavant J. M.; Cooper B. A.; Doty J. L.; et al. Cross-coupling methods for the large-scale preparation of an imidazole - Thienopyridine: Synthesis of [2-(3-methyl-3H-imidazol-4-yl)-thieno[3,2-b]pyridin-7-yl]-(2-methyl-1H-indol-5-yl)-amine. Org. Process Res. Dev. 2003, 7, 676–683. 10.1021/op0340457. [DOI] [Google Scholar]
- Ananikov V. P.; Khemchyan L. L.; Ivanova Y. V.; Bukhtiyarov V. I.; Sorokin A. M.; Prosvirin I. P.; Vatsadze S. Z.; Medved’ko A. V.; Nuriev V. N.; Dilman A. D.; et al. Development of new methods in modern selective organic synthesis: preparation of functionalized molecules with atomic precision. Russ. Chem. Rev. 2014, 83, 885–985. 10.1070/RC2014v83n10ABEH004471. [DOI] [Google Scholar]
- Ananikov V. P. Nickel: The ″spirited horse″ of transition metal catalysis. ACS Catal. 2015, 5, 1964–1971. 10.1021/acscatal.5b00072. [DOI] [Google Scholar]
- Holland P. L. Distinctive Reaction Pathways at Base Metals in High-Spin Organometallic Catalysts. Acc. Chem. Res. 2015, 48, 1696–1702. 10.1021/acs.accounts.5b00036. [DOI] [PubMed] [Google Scholar]
- Deraedt C.; Astruc D. Homeopathic” Palladium Nanoparticle Catalysis of Cross Carbon–Carbon Coupling Reactions. Acc. Chem. Res. 2014, 47, 494–503. 10.1021/ar400168s. [DOI] [PubMed] [Google Scholar]
- Hazari N.; Melvin P. R.; Beromi M. M. Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem. 2017, 1, 0025. 10.1038/s41570-017-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrion M.; Ritleng V.; Chetcuti M. J. Nickel N-Heterocyclic Carbene-Catalyzed C–C Bond Formation: Reactions and Mechanistic Aspects. ACS Catal. 2015, 5, 1283–1302. 10.1021/cs5014927. [DOI] [Google Scholar]
- Riener K.; Haslinger S.; Raba A.; Högerl M. P.; Cokoja M.; Herrmann W. A.; Kühn F. E. Chemistry of Iron N-heterocyclic carbene complexes: Syntheses, structures, reactivities, and catalytic applications. Chem. Rev. 2014, 114, 5215–5272. 10.1021/cr4006439. [DOI] [PubMed] [Google Scholar]
- Bauer I.; Knölker H. J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- Przyojski J. A.; Veggeberg K. P.; Arman H. D.; Tonzetich Z. J. Mechanistic Studies of Catalytic Carbon-Carbon Cross-Coupling by Well-Defined Iron NHC Complexes. ACS Catal. 2015, 5, 5938–5946. 10.1021/acscatal.5b01445. [DOI] [Google Scholar]
- Cassani C.; Bergonzini G.; Wallentin C. J. Active Species and Mechanistic Pathways in Iron-Catalyzed C-C Bond-Forming Cross-Coupling Reactions. ACS Catal. 2016, 6, 1640–1648. 10.1021/acscatal.5b02441. [DOI] [Google Scholar]
- Phapale V. B.; Cárdenas D. J. Nickel-catalysed Negishi cross-coupling reactions: scope and mechanisms. Chem. Soc. Rev. 2009, 38, 1598–1598. 10.1039/b805648j. [DOI] [PubMed] [Google Scholar]
- Harada T.; Ueda Y.; Iwai T.; Sawamura M. Nickel-catalyzed amination of aryl fluorides with primary amines. Chem. Commun. 2018, 54, 1718–1721. 10.1039/C7CC08181B. [DOI] [PubMed] [Google Scholar]
- Tasker S. Z.; Standley E. A.; Jamison T. F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z.; Zhang Q.; Deng L. Dinuclear Iron Complex-Catalyzed Cross-Coupling of Primary Alkyl Fluorides with Aryl Grignard Reagents. Organometallics 2012, 31, 6518–6521. 10.1021/om300722g. [DOI] [Google Scholar]
- Zhang J.; Xu J.; Xu Y.; Sun H.; Shen Q.; Zhang Y. Mixed NHC/Phosphine Ni(II) Complexes: Synthesis and Their Applications as Versatile Catalysts for Selective Cross-Couplings of ArMgX with Aryl Chlorides, Fluorides, and Methyl Ethers. Organometallics 2015, 34, 5792–5800. 10.1021/acs.organomet.5b00874. [DOI] [Google Scholar]
- Quasdorf K. W.; Antoft-Finch A.; Liu P.; Silberstein A. L.; Komaromi A.; Blackburn T.; Ramgren S. D.; Houk K. N.; Snieckus V.; Garg N. K. Suzuki–Miyaura Cross-Coupling of Aryl Carbamates and Sulfamates: Experimental and Computational Studies. J. Am. Chem. Soc. 2011, 133, 6352–6363. 10.1021/ja200398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kathriarachchi K. K. A. D. S.; Zenmyo T.; Seike H.; Nakamura M. Iron-catalyzed alkyl-alkyl Suzuki-Miyaura coupling. Angew. Chem., Int. Ed. 2012, 51, 8834–8837. 10.1002/anie.201202797. [DOI] [PubMed] [Google Scholar]
- Holzwarth M. S.; Plietker B. Biorelevant Metals in Sustainable Metal Catalysis-A Survey. ChemCatChem 2013, 5, 1650–1679. 10.1002/cctc.201200592. [DOI] [Google Scholar]
- Romashov L. V.; Rukhovich G. D.; Ananikov V. P. Analysis of model Pd- and Pt-containing contaminants in aqueous media using ESI-MS and the fragment partitioning approach. RSC Adv. 2015, 5, 107333–107339. 10.1039/C5RA22257E. [DOI] [Google Scholar]
- Q3D Elemental Impurities. Guidance for Industry; U.S. Department of Health, 2015. [Google Scholar]
- Pototschnig G.; Maulide N.; Schnürch M. Direct Functionalization of C–H Bonds by Iron, Nickel, and Cobalt Catalysis. Chem. - Eur. J. 2017, 23, 9206–9232. 10.1002/chem.201605657. [DOI] [PubMed] [Google Scholar]
- Egorova K. S.; Ananikov V. P. Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts. Angew. Chem., Int. Ed. 2016, 55, 12150–12162. 10.1002/anie.201603777. [DOI] [PubMed] [Google Scholar]
- Egorova K. S.; Ananikov V. P. Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. 10.1021/acs.organomet.7b00605. [DOI] [Google Scholar]
