

Summary
Objective
To ascertain the clinical and genetic factors contributing to carbamazepine‐ and oxcarbazepine‐induced hyponatremia (COIH), and to carbamazepine (CBZ) metabolism, in a retrospectively collected, cross‐sectional cohort of people with epilepsy.
Methods
We collected data on serum sodium levels and antiepileptic drug levels in people with epilepsy attending a tertiary epilepsy center while on treatment with CBZ or OXC. We defined hyponatremia as Na+ ≤134 mEq/L. We estimated the CBZ metabolic ratio defined as the log transformation of the ratio of metabolite CBZ‐diol to unchanged drug precursor substrate as measured in serum.
Results
Clinical and genetic data relating to carbamazepine and oxcarbazepine trials were collected in 1141 patients. We did not observe any genome‐wide significant associations with sodium level in a linear trend or hyponatremia as a dichotomous trait. Age, sex, number of comedications, phenytoin use, phenobarbital use, and sodium valproate use were significant predictors of CBZ metabolic ratio. No genome‐wide significant associations with CBZ metabolic ratio were found.
Significance
Although we did not detect a genetic predictor of hyponatremia or CBZ metabolism in our cohort, our findings suggest that the determinants of CBZ metabolism are multifactorial.
Keywords: adverse effects, antiepileptic drugs, EpiPGX Consortium, GWAS, hyponatremia
1.
Key Points.
No large, clinically relevant genetic predictors of carbamazepine‐ or oxcarbazepine‐induced hyponatremia
Age and concurrent phenytoin, phenobarbital, or sodium valproate use was significantly associated with carbamazepine metabolic ratio
No large, clinically relevant genetic predictors of carbamazepine metabolic ratio
2. INTRODUCTION
Carbamazepine (CBZ) and its keto‐analogue, oxcarbazepine (OXC), are routinely used as antiepileptic drugs (AEDs) and are also used in the treatment of chronic pain conditions and bipolar disorder. Although effective, their use is limited by adverse drug reactions (ADRs), including hyponatremia and hypersensitivity. Carbamazepine‐ and oxcarbazepine‐induced hyponatremia (COIH) is reported in up to half of drug exposures. This is often assumed to be asymptomatic but it can lead to difficulties ranging from unsteadiness and mild confusion to seizures and coma. Careful dose titration and monitoring of sodium levels are recommended for reducing the risk of COIH, while individual differences in drug metabolism can make titration difficult.
HLA‐B*1502 is strongly associated with CBZ‐induced Stevens‐Johnson syndrome (SJS) in people of Han Chinese ethnicity, increasing the risk about 100‐fold.1 In individuals of European descent, HLA‐A*3101 is a clinically relevant predictor of the full spectrum of CBZ‐induced hypersensitivity reactions.2, 3 To date, no genetic risk factors have been associated with COIH. Thiazide‐induced hyponatremia is associated with 2 polymorphisms in the KCNJ1 gene, encoding the renal outer medullary potassium channel (ROMK), which plays an important role in sodium reabsorption along the thick ascending limb of the loop of Henle.4 CBZ and OXC seem to influence water reabsorption, independent of salt retention, via stimulation of the vasopressin 2 receptor/aquaporin (AVPR2) pathway.5 Mutations in the AVPR2 gene, a regulator of water reabsorption, can cause a nephrogenic syndrome of inappropriate antidiuresis (NSIAD) with physiologic similarities to the inappropriate antidiuresis induced by CBZ and OXC.6 Studies of AVPR2 copy number variation, however, did not explain variation in sodium levels in non‐Hispanic Caucasian populations.7
We attempted to determine the clinical and genetic factors contributing to COIH and drug metabolism in a retrospectively collected, cross‐sectional cohort of people with epilepsy of European descent treated with CBZ and OXC, which characteristics were previously described.8
3. METHODS
3.1. Study design and phenotypes
We followed a retrospective cohort study design. The majority of the patients were recruited at a Dutch tertiary epilepsy referral center (SEIN), whereas the remainder were recruited around European tertiary referral clinics associated with the EpiPGX Consortium. Clinical information from medical records, with an emphasis on AED history, was recorded in an electronic database designed for retrospective pharmacogenomics studies.9 The database was used to identify all individuals who were prescribed CBZ or OXC and who had a recorded serum sodium level during therapy. Most individuals had several measurements, and the lowest sodium level recorded was selected for analysis. Our primary analyses were structured to test genetic variants for association with this lowest recorded sodium level per subject (mEql/L). Secondary analyses tested for genetic association with the following: (a) COIH (combined and per causal drug) and (b) CBZ metabolic ratio. In a subset of CBZ users for which we had concurrently measured CBZ‐10,11‐diol (CBZ‐diol) levels, we calculated the metabolic ratio defined as the log transformation of the ratio of metabolite CBZ‐diol to unchanged drug precursor substrate as measured in serum. COIH cases were defined as having a blood sodium level ≤134 mmol attributed to CBZ or OXC as determined by their clinician. COIH controls trialed CBZ or OXC for at least 3 months with a sodium level ≥135 mmol. Epilepsy‐specific cohort demographics are presented in Table 1.
Table 1.
CBZ and OXC cohort characteristics
| Description | CBZ | OXC | Combined |
|---|---|---|---|
| Subjects | 1031 | 297 | 1252a |
| % male | 51.4% | 48.1% | 51.2% |
| Mean age (±SD) | 42.9 ± 15.1 | 38.1 ± 15.9 | 41.9 ± 15.6 |
| No. AED comedications (max)b | 1.0 (5) | 0.9 (4) | 1.0 (5) |
| Hyponatremia (Na <135 mEq/L) | 331 (32%) | 170 (57%) | 448c |
| Mean case serum sodium (mEq/L) | 129.5 ± 4.1 | 127.5 ± 4.2 | 129.0 ± 4.2 |
| Mean control serum sodium (mEq/L) | 139.8 ± 2.5 | 139.4 ± 2.7 | 139.7 ± 2.5 |
| Mean serum AED level (mg/L) (±SD)b | 8.7 ± 2.3 | 17.8 ± 8.2 | — |
| Metabolic ratio (±SD)d | 0.35 ± 0.21 | — | — |
Seventy‐nine subjects trialed both CBZ and OXC.
Calculated from SEIN subcohort (n = 1074).
Fifty‐three subjects experienced hyponatremia on both CBZ and OXC.
Metabolic ratio calculated on a subset of subjects with serum CBZ‐diol level readings (n = 468).
3.2. Sampling and genotype analysis
Serum drug and metabolite concentrations were measured during the course of routine monitoring in the morning before drug intake. For each sodium level measurement, we recorded patient age, serum level of CBZ or OXC, and concomitant use of other drugs. Genotyping of all patients was performed at deCODE Genetics on Illumina OmniExpress‐12 v1.1 and OmniExpress‐24 v1.1 single nucleotide polymorphism (SNP) arrays. Genotyping quality control was performed as described previously.10 Principal components analysis (PCA) was performed with European‐ancestral samples from the HapMap Project to assess cohort substructure and identify population outliers (Figure S1). Eigenvectors were computed in the genome‐wide complex trait analysis tool (GCTA) for each subject for inclusion as covariates in genetic‐association testing.11 Subjects were identified as outliers and removed if greater than 3 standard deviations (SD) from the first 8 principal components. We used the functional mapping and annotation of genome‐wide association studies platform (FUMA) to generate Manhattan and quantile‐quantile (Q‐Q) plots.12
3.3. Study power
We estimated from our recruited sample size that our study had 80% power to detect a genetic predictor of relative risk (approximated to odds ratio) ≥3 with an allele frequency ≥2% and an alpha level of 1.0 × 10−8, using the power calculator for case‐control genetic association analyses PGA.13
3.4. Statistical analyses
Clinical cofactors influencing sodium levels and COIH in this cohort were reported previously and used as covariates in our models.8 Association analyses were conducted using additive linear or logistic regression models in PLINK, including clinical covariates where appropriate and 8 principal components from PCA. Dosage, number of comedications, and AED levels were excluded from the genetic analyses due to missing information in the EpiPGX subcohort. We also analyzed the significance of clinical variables influencing CBZ metabolic ratio using a stepwise linear regression model in SPSS statistical software. As before, significant clinical cofactors from the linear regression model were included as covariates along with 8 principal components from PCA. For each association test, SNPs with <90% call rate were excluded. The threshold for genome‐wide statistical significance was set at 1.0 × 10−8, reflecting an empirical Bonferroni correction for 5 tests, of the standard 5 × 10−8 genome‐wide significance threshold.