- Liu W.; Cera G.; Oliveira J. C. A.; Shen Z.; Ackermann L. MnCl2-Catalyzed C–H Alkylations with Alkyl Halides. Chem. - Eur. J. 2017, 23, 11524–11528. 10.1002/chem.201703191. [DOI] [PubMed] [Google Scholar]
- Wang S.-S.; Yang G.-Y. Recent developments in low-cost TM-catalyzed Heck-type reactions (TM = transition metal, Ni, Co, Cu, and Fe). Catal. Sci. Technol. 2016, 6, 2862–2876. 10.1039/C5CY02235E. [DOI] [Google Scholar]
- Lin C.-Y.; Power P. P. Complexes of Ni(I): a ‘‘rare’’ oxidation state of growing importance. Chem. Soc. Rev. 2017, 46, 5347–5399. 10.1039/C7CS00216E. [DOI] [PubMed] [Google Scholar]
- Marín M.; Rama R. J.; Nicasio M. C. Ni-Catalyzed Amination Reactions: An Overview. Chem. Rec. 2016, 16, 1819–1832. 10.1002/tcr.201500305. [DOI] [PubMed] [Google Scholar]
- Cahiez G.; Moyeux A. Cobalt-catalyzed cross-coupling reactions. Chem. Rev. 2010, 110, 1435–1462. 10.1021/cr9000786. [DOI] [PubMed] [Google Scholar]
- Jana R.; Pathak T. P.; Sigman M. S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Hong X. Computational studies on Ni-catalyzed C-O bond activation of esters. J. Organomet. Chem. 2018, 864, 68–80. 10.1016/j.jorganchem.2018.01.019. [DOI] [Google Scholar]
- Mata R. A.; Suhm M. A. Benchmarking Quantum Chemical Methods: Are We Heading in the Right Direction?. Angew. Chem., Int. Ed. 2017, 56, 11011–11018. 10.1002/anie.201611308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong H. A.; Wu W.; Teo Y.-Y. Cobalt-Catalyzed Cross-Coupling Reactions of Arylboronic Esters and Aryl Halides. Organometallics 2017, 36, 4363–4366. 10.1021/acs.organomet.7b00726. [DOI] [Google Scholar]
- Asghar S.; Tailor S. B.; Elorriaga D.; Bedford R. B. Cobalt-Catalyzed Suzuki Biaryl Coupling of Aryl Halides. Angew. Chem., Int. Ed. 2017, 56, 16367–16370. 10.1002/anie.201710053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammann J. M.; Haas D.; Knochel P. Cobalt-Catalyzed Negishi Cross-Coupling Reactions of (Hetero)Arylzinc Reagents with Primary and Secondary Alkyl Bromides and Iodides. Angew. Chem., Int. Ed. 2015, 54, 4478–4481. 10.1002/anie.201411960. [DOI] [PubMed] [Google Scholar]
- Al-Afyouni M. H.; Krishnan V. M.; Arman H. D.; Tonzetich Z. J. Synthesis and Reactivity of Manganese(II) Complexes Containing N-Heterocyclic Carbene Ligands. Organometallics 2015, 34, 5088–5094. 10.1021/acs.organomet.5b00684. [DOI] [Google Scholar]
- Inatomi T.; Koga Y.; Matsubara K. Dinuclear Nickel(I) and Palladium(I) Complexes for Highly Active Transformations of Organic Compounds. Molecules 2018, 23, 140–140. 10.3390/molecules23010140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohadjer Beromi M.; Nova A.; Balcells D.; Brasacchio A. M.; Brudvig G. W.; Guard L. M.; Hazari N.; Vinyard D. J. Mechanistic Study of an Improved Ni Precatalyst for Suzuki–Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc. 2017, 139, 922–936. 10.1021/jacs.6b11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley E. A.; Jamison T. F. Simplifying Nickel(0) Catalysis: An Air-Stable Nickel Precatalyst for the Internally Selective Benzylation of Terminal Alkenes. J. Am. Chem. Soc. 2013, 135, 1585–1592. 10.1021/ja3116718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields J. D.; Gray E. E.; Doyle A. G. A Modular, Air-Stable Nickel Precatalyst. Org. Lett. 2015, 17, 2166–2169. 10.1021/acs.orglett.5b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magano J.; Monfette S. Development of an Air-Stable, Broadly Applicable Nickel Source for Nickel-Catalyzed Cross-Coupling. ACS Catal. 2015, 5, 3120–3123. 10.1021/acscatal.5b00498. [DOI] [Google Scholar]
- Meconi G. M.; Vummaleti S. V. C.; Luque-Urrutia J. A.; Belanzoni P.; Nolan S. P.; Jacobsen H.; Cavallo L.; Solà M.; Poater A. Mechanism of the Suzuki–Miyaura Cross-Coupling Reaction Mediated by [Pd(NHC)(allyl)Cl] Precatalysts. Organometallics 2017, 36, 2088–2095. 10.1021/acs.organomet.7b00114. [DOI] [Google Scholar]
- Hedström A.; Izakian Z.; Vreto I.; Wallentin C.-J.; Norrby P.-O. On the Radical Nature of Iron-Catalyzed Cross-Coupling Reactions. Chem. - Eur. J. 2015, 21, 5946–5953. 10.1002/chem.201406096. [DOI] [PubMed] [Google Scholar]
- Mesganaw T.; Garg N. K. Ni- and Fe-Catalyzed Cross-Coupling Reactions of Phenol Derivatives. Org. Process Res. Dev. 2013, 17, 29–39. 10.1021/op300236f. [DOI] [Google Scholar]
- Cong X.; Fan F.; Ma P.; Luo M.; Chen H.; Zeng X. Low-Valent, High-Spin Chromium-Catalyzed Cleavage of Aromatic Carbon–Nitrogen Bonds at Room Temperature: A Combined Experimental and Theoretical Study. J. Am. Chem. Soc. 2017, 139, 15182–15190. 10.1021/jacs.7b08579. [DOI] [PubMed] [Google Scholar]
- Chirik P. J. Carbon-Carbon Bond Formation in a Weak Ligand Field: Leveraging Open-Shell First-Row Transition-Metal Catalysts. Angew. Chem., Int. Ed. 2017, 56, 5170–5181. 10.1002/anie.201611959. [DOI] [PubMed] [Google Scholar]
- Craciun R.; Vincent A. J.; Shaughnessy K. H.; Dixon D. A. Prediction of reliable metal-PH3 bond energies for Ni, Pd, and Pt in the 0 and + 2 oxidation states. Inorg. Chem. 2010, 49, 5546–5553. 10.1021/ic1004853. [DOI] [PubMed] [Google Scholar]
- Carrow B. P.; Hartwig J. F. Ligandless, Anionic, Arylpalladium Halide Intermediates in the Heck Reaction. J. Am. Chem. Soc. 2010, 132, 79–81. 10.1021/ja909306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127–150. 10.1021/cr5004375. [DOI] [PubMed] [Google Scholar]
- Szilvási T.; Veszprémi T. Internal Catalytic Effect of Bulky NHC Ligands in Suzuki–Miyaura Cross-Coupling Reaction. ACS Catal. 2013, 3, 1984–1991. 10.1021/cs400429j. [DOI] [Google Scholar]
- Zhang Y.; Lavigne G.; Lugan N.; César V. Buttressing Effect as a Key Design Principle towards Highly Efficient Palladium/N-Heterocyclic Carbene Buchwald-Hartwig Amination Catalysts. Chem. - Eur. J. 2017, 23, 13792–13801. 10.1002/chem.201702859. [DOI] [PubMed] [Google Scholar]
- Gordeev E. G.; Eremin D. B.; Chernyshev V. M.; Ananikov V. P. Influence of R–NHC Coupling on the Outcome of R–X Oxidative Addition to Pd/NHC Complexes (R = Me, Ph, Vinyl, Ethynyl). Organometallics 2018, 37, 787–796. 10.1021/acs.organomet.7b00669. [DOI] [Google Scholar]
- Astakhov A. V.; Khazipov O. V.; Chernenko A. Y.; Pasyukov D. V.; Kashin A. S.; Gordeev E. G.; Khrustalev V. N.; Chernyshev V. M.; Ananikov V. P. A New Mode of Operation of Pd-NHC Systems Studied in a Catalytic Mizoroki-Heck Reaction. Organometallics 2017, 36, 1981–1992. 10.1021/acs.organomet.7b00184. [DOI] [Google Scholar]
- Astakhov A. V.; Khazipov O. V.; Degtyareva E. S.; Khrustalev V. N.; Chernyshev V. M.; Ananikov V. P. Facile Hydrolysis of Nickel(II) Complexes with N-Heterocyclic Carbene Ligands. Organometallics 2015, 34, 5759–5766. 10.1021/acs.organomet.5b00856. [DOI] [Google Scholar]
- Zhang S.-Q.; Taylor B. L. H.; Ji C.-L.; Gao Y.; Harris M. R.; Hanna L. E.; Jarvo E. R.; Houk K. N.; Hong X. Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni-Catalyzed Suzuki–Miyaura Coupling with Benzylic Esters: A Computational Study. J. Am. Chem. Soc. 2017, 139, 12994–13005. 10.1021/jacs.7b04973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Li Y.; Wang L.; Kuang J.; Lei Q.; Fang W. Why different ligands can control stereochemistry selectivity of Ni-catalyzed Suzuki–Miyaura cross-coupling of benzylic carbamates with arylboronic esters: a mechanistic study. Dalton Trans 2017, 46, 13010–13019. 10.1039/C7DT02532G. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Sameera W. M. C.; Jin M.; Adak L.; Okuzono C.; Iwamoto T.; Kato M.; Nakamura M.; Morokuma K. DFT and AFIR Study on the Mechanism and the Origin of Enantioselectivity in Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2017, 139, 16117–16125. 10.1021/jacs.7b05917. [DOI] [PubMed] [Google Scholar]
- Kleimark J.; Hedström A.; Larsson P.-F.; Johansson C.; Norrby P.-O. Mechanistic Investigation of Iron-Catalyzed Coupling Reactions. ChemCatChem 2009, 1, 152–161. 10.1002/cctc.200900061. [DOI] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kondo Y.; Fujiwara Y.; Seike H.; Takaya H.; Tamada Y.; Ono T.; Nakamura M. Iron-Catalyzed Suzuki–Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc. 2010, 132, 10674–10676. 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- Noverges Pedro B.; Medio-Simón M.; Jutand A. Influence of the Ligand of Palladium(0) Complexes on the Rate of the Oxidative Addition of Aryl and Activated Alkyl Bromides: Csp2–Br versus Csp3–Br Reactivity and Selectivity. ChemCatChem 2017, 9, 2136–2144. 10.1002/cctc.201700041. [DOI] [Google Scholar]
- Maes B. U. W.; Verbeeck S.; Verhelst T.; Ekomié A.; Von Wolff N.; Lefèvre G.; Mitchell E. A.; Jutand A. Oxidative addition of haloheteroarenes to palladium(0): Concerted versus SNAr-type mechanism. Chem. - Eur. J. 2015, 21, 7858–7865. 10.1002/chem.201406210. [DOI] [PubMed] [Google Scholar]
- Lin B. L.; Liu L.; Fu Y.; Luo S. W.; Chen Q.; Guo Q. X. Comparing nickel- and palladium-catalyzed Heck reactions. Organometallics 2004, 23, 2114–2123. 10.1021/om034067h. [DOI] [Google Scholar]
- Menezes da Silva V. H.; Braga A. A. C.; Cundari T. R. N-Heterocyclic Carbene Based Nickel and Palladium Complexes: A DFT Comparison of the Mizoroki–Heck Catalytic Cycles. Organometallics 2016, 35, 3170–3181. 10.1021/acs.organomet.6b00532. [DOI] [Google Scholar]
- Lavoie C. M.; McDonald R.; Johnson E. R.; Stradiotto M. Bisphosphine-Ligated Nickel Pre-catalysts in C(sp2)–N Cross-Couplings of Aryl Chlorides: A Comparison of Nickel(I) and Nickel(II). Adv. Synth. Catal. 2017, 359, 2972–2980. 10.1002/adsc.201700672. [DOI] [Google Scholar]
- Bajo S.; Laidlaw G.; Kennedy A. R.; Sproules S.; Nelson D. J. Oxidative Addition of Aryl Electrophiles to a Prototypical Nickel(0) Complex: Mechanism and Structure/Reactivity Relationships. Organometallics 2017, 36, 1662–1672. 10.1021/acs.organomet.7b00208. [DOI] [Google Scholar]
- Xu H.; Muto K.; Yamaguchi J.; Zhao C.; Itami K.; Musaev D. G. Key Mechanistic Features of Ni-Catalyzed C–H/C–O Biaryl Coupling of Azoles and Naphthalen-2-yl Pivalates. J. Am. Chem. Soc. 2014, 136, 14834–14844. 10.1021/ja5071174. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang S.-L.; Fu Y.; Guo Q.-X.; Liu L. Mechanism of Ni-Catalyzed Selective C–O Bond Activation in Cross-Coupling of Aryl Esters. J. Am. Chem. Soc. 2009, 131, 8815–8823. 10.1021/ja810157e. [DOI] [PubMed] [Google Scholar]