3.5. Ethical considerations
All study participants provided written, informed consent for genetic analysis. Study protocols were approved by the research ethics committees listed in Table S1.
4. RESULTS
We collected clinical and genetic data relating to CBZ (n = 1031 subjects) and OXC (n = 297 subjects) trials. A subset (n = 79 subjects) were trialed on CBZ and OXC. Of the total 1252 patients, 1047 were recruited at SEIN while 201 were recruited through EpiPGX partner sites. Data on drug levels and compliance were available for 98% of the SEIN cohort, but not for the EpiPGX partner sites. In 5% of our SEIN cohort the drug levels or dosage was below therapeutic values (for CBZ <4 mmol/L or <400 mg/d, for OXC <10 mmol/L or <900 mg/d). We report 448 cases with COIH and 804 controls with normal serum sodium measurement. Within our cases there was a subset of 61 subjects with extreme hyponatremia. The incidence of OXC‐induced hyponatremia (57%) was almost twofold higher than that of CBZ (32%). Characteristics of our cohort are described in Table 1. A total of 25 subjects were removed after genotyping quality control.
To test whether common genetic variants predict sodium levels, we performed a genome‐wide linear regression adjusted for age, clobazam use, sex, plus 8 principal components. We did not observe any genome‐wide significant associations with sodium level (Figure 1).
Figure 1.
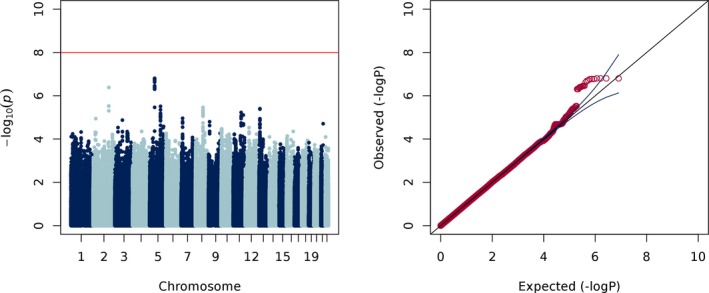
Manhattan and Q‐Q plots of sodium levels (λ = 1.00)
To test whether common genetic variants predict COIH, defined as a serum sodium level <135 mEq/L, we performed a case‐control genome‐wide logistic regression adjusted for sex, age <40, plus 8 principal components. We did not observe any genome‐wide significant associations when we considered COIH as a dichotomous trait (Figure 2). There was a suggestive association signal (P < 1 × 10−6) from chromosome 5, in an intergenic region approximately 500 Mb downstream of the gene ANKRD55, evident in the quantitative and dichotomous analyses of sodium. Furthermore, we did not detect evidence for a genetic signal in a subset of 61 severe COIH cases, defined as a serum sodium ≤125 mEq/L, when compared to controls. Neither did we did observe any significant associations when we differentiated hyponatremia by causal drug (see Figures S2‐S4). Given prior reports of an association with thiazide‐induced hyponatremia, we looked closely within the KCNJ1 and AVPR2 genes, yet we did not observe any signals of association in our data.
Figure 2.
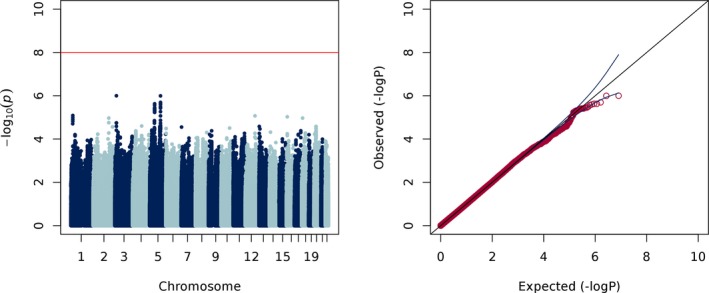
Manhattan and Q‐Q plots of COIH (λ = 1.00)
Next, we explored whether clinical cofactors or genetic variants could predict the ratio of active drug to metabolite in our CBZ‐exposed subjects. We modeled the contribution of clinical cofactors to CBZ metabolic ratio and found that age, sex, number of comedications, phenytoin use, phenobarbital use, and sodium valproate use were significantly predictive of outcome (adjusted r 2 = 0.236, Model 5 in Table 2).
Table 2.
Stepwise linear regression of carbamazepine metabolic ratio
| Model | Factors | R | R 2 | Adjusted R 2 | Std. error |
|---|---|---|---|---|---|
| 1 | PHT | .363a | 0.132 | 0.130 | 0.196 |
| 2 | PHT, NoCoMed | .421b | 0.177 | 0.174 | 0.191 |
| 3 | PHT, NoCoMed, age | .458c | 0.210 | 0.205 | 0.188 |
| 4 | PHT, NoCoMed, age, PHB | .486d | 0.236 | 0.230 | 0.185 |
| 5 | PHT, NoCoMed, age, PHB, VPA | .493e | 0.243 | 0.236 | 0.184 |
NoCoMed, number of comedications; PHB, phenobarbital; PHT, phenytoin; VPA, sodium valproate.
To test whether common genetic variants predict CBZ metabolic ratio, we then performed a genome‐wide linear regression adjusted for the covariates in Model 5, plus 8 principal components. We did not observe any genome‐wide significant associations with CBZ metabolic ratio (Figure 3). The top 10 most significant GWAS markers for each analysis are listed in Tables S1‐S5. Polymorphisms in CYP3A4 and EPHX1 have been shown to associate with interindividual variability of CBZ metabolism.14, 15 We did not observe even nominally significant associations between SNPs in CYP3A4 and CBZ metabolic ratio. It had been reported that homozygous carriers of the EPHX1 c.416A>G SNP (rs2234922) seemingly show a reduced CBZ metabolism, as measured by a significantly decreased metabolic ratio.16 We did not replicate this finding (rs2234922; P = 0.303) but we observed a nominally significant association between an intronic EPHX1 SNP (c.365‐2139T>C) and CBZ metabolic ratio (rs4653689; P = 1.1 × 10‐4).
Figure 3.
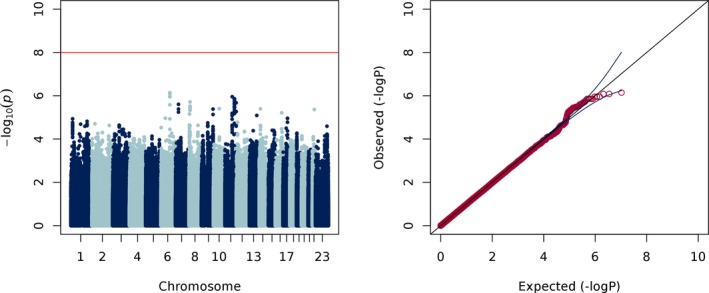
Manhattan and Q‐Q plots of carbamazepine metabolic ratio (λ = 0.98)
5. DISCUSSION
Although we did not detect a genetic predictor of hyponatremia in our cohort, we have demonstrated that the determinants of CBZ metabolism are multifactorial. Modeling the contribution of clinical variables showed there were strong nongenetic predictors of CBZ metabolism. Subject age, total number of comedications, and the concurrent use of phenytoin, phenobarbital, or sodium valproate were significantly associated with a higher CBZ‐diol to CBZ ratio. Much of this can be explained by the induction of the cytochrome P450 enzyme CYP3A4. CBZ is metabolized in the liver by CYP3A4 to carbamazepine‐10,11‐epoxide, which is further metabolized by microsomal epoxide hydrolase (mEH) to carbamazepine‐10,11‐diol.17 Phenytoin and phenobarbital induce CYP3A4 and thus can lower plasma CBZ levels but leave the metabolite levels unaltered, which results in the observed higher metabolic ratio.18, 19 Sodium valproate inhibits epoxide hydrolase, potentiating higher levels of the active metabolite CBZ‐10,11‐epoxide, which is associated with toxicity and adverse events,20 but this was not directly measured in this study. Valproate has been shown to increase dose ratios between CBZ and its metabolites, for both the diol and epoxide forms.21 Age has been found previously to contribute to pharmacokinetic variability in individuals using CBZ with increasing clearance until age 33 and a gradual decrease toward older age.22
A limitation of the study is that AED dosage was not reliably recorded in our cohort and we had to make an assumption that the measurement of CBZ metabolic ratio was independent of the individual subject's dosage. In the subset of subjects for whom serum CBZ‐diol and CBZ dosage information was available (n = 40) we did not detect a significant effect of CBZ dose on the metabolic ratio (P = 0.41).