- Dürr A. B.; Yin G.; Kalvet I.; Napoly F.; Schoenebeck F. Nickel-catalyzed trifluoromethylthiolation of Csp2–O bonds. Chem. Sci. 2016, 7, 1076–1081. 10.1039/C5SC03359D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell D. G.; Kochi J. K. Mechanistic studies of nickel catalysis in the cross coupling of aryl halides with alkylmetals. Role of arylalkylnickel(II) species as intermediates. J. Am. Chem. Soc. 1975, 97, 7262–7270. 10.1021/ja00858a011. [DOI] [Google Scholar]
- Li Z.; Jiang Y.-Y.; Fu Y. Theoretical Study on the Mechanism of Ni-Catalyzed Alkyl-Alkyl Suzuki Cross-Coupling. Chem. - Eur. J. 2012, 18, 4345–4357. 10.1002/chem.201103882. [DOI] [PubMed] [Google Scholar]
- Tang S.; Eisenstein O.; Nakao Y.; Sakaki S. Aromatic C–H σ-Bond Activation by Ni0, Pd0, and Pt0 Alkene Complexes: Concerted Oxidative Addition to Metal vs Ligand-to-Ligand H Transfer Mechanism. Organometallics 2017, 36, 2761–2771. 10.1021/acs.organomet.7b00256. [DOI] [Google Scholar]
- Chatt J.; Duncanson L. A. Olefin co-ordination compounds. Part III. Infrared spectra and structure: attempted preparation of acetylene complexes. J. Chem. Soc. 1953, 2939–2939. 10.1039/jr9530002939. [DOI] [Google Scholar]
- Chatt J.; Duncanson L. A.; Venanzi L. M. Directing effects in inorganic substitution reactions. Part I. A hypothesis to explain the trans-effect. J. Chem. Soc. 1955, 0, 4456–4456. 10.1039/jr9550004456. [DOI] [Google Scholar]
- Sontag S. K.; Bilbrey J. A.; Huddleston N. E.; Sheppard G. R.; Allen W. D.; Locklin J. π-Complexation in Nickel-Catalyzed Cross-Coupling Reactions. J. Org. Chem. 2014, 79, 1836–1841. 10.1021/jo402259z. [DOI] [PubMed] [Google Scholar]
- Bilbrey J. A.; Bootsma A. N.; Bartlett M. A.; Locklin J.; Wheeler S. E.; Allen W. D. Ring-Walking of Zerovalent Nickel on Aryl Halides. J. Chem. Theory Comput. 2017, 13, 1706–1711. 10.1021/acs.jctc.6b01143. [DOI] [PubMed] [Google Scholar]
- Massera C.; Frenking G. Energy partitioning analysis of the bonding in L2TM-C2H2 and L2TM-C2H4 (TM = Ni, Pd, Pt; L2= (PH3)2, (PMe3)2, H2PCH2PH2, H2P(CH2)2PH2). Organometallics 2003, 22, 2758–2765. 10.1021/om0301637. [DOI] [Google Scholar]
- Hering F.; Nitsch J.; Paul U.; Steffen A.; Bickelhaupt F. M.; Radius U. Bite-angle bending as a key for understanding group-10 metal reactivity of d10-[M(NHC)2] complexes with sterically modest NHC ligands. Chem. Sci. 2015, 6, 1426–1432. 10.1039/C4SC02998D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M. S.; Sheong F. K.; Marder T. B.; Lin Z. Computational Insight into Nickel-Catalyzed Carbon-Carbon versus Carbon-Boron Coupling Reactions of Primary, Secondary, and Tertiary Alkyl Bromides. Chem. - Eur. J. 2015, 21, 7480–7488. 10.1002/chem.201500110. [DOI] [PubMed] [Google Scholar]
- Pudasaini B.; Janesko B. G. Agostic Interactions in Nickel(II) Complexes: Trans Influence of Ancillary Ligands on the Strength of the Bond. Organometallics 2014, 33, 84–93. 10.1021/om400731j. [DOI] [Google Scholar]
- Kozuch S. A refinement of everyday thinking: the energetic span model for kinetic assessment of catalytic cycles. WIREs Comput. Mol. Sci. 2012, 2, 795–815. 10.1002/wcms.1100. [DOI] [Google Scholar]
- Kozuch S.; Martin J. M. L. What Makes for a Bad Catalytic Cycle? A Theoretical Study on the Suzuki–Miyaura Reaction within the Energetic Span Model. ACS Catal. 2011, 1, 246–253. 10.1021/cs100129u. [DOI] [Google Scholar]
- Kozuch S.; Martin J. M. L. What makes for a good catalytic cycle? A theoretical study of the SPhos ligand in the Suzuki–Miyaura reaction. Chem. Commun. 2011, 47, 4935–4935. 10.1039/c1cc10717h. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Shaik S. A combined kinetic-quantum mechanical model for assessment of catalytic cycles: Application to cross-coupling and Heck reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. 10.1021/ja0559146. [DOI] [PubMed] [Google Scholar]
- Lin X.; Sun J.; Xi Y.; Lin D. How Racemic Secondary Alkyl Electrophiles Proceed to Enantioselective Products in Negishi Cross-Coupling Reactions. Organometallics 2011, 30, 3284–3292. 10.1021/om1012049. [DOI] [Google Scholar]
- Kozuch S.; Lee S. E.; Shaik S. Theoretical Analysis of the Catalytic Cycle of a Nickel Cross-Coupling Process: Application of the Energetic Span Model. Organometallics 2009, 28, 1303–1308. 10.1021/om800772g. [DOI] [Google Scholar]
- Mesganaw T.; Silberstein A. L.; Ramgren S. D.; Nathel N. F. F.; Hong X.; Liu P.; Garg N. K. Nickel-catalyzed amination of aryl carbamates and sequential site-selective cross-couplings. Chem. Sci. 2011, 2, 1766–1766. 10.1039/c1sc00230a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas-Vives A.; González-Arellano C.; Corma A.; Iglesias M.; Sánchez F.; Ujaque G. Single-site homogeneous and heterogeneized gold(III) hydrogenation catalysts: Mechanistic implications. J. Am. Chem. Soc. 2006, 128, 4756–4765. 10.1021/ja057998o. [DOI] [PubMed] [Google Scholar]
- Bourrez M.; Gloaguen F. Application of the energetic span model to the electrochemical catalysis of proton reduction by a diiron azadithiolate complex. New J. Chem. 2015, 39, 8073–8079. 10.1039/C5NJ00770D. [DOI] [Google Scholar]
- Stegelmann C.; Andreasen A.; Campbell C. T. Degree of Rate Control: How Much the Energies of Intermediates and Transition States Control Rates. J. Am. Chem. Soc. 2009, 131, 13563–13563. 10.1021/ja9065199. [DOI] [PubMed] [Google Scholar]
- Campbell C. T. The Degree of Rate Control: A Powerful Tool for Catalysis Research. ACS Catal. 2017, 7, 2770–2779. 10.1021/acscatal.7b00115. [DOI] [Google Scholar]
- Kozuch S. Steady State Kinetics of Any Catalytic Network: Graph Theory, the Energy Span Model, the Analogy between Catalysis and Electrical Circuits, and the Meaning of “Mechanism. ACS Catal. 2015, 5, 5242–5255. 10.1021/acscatal.5b00694. [DOI] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters L. P.; Bickelhaupt F. M. The activation strain model and molecular orbital theory. WIREs Comput. Mol. Sci. 2015, 5, 324–343. 10.1002/wcms.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong G. T.; Bickelhaupt F. M. Transition-State Energy and Position along the Reaction Coordinate in an Extended Activation Strain Model. ChemPhysChem 2007, 8, 1170–1181. 10.1002/cphc.200700092. [DOI] [PubMed] [Google Scholar]
- Al-Afyouni M. H.; Suturina E.; Pathak S.; Atanasov M.; Bill E.; DeRosha D. E.; Brennessel W. W.; Neese F.; Holland P. L. Spin Isomers and Ligand Isomerization in a Three-Coordinate Cobalt(I) Carbonyl Complex. J. Am. Chem. Soc. 2015, 137, 10689–10699. 10.1021/jacs.5b06078. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Tang H.; Chen K.; Hu L.; Yao J.; Shaik S.; Chen H. Two-State Reactivity in Low-Valent Iron-Mediated C–H Activation and the Implications for Other First-Row Transition Metals. J. Am. Chem. Soc. 2016, 138, 3715–3730. 10.1021/jacs.5b12150. [DOI] [PubMed] [Google Scholar]
- Poli R. Open-shell organometallics as a bridge between Werner-type and low-valent organometallic complexes. The effect of the spin state on the stability, reactivity, and structure. Chem. Rev. 1996, 96, 2135–2204. 10.1021/cr9500343. [DOI] [PubMed] [Google Scholar]
- Harvey J. N.; Poli R.; Smith K. M. Understanding the reactivity of transition metal complexes involving multiple spin states. Coord. Chem. Rev. 2003, 238–239, 347–361. 10.1016/S0010-8545(02)00283-7. [DOI] [Google Scholar]
- Bellows S. M.; Cundari T. R.; Holland P. L. Spin Crossover during β-Hydride Elimination in High-Spin Iron(II)– and Cobalt(II)–Alkyl Complexes. Organometallics 2013, 32, 4741–4751. 10.1021/om400325x. [DOI] [Google Scholar]
- Camasso N. M.; Canty A. J.; Ariafard A.; Sanford M. S. Experimental and Computational Studies of High-Valent Nickel and Palladium Complexes. Organometallics 2017, 36, 4382–4393. 10.1021/acs.organomet.7b00613. [DOI] [Google Scholar]
- Nicolas E.; Ohleier A.; D’Accriscio F.; Pécharman A.-F.; Demange M.; Ribagnac P.; Ballester J.; Gosmini C.; Mézailles N. (Diphosphine)Nickel”-Catalyzed Negishi Cross-Coupling: An Experimental and Theoretical Study. Chem. - Eur. J. 2015, 21, 7690–7694. 10.1002/chem.201500192. [DOI] [PubMed] [Google Scholar]
- Iffland L.; Petuker A.; van Gastel M.; Apfel U.-P. Mechanistic Implications for the Ni(I)-Catalyzed Kumada Cross-Coupling Reaction. Inorganics 2017, 5, 78–78. 10.3390/inorganics5040078. [DOI] [Google Scholar]
- Matsubara K.; Yamamoto H.; Miyazaki S.; Inatomi T.; Nonaka K.; Koga Y.; Yamada Y.; Veiros L. F.; Kirchner K. Dinuclear Systems in the Efficient Nickel-Catalyzed Kumada–Tamao–Corriu Cross-Coupling of Aryl Halides. Organometallics 2017, 36, 255–265. 10.1021/acs.organomet.6b00451. [DOI] [Google Scholar]
- Dürr A. B.; Fisher H. C.; Kalvet I.; Truong K. N.; Schoenebeck F. Divergent Reactivity of a Dinuclear (NHC)Nickel(I) Catalyst versus Nickel(0) Enables Chemoselective Trifluoromethylselenolation. Angew. Chem., Int. Ed. 2017, 56, 13431–13435. 10.1002/anie.201706423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H.; Minami H.; Ozaki T.; Komagawa S.; Wang C.; Uchiyama M. How and Why Does Ni0 Promote Smooth Etheric C–O Bond Cleavage and C–C Bond Formation? A Theoretical Study. Chem. - Eur. J. 2015, 21, 13904–13908. 10.1002/chem.201502114. [DOI] [PubMed] [Google Scholar]
- Eremin D. B.; Ananikov V. P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. 10.1016/j.ccr.2016.12.021. [DOI] [Google Scholar]
- Janthon P.; Luo S.; Kozlov S. M.; Viñes F.; Limtrakul J.; Truhlar D. G.; Illas F. Bulk properties of transition metals: A challenge for the design of universal density functionals. J. Chem. Theory Comput. 2014, 10, 3832–3839. 10.1021/ct500532v. [DOI] [PubMed] [Google Scholar]
- Ma S.; Wang H.; Gao K.; Zhao F. Nickel complexes catalyzed Heck reaction of iodobenzene and methyl acrylate. J. Mol. Catal. A: Chem. 2006, 248, 17–20. 10.1016/j.molcata.2005.12.013. [DOI] [Google Scholar]
- Briggs B. D.; Bedford N. M.; Seifert S.; Koerner H.; Ramezani-Dakhel H.; Heinz H.; Naik R. R.; Frenkel A. I.; Knecht M. R.; Knecht M. R. Atomic-scale identification of Pd leaching in nanoparticle catalyzed C–C coupling: effects of particle surface disorder. Chem. Sci. 2015, 6, 6413–6419. 10.1039/C5SC01424G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa S.; Slack E. D.; Lipshutz B. H. Nanonickel-Catalyzed Suzuki-Miyaura Cross-Couplings in Water. Angew. Chem., Int. Ed. 2015, 54, 11994–11998. 10.1002/anie.201505136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz B. H.; Frieman B. A.; Lee C.-T.; Lower A.; Nihan D. M.; Taft B. R. Microwave-Assisted Heterogeneous Cross-Coupling Reactions Catalyzed by Nickel-in-Charcoal (Ni/C). Chem. - Asian J. 2006, 1, 417–429. 10.1002/asia.200600031. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Ueda H. Aromatic aminations by heterogeneous Ni0/C catalysis. Angew. Chem., Int. Ed. 2000, 39, 4492–4494. . [DOI] [PubMed] [Google Scholar]
- Tasler S.; Lipshutz B. H. Nickel-on-charcoal-catalyzed aromatic aminations and Kumada couplings: Mechanistic and synthetic aspects. J. Org. Chem. 2003, 68, 1190–1199. 10.1021/jo020297e. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Tasler S.; Chrisman W.; Spliethoff B.; Tesche B. On the nature of the ’heterogeneous’ catalyst: Nickel-on-charcoal. J. Org. Chem. 2003, 68, 1177–1189. 10.1021/jo020296m. [DOI] [PubMed] [Google Scholar]
- Collins G.; Schmidt M.; O’Dwyer C.; McGlacken G.; Holmes J. D. Enhanced Catalytic Activity of High-Index Faceted Palladium Nanoparticles in Suzuki–Miyaura Coupling Due to Efficient Leaching Mechanism. ACS Catal. 2014, 4, 3105–3111. 10.1021/cs5008014. [DOI] [Google Scholar]
- Collins G.; Schmidt M.; O’Dwyer C.; Holmes J. D.; McGlacken G. P. The origin of shape sensitivity in palladium-catalyzed Suzuki-Miyaura cross coupling reactions. Angew. Chem., Int. Ed. 2014, 53, 4142–4145. 10.1002/anie.201400483. [DOI] [PubMed] [Google Scholar]
- Lewis E. A.; Murphy C. J.; Pronschinske A.; Liriano M. L.; Sykes E. C. H. Nanoscale insight into C–C coupling on cobalt nanoparticles. Chem. Commun. 2014, 50, 10035–10035. 10.1039/C4CC03678F. [DOI] [PubMed] [Google Scholar]
- Iwasaki T.; Akimoto R.; Kuniyasu H.; Kambe N. Fe-Catalyzed Cross-Coupling Reaction of Vinylic Ethers with Aryl Grignard Reagents. Chem. - Asian J. 2016, 11, 2834–2837. 10.1002/asia.201600972. [DOI] [PubMed] [Google Scholar]
- Haas D.; Hammann J. M.; Lutter F. H.; Knochel P. Mild Cobalt-Catalyzed Negishi Cross-Couplings of (Hetero)arylzinc Reagents with (Hetero)aryl Halides. Angew. Chem., Int. Ed. 2016, 55, 3809–3812. 10.1002/anie.201510665. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Amatore C.; Jutand A.; Shaik S. What makes for a good catalytic cycle? a theoretical study of the role of an anionic palladium(0) complex in the cross-coupling of an aryl halide with an anionic nucleophile. Organometallics 2005, 24, 2319–2330. 10.1021/om050160p. [DOI] [Google Scholar]
- Henriksen S. T.; Tanner D.; Cacchi S.; Norrby P.-O. DFT-Based Explanation of the Effect of Simple Anionic Ligands on the Regioselectivity of the Heck Arylation of Acrolein Acetals. Organometallics 2009, 28, 6201–6205. 10.1021/om900674y. [DOI] [Google Scholar]
- Gøgsig T. M.; Kleimark J.; Nilsson Lill S. O.; Korsager S.; Lindhardt A. T.; Norrby P.-O.; Skrydstrup T. Mild and Efficient Nickel-Catalyzed Heck Reactions with Electron-Rich Olefins. J. Am. Chem. Soc. 2012, 134, 443–452. 10.1021/ja2084509. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. Kinetic Data for the Transmetalation/Reductive Elimination in Palladium-Catalyzed Suzuki-Miyaura Reactions: Unexpected Triple Role of Hydroxide Ions Used as Base. Chem. - Eur. J. 2011, 17, 2492–2503. 10.1002/chem.201001911. [DOI] [PubMed] [Google Scholar]
- Hervé M.; Lefèvre G.; Mitchell E. A.; Maes B. U. W.; Jutand A. On the Triple Role of Fluoride Ions in Palladium-Catalyzed Stille Reactions. Chem. - Eur. J. 2015, 21, 18401–18406. 10.1002/chem.201503309. [DOI] [PubMed] [Google Scholar]
- Grisorio R.; Suranna G. P. Impact of Precatalyst Activation on Suzuki-Miyaura Catalyst-Transfer Polymerizations: New Mechanistic Scenarios for Pre-transmetalation Events. ACS Macro Lett. 2017, 6, 1251–1256. 10.1021/acsmacrolett.7b00696. [DOI] [PubMed] [Google Scholar]
- Proutiere F.; Schoenebeck F. Solvent Effect on Palladium-Catalyzed Cross-Coupling Reactions and Implications on the Active Catalytic Species. Angew. Chem., Int. Ed. 2011, 50, 8192–8195. 10.1002/anie.201101746. [DOI] [PubMed] [Google Scholar]
- Sunoj R. B.; Anand M. Microsolvated transition state models for improved insight into chemical properties and reaction mechanisms. Phys. Chem. Chem. Phys. 2012, 14, 12715–12715. 10.1039/c2cp41719g. [DOI] [PubMed] [Google Scholar]
- Wang B.; Cao Z. Mechanism of Acid-Catalyzed Hydrolysis of Formamide from Cluster-Continuum Model Calculations: Concerted versus Stepwise Pathway. J. Phys. Chem. A 2010, 114, 12918–12927. 10.1021/jp106560s. [DOI] [PubMed] [Google Scholar]
- del Pozo J.; Pérez-Iglesias M.; Álvarez R.; Lledós A.; Casares J. A.; Espinet P. Speciation of ZnMe2, ZnMeCl, and ZnCl2 in Tetrahydrofuran (THF), and Its Influence on Mechanism Calculations of Catalytic Processes. ACS Catal. 2017, 7, 3575–3583. 10.1021/acscatal.6b03636. [DOI] [Google Scholar]
- Harvey J. N. Ab initio transition state theory for polar reactions in solution. Faraday Discuss. 2010, 145, 487–505. 10.1039/B907340J. [DOI] [Google Scholar]
- General I. J. A. Note on the Standard State’s Binding Free Energy. J. Chem. Theory Comput. 2010, 6, 2520–2524. 10.1021/ct100255z. [DOI] [PubMed] [Google Scholar]
- Affo W.; Ohmiya H.; Fujioka T.; Ikeda Y.; Nakamura T.; Yorimitsu H.; Oshima K.; Imamura Y.; Mizuta T.; Miyoshi K. Cobalt-Catalyzed Trimethylsilylmethylmagnesium-Promoted Radical Alkenylation of Alkyl Halides: A Complement to the Heck Reaction. J. Am. Chem. Soc. 2006, 128, 8068–8077. 10.1021/ja061417t. [DOI] [PubMed] [Google Scholar]
- Smith M. B., March J.. March’s Advanced Organic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, 2006. [Google Scholar]
- Lee W.; Zhou J.; Gutierrez O. Mechanism of Nakamura’s Bisphosphine-Iron-Catalyzed Asymmetric C(sp2)-C(sp3) Cross-Coupling Reaction: The Role of Spin in Controlling Arylation Pathways. J. Am. Chem. Soc. 2017, 139, 16126–16133. 10.1021/jacs.7b06377. [DOI] [PubMed] [Google Scholar]
- Qi Z.-H.; Ma J. Dual Role of a Photocatalyst: Generation of Ni(0) Catalyst and Promotion of Catalytic C–N Bond Formation. ACS Catal. 2018, 8, 1456–1463. 10.1021/acscatal.7b03024. [DOI] [Google Scholar]
- Marenich A. V.; Ho J.; Coote M. L.; Cramer C. J.; Truhlar D. G. Computational electrochemistry: prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. 10.1039/C4CP01572J. [DOI] [PubMed] [Google Scholar]
- Demissie T. B.; Ruud K.; Hansen J. H. DFT as a Powerful Predictive Tool in Photoredox Catalysis: Redox Potentials and Mechanistic Analysis. Organometallics 2015, 34, 4218–4228. 10.1021/acs.organomet.5b00582. [DOI] [Google Scholar]
- Acevedo O.; Jorgensen W. L. Advances in quantum and molecular mechanical (QM/MM) simulations for organic and enzymatic reactions. Acc. Chem. Res. 2010, 43, 142–151. 10.1021/ar900171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley K. E.; Pitonak M.; Jurecka P.; Hobza P. Stabilization and Structure Calculations for Noncovalent Interactions in Extended Molecular Systems Based on Wave Function and Density Functional Theories. Chem. Rev. 2010, 110, 5023–5063. 10.1021/cr1000173. [DOI] [PubMed] [Google Scholar]
- Lam Y.-h.; Grayson M. N.; Holland M. C.; Simon A.; Houk K. N. Theory and Modeling of Asymmetric Catalytic Reactions. Acc. Chem. Res. 2016, 49, 750–762. 10.1021/acs.accounts.6b00006. [DOI] [PubMed] [Google Scholar]
- Sperger T.; Sanhueza I. A.; Schoenebeck F. Computation and Experiment: A Powerful Combination to Understand and Predict Reactivities. Acc. Chem. Res. 2016, 49, 1311–1319. 10.1021/acs.accounts.6b00068. [DOI] [PubMed] [Google Scholar]
- Lernmark Å. The Streetlight Effect—Is There Light at the End of the Tunnel?. Diabetes 2015, 64, 1105–1107. 10.2337/db15-0011. [DOI] [PubMed] [Google Scholar]
- Haynes W. A.; Tomczak A.; Khatri P. Gene annotation bias impedes biomedical research. Sci. Rep. 2018, 8, 1362. 10.1038/s41598-018-19333-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix C. S. Searching for climate–conflict links. Nat. Clim. Change 2018, 8, 190–191. 10.1038/s41558-018-0083-3. [DOI] [Google Scholar]
- Dewyer A. L.; Zimmerman P. M. Finding reaction mechanisms, intuitive or otherwise. Org. Biomol. Chem. 2017, 15, 501–504. 10.1039/C6OB02183B. [DOI] [PubMed] [Google Scholar]
- Dewyer A. L.; Argüelles A. J.; Zimmerman P. M. Methods for exploring reaction space in molecular systems. WIREs Comput. Mol. Sci. 2018, 8, e1354. 10.1002/wcms.1354. [DOI] [Google Scholar]
- Ensing B.; De Vivo M.; Liu Z.; Moore P.; Klein M. L. Metadynamics as a Tool for Exploring Free Energy Landscapes of Chemical Reactions. Acc. Chem. Res. 2006, 39, 73–81. 10.1021/ar040198i. [DOI] [PubMed] [Google Scholar]
- Laio A.; Gervasio F. L. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]
- van Speybroeck V.; Meier R. J. A recent development in computational chemistry: chemical reactions from first principles molecular dynamics simulations. Chem. Soc. Rev. 2003, 32, 151–157. 10.1039/b210410p. [DOI] [PubMed] [Google Scholar]
- Wang L.-P.; McGibbon R. T.; Pande V. S.; Martinez T. J. Automated Discovery and Refinement of Reactive Molecular Dynamics Pathways. J. Chem. Theory Comput. 2016, 12, 638–649. 10.1021/acs.jctc.5b00830. [DOI] [PubMed] [Google Scholar]
- Schlegel H. B. Geometry optimization. WIREs Comput. Mol. Sci. 2011, 1, 790–809. 10.1002/wcms.34. [DOI] [Google Scholar]
- Sameera W. M. C.; Maeda S.; Morokuma K. Computational Catalysis Using the Artificial Force Induced Reaction Method. Acc. Chem. Res. 2016, 49, 763–773. 10.1021/acs.accounts.6b00023. [DOI] [PubMed] [Google Scholar]
- Martinez-Nunez E. An Automated Method to Find Transition States Using Chemical Dynamics Simulations. J. Comput. Chem. 2015, 36, 222–234. 10.1002/jcc.23790. [DOI] [PubMed] [Google Scholar]
- Dewyer A. L.; Zimmerman P. M. Simulated Mechanism for Palladium-Catalyzed, Directed gamma-Arylation of Piperidine. ACS Catal. 2017, 7, 5466–5477. 10.1021/acscatal.7b01390. [DOI] [Google Scholar]
- Ludwig J. R.; Zimmerman P. M.; Gianino J. B.; Schindler C. S. Iron(III)-catalysed carbonyl-olefin metathesis. Nature 2016, 533, 374–379. 10.1038/nature17432. [DOI] [PubMed] [Google Scholar]
- Ludwig J. R.; Phan S.; McAtee C. C.; Zimmerman P. M.; Devery J. J. III; Schindler C. S. Mechanistic Investigations of the Iron(III)-Catalyzed Carbonyl-Olefin Metathesis Reaction. J. Am. Chem. Soc. 2017, 139, 10832–10842. 10.1021/jacs.7b05641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. L.; Leone A. K.; Zimmerman P. M.; McNeil A. J. Impact of Preferential pi-Binding in Catalyst-Transfer Polycondensation of Thiazole Derivatives. ACS Macro Lett. 2016, 5, 1411–1415. 10.1021/acsmacrolett.6b00886. [DOI] [PubMed] [Google Scholar]
- Hale L. V. A.; Malakar T.; Tseng K.-N. T.; Zimmerman P. M.; Paul A.; Szymczak N. K. The Mechanism of Acceptorless Amine Double Dehydrogenation by N,N,N-Amide Ruthenium(II) Hydrides: A Combined Experimental and Computational Study. ACS Catal. 2016, 6, 4799–4813. 10.1021/acscatal.6b01465. [DOI] [Google Scholar]
- Ellington B. R.; Paul B.; Das D.; Vitek A. K.; Zimmerman P. M.; Marsh E. N. G. An Unusual Iron-Dependent Oxidative Deformylation Reaction Providing Insight into Hydrocarbon Biosynthesis in Nature. ACS Catal. 2016, 6, 3293–3300. 10.1021/acscatal.6b00592. [DOI] [Google Scholar]
- Hatanaka M.; Maeda S.; Morokuma K. Sampling of Transition States for Predicting Diastereoselectivity Using Automated Search Method-Aqueous Lanthanide-Catalyzed Mukaiyama Aldol Reaction. J. Chem. Theory Comput. 2013, 9, 2882–2886. 10.1021/ct4002637. [DOI] [PubMed] [Google Scholar]
- Hatanaka M.; Morokuma K. Role of Water in Mukaiyama-Aldol Reaction Catalyzed by Lanthanide Lewis Acid: A Computational Study. J. Am. Chem. Soc. 2013, 135, 13972–13979. 10.1021/ja407357c. [DOI] [PubMed] [Google Scholar]
- Hatanaka M.; Morokuma K. How Can Fluctional Chiral Lanthanide (III) Complexes Achieve a High Stereoselectivity in Aqueous Mukaiyama-Aldol Reaction?. ACS Catal. 2015, 5, 3731–3739. 10.1021/acscatal.5b00438. [DOI] [Google Scholar]
- Varela J. A.; Vazquez S. A.; Martinez-Nunez E. An automated method to find reaction mechanisms and solve the kinetics in organometallic catalysis. Chem. Sci. 2017, 8, 3843–3851. 10.1039/C7SC00549K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck R. F.; Breslow D. S. Reaction of cobalt hydrotetracarbonyl with olefins. J. Am. Chem. Soc. 1961, 83, 4023. 10.1021/ja01480a017. [DOI] [Google Scholar]
- Martínez-Núñez E.; Shalashilin D. V. Acceleration of classical mechanics by phase space constraints. J. Chem. Theory Comput. 2006, 2, 912–919. 10.1021/ct060042z. [DOI] [PubMed] [Google Scholar]
- Bergeler M.; Simm G. N.; Proppe J.; Reiher M. Heuristics-Guided Exploration of Reaction Mechanisms. J. Chem. Theory Comput. 2015, 11, 5712–5722. 10.1021/acs.jctc.5b00866. [DOI] [PubMed] [Google Scholar]
- Petrosko S. H.; Johnson R.; White H.; Mirkin C. A. Nanoreactors: Small Spaces, Big Implications in Chemistry. J. Am. Chem. Soc. 2016, 138, 7443–7445. 10.1021/jacs.6b05393. [DOI] [PubMed] [Google Scholar]
- Wang L.-P.; Titov A.; McGibbon R.; Liu F.; Pande V. S.; Martínez T. J. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 2014, 6, 1044. 10.1038/nchem.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Harabuchi Y.; Takagi M.; Taketsugu T.; Morokuma K. Artificial Force Induced Reaction (AFIR) Method for Exploring Quantum Chemical Potential Energy Surfaces. Chem. Rec. 2016, 16, 2232–2248. 10.1002/tcr.201600043. [DOI] [PubMed] [Google Scholar]
- Martinez-Nunez E. An automated transition state search using classical trajectories initialized at multiple minima. Phys. Chem. Chem. Phys. 2015, 17, 14912–14921. 10.1039/C5CP02175H. [DOI] [PubMed] [Google Scholar]
- Bhoorasingh P. L.; West R. H. Transition state geometry prediction using molecular group contributions. Phys. Chem. Chem. Phys. 2015, 17, 32173–32182. 10.1039/C5CP04706D. [DOI] [PubMed] [Google Scholar]
- Zimmerman P. M. Growing string method with interpolation and optimization in internal coordinates: Method and examples. J. Chem. Phys. 2013, 138, 184102. 10.1063/1.4804162. [DOI] [PubMed] [Google Scholar]
- Suleimanov Y. V.; Green W. H. Automated Discovery of Elementary Chemical Reaction Steps Using Freezing String and Berny Optimization Methods. J. Chem. Theory Comput. 2015, 11, 4248–4259. 10.1021/acs.jctc.5b00407. [DOI] [PubMed] [Google Scholar]
- Habershon S. Automated Prediction of Catalytic Mechanism and Rate Law Using Graph-Based Reaction Path Sampling. J. Chem. Theory Comput. 2016, 12, 1786–1798. 10.1021/acs.jctc.6b00005. [DOI] [PubMed] [Google Scholar]
- Wang L.-P.; Titov A.; McGibbon R.; Liu F.; Pande V. S.; Martinez T. J. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 2014, 6, 1044–1048. 10.1038/nchem.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Zou J.; Wang G.; Li S. Automatic Reaction Pathway Search via Combined Molecular Dynamics and Coordinate Driving Method. J. Phys. Chem. A 2017, 121, 1351–1361. 10.1021/acs.jpca.6b12195. [DOI] [PubMed] [Google Scholar]
- Janet J. P.; Kulik H. J. Predicting electronic structure properties of transition metal complexes with neural networks. Chem. Sci. 2017, 8, 5137–5152. 10.1039/C7SC01247K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahneman D. T.; Estrada J. G.; Lin S.; Dreher S. D.; Doyle A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. 10.1126/science.aar5169. [DOI] [PubMed] [Google Scholar]
- Kitchin J. R. Machine learning in catalysis. Nat. Catal. 2018, 1, 230–232. 10.1038/s41929-018-0056-y. [DOI] [Google Scholar]