CBZ and OXC are widely prescribed but their use coincides with a high prevalence of COIH. Within our cohort OXC‐induced hyponatremia has a much higher prevalence than that of CBZ, which is consistent with previous estimates.23, 24 From clinical experience, susceptibility to COIH is individually variable. Experimental studies and a recent clinical report suggest that COIH is caused by a direct effect of CBZ/OXC on the kidney by stimulating the vasopressin receptor.25, 26 Mutations in the V2R/AQP2 pathway regulating water reabsorption can cause disorders clinically similar to the syndromes of inappropriate secretion of antidiuresis associated with CBZ/OXC use. Meanwhile thiazide‐induced hyponatremia has been associated with polymorphisms in the gene KCNJ1 and a suggestive association with a variant in SLCO2A1, encoding a prostaglandin transporter; these signals did not show even nominal significance in our data.4, 27 We further looked for an effect from these markers within the subset of 53 subjects who experienced hyponatremia on both CBZ and OXC independently, but there was no significant enrichment.
Previously described clinical predictors of serum sodium levels explain only 11%‐14% of the variance in the SEIN cohort.8 Therefore, it is hypothesized that genetic variation could in part explain the variation in susceptibility to COIH. Yet, after analyzing sodium levels in a linear trend and hyponatremia as a dichotomous trait, we did not find genetic predictors for COIH. A recent report of variants in NFAT5 and SLC4A10 with suggestive association with plasma osmolality further imply a genetic component to hyponatremia, but these variants showed no evidence for effect on serum sodium measurements in our study, albeit we were not as powered as the original discovery cohort.28
In summary, our study rules out common genetic variants of clinically relevant effect size; however, genetic susceptibility for COIH cannot be ruled out completely, as rare variants and combinations of genetic variants of smaller effect size (polygenic risk) may contribute to overall risk. Further study, ideally in a prospective cohort with baseline sodium level and CBZ‐diol measurements, is warranted to investigate the genetic contribution to CBZ‐ and OXC‐induced hyponatremia.
DISCLOSURE
None of the authors has any conflict of interest to disclose in relation to this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
Parts of the computational analysis were performed on the high‐performance computer system of the University of Luxembourg (https://hpc.uni.lu). BB was supported by Christelijke Vereniging voor de Verpleging van Lijders aan Epilepsie, The Netherlands. MMC and GLC are supported by Science Foundation Ireland, grant 13/CDA/2223. MMC is supported by a Marie‐Curie Individual Fellowship (No. 751761) from the European Commission. The EpiPGX Consortium was funded by FP7 Grant 279062 “EpiPGX” from the European Commission. SMS and JWS are based at the NIHR University College London Hospitals Comprehensive Biomedical Research Centre, which receives a proportion of funding from the UK Department of Health's Biomedical Research Centres’ funding scheme. SMS and JWS receive support the UK Epilepsy Society. JWS receives support from the Dr. Marvin Weil Epilepsy Research Fund. CPS receives support from the Irish Research Council and Punchestown Kidney Research fund (grant number EPSPG2015).
APPENDIX 1.
1.1.
Andreja Avbersek, Costin Leu, Kristin Heggeli, Rita Demurtas, Joseph Willis, Douglas Speed, Narek Sargsyan, Krishna Chinthapalli, Mojgansadat Borghei, Antonietta Coppola, Antonio Gambardella, Stefan Wolking, Felicitas Becker, Sarah Rau, Christian Hengsbach, Yvonne G. Weber, Bianca Berghuis, Wolfram S. Kunz, Mark McCormack, Norman Delanty, Ellen Campbell, Lárus J. Gudmundsson, Andres Ingason, Kári Stefánsson, Reinhard Schneider, Rudi Balling, Pauls Auce, Ben Francis, Andrea Jorgensen, Andrew Morris, Sarah Langley, Prashant Srivastava, Martin Brodie, Marian Todaro, Slave Petrovski, Jane Hutton, Fritz Zimprich, Martin Krenn, Hiltrud Muhle, Karl Martin Klein, Rikke Moller, Marina Nikanorova, Sarah Weckhuysen, Zvonka Rener‐Primec, Gianpiero L. Cavalleri, John Craig, Chantal Depondt, Michael R. Johnson, Bobby P. C. Koeleman, Roland Krause, Holger Lerche, Anthony G. Marson, Terence J. O'Brien, Josemir W. Sander, Graeme J. Sills, Hreinn Stefansson, Pasquale Striano, Federico Zara and Sanjay M. Sisodiya
Berghuis B, Stapleton C, Sonsma ACM, et al. A genome‐wide association study of sodium levels and drug metabolism in an epilepsy cohort treated with carbamazepine and oxcarbazepine. Epilepsia Open. 2019;4:102–109. 10.1002/epi4.12297
Contributor Information
Bobby P. C. Koeleman, Email: b.p.c.koeleman@umcutrecht.nl
The EpiPGX Consortium:
Andreja Avbersek, Costin Leu, Kristin Heggeli, Joseph Willis, Douglas Speed, Narek Sargsyan, Krishna Chinthapalli, Mojgansadat Borghei, Antonietta Coppola, Antonio Gambardella, Felicitas Becker, Sarah Rau, Christian Hengsbach, Yvonne G. Weber, Norman Delanty, Ellen Campbell, Lárus J. Gudmundsson, Andres Ingason, Kári Stefánsson, Reinhard Schneider, Rudi Balling, Ben Francis, Andrea Jorgensen, Andrew Morris, Sarah Langley, Prashant Srivastava, Martin Brodie, Marian Todaro, Slave Petrovski, Jane Hutton, Fritz Zimprich, Martin Krenn, Hiltrud Muhle, Karl Martin Klein, Rikke Moller, Marina Nikanorova, Sarah Weckhuysen, and Zvonka Rener‐Primec
REFERENCES
- 1. Chung W‐H, Hung S‐I, Hong H‐S, et al. Medical genetics: a marker for Stevens–Johnson syndrome. Nature. 2004;428:486. [DOI] [PubMed] [Google Scholar]
- 2. McCormack M, Alfirevic A, Bourgeois S, et al. HLA‐A*3101 and carbamazepine‐induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364:1134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ozeki T, Mushiroda T, Yowang A, et al. Genome‐wide association study identifies HLA‐A*3101 allele as a genetic risk factor for carbamazepine‐induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20:1034–41. [DOI] [PubMed] [Google Scholar]
- 4. Huang C‐C, Chung C‐M, Hung S‐I, et al. Clinical and genetic factors associated with thiazide‐induced hyponatremia. Medicine (Baltimore). 2015;94:e1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berghuis B, de Haan GJ, van den Broek MPH, et al. Epidemiology, pathophysiology and putative genetic basis of carbamazepine‐ and oxcarbazepine‐induced hyponatremia. Eur J Neurol. 2016;23:1393–9. [DOI] [PubMed] [Google Scholar]
- 6. Levtchenko EN, Monnens LAH. Nephrogenic syndrome of inappropriate antidiuresis. Nephrol Dial Transplant. 2010;25:2839–43. [DOI] [PubMed] [Google Scholar]
- 7. Fu Y, Chen Z, Blakemore AIF, et al. Absence of AVPR2 copy number variation in eunatremic and dysnatremic subjects in non‐Hispanic Caucasian populations. Physiol Genomics. 2010;40:121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berghuis B, van der Palen J, de Haan GJ, et al. Carbamazepine‐ and oxcarbazepine‐induced hyponatremia in people with epilepsy. Epilepsia. 2017;58:1227–33. [DOI] [PubMed] [Google Scholar]
- 9. Androsova G, Krause R, Borghei M, et al. Comparative effectiveness of antiepileptic drugs in patients with mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2017;58:1734–41. [DOI] [PubMed] [Google Scholar]
- 10. McCormack M, Gui H, Ingason A, et al. Genetic variation in CFH predicts phenytoin‐induced maculopapular exanthema in European‐descent patients. Neurology. 2018;90:e332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome‐wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Menashe I, Rosenberg PS, Chen BE. PGA: power calculator for case‐control genetic association analyses. BMC Genet. 2008;9:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chbili C, Fathallah N, Laouani A, et al. Effects of EPHX1 and CYP3A4*22 genetic polymorphisms on carbamazepine metabolism and drug response among Tunisian epileptic patients. J Neurogenet. 2016;30:16–21. [DOI] [PubMed] [Google Scholar]
- 15. Nakajima Y, Saito Y, Shiseki K, et al. Haplotype structures of EPHX1 and their effects on the metabolism of carbamazepine‐10,11‐epoxide in Japanese epileptic patients. Eur J Clin Pharmacol. 2005;61:25–34. [DOI] [PubMed] [Google Scholar]
- 16. Daci A, Beretta G, Vllasaliu D, et al. Polymorphic variants of SCN1A and EPHX1 influence plasma carbamazepine concentration, metabolism and pharmacoresistance in a population of kosovar albanian epileptic patients. PLoS ONE. 2015;10:e0142408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pirmohamed M, Kitteringham NR, Breckenridge AM, et al. The effect of enzyme induction on the cytochrome P450‐mediated bioactivation of carbamazepine by mouse liver microsomes. Biochem Pharmacol. 1992;44:2307–14. [DOI] [PubMed] [Google Scholar]
- 18. McKauge L, Tyrer JH, Eadie MJ. Factors influencing simultaneous concentrations of carbamazepine and its epoxide in plasma. Ther Drug Monit. 1981;3:63–70. [PubMed] [Google Scholar]
- 19. Riva R, Contin M, Albani F, et al. Free and total serum concentrations of carbamazepine and carbamazepine‐10,11‐epoxide in infancy and childhood. Epilepsia. 1985;26:320–2. [DOI] [PubMed] [Google Scholar]
- 20. Fricke‐Galindo I, LLerena A, Jung‐Cook H, López‐López M. Carbamazepine adverse drug reactions. Expert Rev Clin Pharmacol. 2018;11:705–18. [DOI] [PubMed] [Google Scholar]
- 21. Svinarov DA, Pippenger CE. Relationships between carbamazepine‐diol, carbamazepine‐epoxide, and carbamazepine total and free steady‐state concentrations in epileptic patients: the influence of age, sex, and comedication. Ther Drug Monit. 1996;18:660–5. [DOI] [PubMed] [Google Scholar]
- 22. Wegner I, Wilhelm AJ, Sander JW, et al. The impact of age on lamotrigine and oxcarbazepine kinetics: a historical cohort study. Epilepsy Behav. 2013;29:217–21. [DOI] [PubMed] [Google Scholar]
- 23. Intravooth T, Staack AM, Juerges K, et al. Antiepileptic drugs‐induced hyponatremia: review and analysis of 560 hospitalized patients. Epilepsy Res. 2018;143:7–10. [DOI] [PubMed] [Google Scholar]
- 24. Lu X, Wang X. Hyponatremia induced by antiepileptic drugs in patients with epilepsy. Expert Opin Drug Saf. 2017;16:77–87. [DOI] [PubMed] [Google Scholar]
- 25. De Bragana AC, Moyses ZP, Magaldi AJ. Carbamazepine can induce kidney water absorption by increasing aquaporin 2 expression. Nephrol Dial Transplant. 2010;25:3840–5. [DOI] [PubMed] [Google Scholar]
- 26. Sekiya N, Awazu M. A case of nephrogenic syndrome of inappropriate antidiuresis caused by carbamazepine. CEN Case Rep. 2018;7:66–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ware JS, Wain LV, Channavajjhala SK, et al. Phenotypic and pharmacogenetic evaluation of patients with thiazide‐induced hyponatremia. J Clin Invest. 2017;127:3367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Böger CA, Gorski M, McMahon GM, et al. NFAT5 and SLC4A10 loci associate with plasma osmolality. J Am Soc Nephrol. 2017;28:2311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials