

Summary
Haemophagocytic lymphohistiocytosis (HLH) constitutes a spectrum of immunological disorders characterized by uncontrolled immune activation and key symptoms such as fever, splenomegaly, pancytopenia, haemophagocytosis, hyperferritinaemia and hepatitis. In genetic or primary HLH, hyperactivated CD8+ T cells are the main drivers of pathology. However, in acquired secondary HLH, the role of lymphocytes remains vague. In the present study the involvement of lymphocytes in the pathogenesis of a cytomegalovirus‐induced model of secondary HLH was explored. We have previously reported CD8+ T cells to be redundant in this model, and therefore focused on CD4+ helper and regulatory T cells. CD4+ T cells were activated markedly and skewed towards a proinflammatory T helper type 1 transcription profile in mice displaying a severe and complete HLH phenotype. Counter to expectations, regulatory T cells were not reduced in numbers and were, in fact, more activated. Therapeutic strategies targeting CD25high hyperactivated T cells were ineffective to alleviate disease, indicating that T cell hyperactivation is not a pathogenic factor in cytomegalovirus‐induced murine HLH. Moreover, even though T cells were essential in controlling viral proliferation, CD4+ T cells, in addition to CD8+ T cells, were dispensable in the development of the HLH‐like syndrome. In fact, no T or B cells were required for induction and propagation of HLH disease, as evidenced by the occurrence of cytomegalovirus‐associated HLH in severe combined immunodeficient (SCID) mice. These data suggest that lymphocyte‐independent mechanisms can underlie virus‐associated secondary HLH, accentuating a clear distinction with primary HLH.
Keywords: haemophagocytic lymphohistiocytosis, macrophage activation syndrome, mouse cytomegalovirus, mouse model, T cells
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is an increasingly recognized but incompletely understood hyperinflammatory syndrome occurring in children and adults. Characteristic symptoms include fever, hepatosplenomegaly, pancytopenia, hyperferritinaemia, coagulopathy and haemophagocytosis, arising in the event of a severe cytokine storm 1, 2. Without a timely diagnosis and appropriate, often aggressive, therapy, the disorder is generally lethal. Hereditary, so‐called primary, HLH is caused by harmful mutations in genes involved in the granule‐mediated cytotoxic function of CD8+ T cells and natural killer (NK) cells 3. Acquired, secondary HLH is clinically similar to primary HLH but associated with various medical conditions, such as infections, malignancies, metabolic disorders, immunodeficiencies and autoimmune or autoinflammatory diseases 4, the latter category often referred to as ‘macrophage activation syndrome’ (MAS). Infections with herpesviruses such as Epstein–Barr virus and cytomegalovirus are considered the principal triggering factors of HLH 5, 6, 7.
Animal models have designated CD8+ T cells and their excess secretion of interferon (IFN)‐γ as the main culprits in the pathogenesis of primary HLH. The aberrantly activated T cells infiltrate multiple organs, inducing life‐threatening immunopathology and driving uncontrolled systemic inflammation 8, 9, 10, 11. Conversely, models of secondary HLH suggest that different mechanisms may underlie the pathogenesis of this subtype. In cytosine–phosphatase–guanosine (CpG)‐mediated secondary HLH, CD8+ T cells were only mildly activated and dispensable for disease development 12. In fact, T cells and adaptive immunity altogether were redundant in this model. The role of IFN‐γ was found to depend upon the activity of interleukin (IL)‐10, being either pathogenic or largely redundant, with the exception of mediating anaemia 12, 13. A similar absence of lymphoid involvement was described in two transgenic mouse models of secondary HLH 14, 15. In a model of virus‐associated secondary HLH that we have described recently (illustrated in Supporting information, Fig. S1), IFN‐γ was not required for HLH development; moreover, disease was worsened and a more complete spectrum of HLH symptoms developed in IFN‐γ‐deficient mice. Thus, the model represents a tool to unravel IFN‐γ‐independent pathways of HLH pathogenesis, as reported recently in IFN‐γ receptor 1/2 (R1/2) or signal transducer and activator of transcription 1 (STAT‐1)‐deficient patients 16, 17, 18, 19. Additionally in this model, CD8+ T cells were highly activated but dispensable in disease development, as their depletion did not mitigate HLH disease 20. In contrast, the absence of CD8+ T cells resulted in augmented viral proliferation, appointing T cells as an essential defence in virus‐associated secondary HLH 20. Also in patients, recent data suggest a discrepancy between primary and secondary HLH on the basis of T cell activation and differentiation patterns. Patients with primary HLH possessed more highly activated effector CD8+ cells and a unique cytotoxic CD4+ T cell signature compared to patients with secondary HLH. Viral infections were shown to shift the pattern of CD8+ T cell activation in secondary HLH to overlap partially with the T cell phenotype observed in primary HLH 21, 22. Thus, both animal and human data indicate that the lymphoid compartment is activated differentially and may play divergent roles in the pathogenesis of primary and secondary HLH. Additionally, secondary HLH has been reported to develop in patients lacking functional T cells, including severe combined immunodeficient (SCID) patients 23. These intriguing findings urged us to investigate further the pathogenic or regulatory roles of different T cell populations in the development and progression of virus‐associated secondary HLH, as well as potential therapeutic strategies to target these cell populations.
Using the mouse cytomegalovirus (MCMV)‐induced animal model of secondary HLH 20, we found the T helper (Th) cell population to be skewed towards a proinflammatory Th1 phenotype. CD4+ T cells, including regulatory T cells (Treg), showed a marked activation profile. As opposed to reports in primary HLH 24, Treg cell numbers were not reduced markedly in secondary HLH. Therapeutic targeting of T cells had no effect on the initiation or progression of the HLH syndrome, but instead affected the rate of viral proliferation negatively, confirming the essential role of T cells in viral control. Not only T cells, but also B cells were dispensable for disease induction. To preserve control of the viral infection while simultaneously attenuating hyperinflammation in HLH, a therapeutic strategy was devised to target the highly activated CD25+ T cells selectively, sparing quiescent T cells. Although this strategy proved unsuccessful to alleviate HLH disease, it conserved viral control. Thus, rather than being lymphocyte‐mediated, murine virus‐associated secondary HLH appeared to operate through innate immune mechanisms, although the precise cell type could not be identified. Within the innate compartment, neutrophils were increased highly in blood and tissue, but their depletion was insufficient to prevent HLH development, suggesting no crucial role for neutrophils in disease pathogenesis.
Materials and methods
Mice, experimental design, viral infection and plaque assay
IFN‐γ knock‐out (KO) BALB/c mice, corresponding wild‐type (WT) BALB/c mice and C.B.−17 Prkdcscid/scid SCID mice were bred under specific pathogen‐free conditions in the Experimental Animal Centre of KU Leuven. Mice, 5–6 weeks old, were age‐ and sex‐matched within each experiment. Experiments were approved by the Animal Ethics Committee of KU Leuven and performed in a conventional animal facility. Per mouse, an inoculum of 5 × 103 plaque‐forming units (pfu) of salivary gland‐derived MCMV (Smith strain, VR‐1399; American Type Culture Collection (ATCC), Manassas, VA, USA) in 100 µl phosphate‐buffered saline (PBS) was injected intraperitoneally (i.p.) on day 0. PBS‐injected mice were included as controls. The resulting HLH model, described in reference 20, is illustrated in Supporting information, Fig. S1. Weight and rectal temperature of the mice was measured daily. Mice were euthanized with Nembutal (Ceva Santé Animale, Libourne, France) when chronic weight loss exceeded 25% of initial body weight or when rectal temperature dropped below 34·5°C (humane end‐points according to institutional ethical policies). This usually occurred at day 5 post‐infection (p.i.), at which characteristic HLH symptoms and viral titres were examined. The amount of infectious virus in spleen was determined by a plaque assay using a 10‐fold titration of the supernatant of disrupted spleen tissue on C127I cells (CRL‐1616; ATCC). Detection limit of the assay was 2 pfu per organ.
In‐vivo depletion of T cells
To deplete CD4+ and/or CD8+ T cells, a monoclonal anti‐CD4‐antibody (clone GK1.5) and/or monoclonal anti‐CD8‐antibody (clone YTS169, both Epirus Biopharmaceuticals, Utrecht, the Netherlands) were injected i.p. as a preventive treatment (300 μg/mouse on day −1 and 200 μg/mouse on day 2 p.i. in 100 μl PBS = early depletion) or as a curative treatment (300 μg/mouse on day 2 p.i., in 100 μl PBS = late depletion). Depletion was verified in blood and lymph nodes via flow cytometry using anti‐CD4 (clone RMA‐4) and anti‐CD8a (clone 53‐6.7). To target activated T cells, anti‐CD25‐antibodies (blocking function, clone 7D4, 500 μg/mouse or depleting function, clone PC61, 300 μg/mouse, both Epirus Biopharmaceuticals) were injected i.p. on day 2 p.i. Depletion was verified in blood and lymph nodes using anti‐CD25 (clone 3C7). Control MCMV‐infected mice were injected with an equal volume of PBS. Antibodies were administered using a randomized design so that animals from different experimental groups were co‐housed to avoid cage effects.
In‐vivo depletion of neutrophils
To deplete neutrophils, a monoclonal anti‐lymphocyte antigen 6 complex, locus G (Ly6G)‐antibody (clone 1A8) and monoclonal anti‐glutathione reductase (Gr1)‐antibody (clone RB6–8C5, both Epirus Biopharmaceuticals) were injected i.p. as a preventive treatment (250 μg/mouse on day −1 and day 2 p.i. in 100 μl PBS). Depletion was verified in blood and lungs via flow cytometry using anti‐Ly6G‐ and anti‐Gr1‐antibodies on anti‐Gr1‐ and anti‐Ly6G‐treated animals, respectively. Control MCMV‐infected mice were injected with an equal volume of PBS or an isotype control [immunoglobulin (Ig)G2a, clone 2A3]. Antibodies were administered using a randomized design, co‐housing animals from different experimental groups to avoid cage effects.
Blood analysis and quantification of liver enzymes
Blood samples were obtained via cardiac puncture with heparin (LEO Pharma, Ballerup, Denmark). Blood cell analysis was performed with a Cell‐Dyn 3700 Hematology Analyzer (Abbott Diagnostics, Lake Forest, IL, USA). Plasma concentrations of alanine transaminase (ALT) were measured spectrophotometrically using an ultraviolet (UV)‐kinetic method according to the manufacturer's instructions [ALT (SGPT) Reagent Set, Teco Diagnostics, Anaheim, CA, USA].
Quantification of cytokines, soluble CD25 (sCD25) and ferritin
IFN‐γ, IL‐2, IL‐4, IL‐5, IL‐10, IL‐12p70, IL‐13, IL‐17A, IL‐18, IL‐21, IL‐22 and IL‐23 were measured in serum using a multiplex assay (ProcartaPlex Mouse Th1/Th2 cytokine panel, Th9/Th17/Th22/Treg cytokine panel with custom added IL‐21; eBioscience, San Diego, CA, USA). Alternatively, IFN‐γ and IL‐10 were determined in plasma (Ready‐Set‐Go; eBioscience and Quantikine, R&D Systems, Minnneapolis, MN, USA, respectively). Transforming growth factor (TGF)‐β was quantified in serum using a mouse latency‐associated peptide (LAP) (TGF‐β1) Ready‐SET‐Go! Enzyme‐linked immunosorbent assay (ELISA); (eBioscience). Plasma levels of sCD25 and granulocyte–macrophage colony‐stimulatory factor (GM‐CSF) were determined with a DuoSet (R&D Systems). A sandwich ELISA detecting the ferritin heavy chain was kindly offered by Dr Paolo Santambrogio (San Raffaele Scientific Institute, Il Dipartimento di Biotecnologie, Milan, Italy) 25.
Quantification of T helper cell transcription factors using quantitative real‐time polymerase chain reaction (PCR)
Total RNA was extracted using a PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using Superscript II reverse transcriptase and random primers (Invitrogen). Quantitative real‐time PCR was performed with a TaqMan Gene Expression Assay on the 7500 Fast Real‐Time PCR System instrument (both Applied Biosystems, Foster City, CA, USA). Expression levels of T‐box transcription factor (Tbet) (assay ID Mm00450960_m1), GATA binding protein 3 (Gata3) (Mm00484683_m1), retinoid‐related orphan receptor gamma t (Rorgt) (Mm01261019_g1) and forkhead box protein 3 (Foxp3) (Mm00475156_m1) were normalized to the expression of housekeeping gene Gapdh (Mm99999915_g1). Housekeeping genes beta glucuronidase (GusB), hypoxanthine guanine phosphoribosyl transferase (Hprt1), eucaryotic 18S ribosomal RNA (18s) and beta‐2‐microglobulin (B2m) were also tested, but Gapdh expression fluctuated the least following viral infection, thus all analyses were performed using Gapdh normalization. Relative gene expression was calculated with the 2–ΔΔCt method 26.
Single‐cell suspensions, cytospins and flow cytometry
For single‐cell suspensions, white blood cells were obtained from blood after lysis of red blood cells with NH4Cl. Lung leucocytes were obtained from density gradient‐centrifuged lung cell suspensions (Percoll 40 and 72%; GE Healthcare, Chicago, IL, USA). Lymph node cells were extracted from both inguinal lymph nodes. For cytospin preparations, single‐cell suspensions were spun on a glass slide and stained with H&E. For flow cytometry, cell suspensions were incubated with anti‐CD16/anti‐CD32 (Miltenyi Biotec, Bergisch Gladbach, Germany) and stained extracellularly with the following monoclonal antibodies: CD3e (clone 145‐2C11), CD4 (RMA‐4), CD8a (53‐6.7), CD25 (PC61.5 or 3C7), CD49b (DX5), CD69 (H1.2F3), CD122 (5H4) and intracellularly with FoxP3 (FJK‐16s) and Ki‐67 (SolA15) (eBioscience, BD Biosciences or BioLegend, San Diego, CA, USA). Dead cells were excluded using propidium iodide (PI) or Zombie Aqua Fixable Viability Dye (BioLegend). Samples were run with a fluorescence activated cell sorter (FACS)Calibur using CellQuest software or an LSR Fortessa X‐20 using FACSDiva software (all BD Biosciences). Live singlet cells (PI– or ZombieAqua–) were analysed with FlowJo version 10 (TreeStar, Inc., Ashland, OR, USA).
Statistical analysis
Data with two experimental groups were analysed via a two‐tailed non‐parametric Mann‐Whitney U‐test. For comparison of three or more groups, a non‐parametric Kruskal–Wallis test was performed, followed by Dunn's multiple comparison post‐test. GraphPad Prism version 5.00 was utilized.
Results
T helper cells are skewed towards a Th1 response in MCMV‐induced secondary HLH
To unravel the role of T cells in the pathogenesis of murine virus‐associated secondary HLH, T helper (Th) cell profiles in MCMV‐infected wild‐type (WT) and IFN‐γ knock‐out (KO) mice, which display a more severe and more complete HLH phenotype (Supporting information, Fig. S1), were investigated in different organs. The expression of specific Th cell lineage‐defining transcription factors was assessed by quantitative real‐time PCR: T‐bet, GATA‐3, ROR‐γt and FoxP3 for, respectively, Th1, Th2, Th17 and Treg. Both in lymph nodes and spleen, the Th cell population appeared to be skewed towards a Th1 profile following MCMV infection, as evidenced by increased T‐bet expression, while Th2‐, Th17‐ and Treg‐defining transcription factors were down‐regulated (Fig. 1a).
Figure 1.
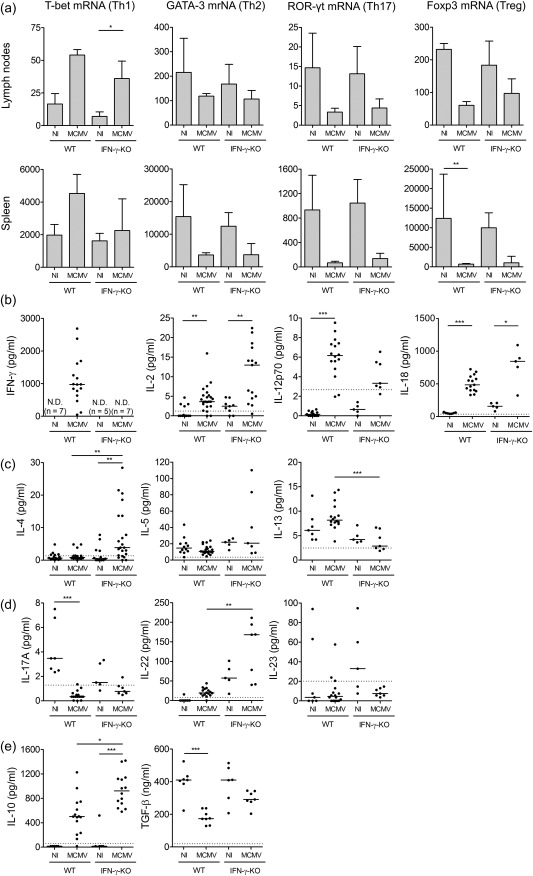
Prevalence of T helper (Th) cell populations in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) mice. (a) mRNA expression of Th cell transcription factors in cells extracted from inguinal lymph nodes and spleen as determined via reverse transcription–quantitative polymerase chain reaction (RT–qPCR). T‐bet for Th1 cells, GATA binding protein 3 (GATA‐3) for Th2 cells, retinoid‐related orphan receptor gamma t (ROR‐γt) for Th17 cells and forkhead box protein 3 (FoxP3) for regulatory T cells (Treg). Bars refer to means, with standard deviation; n = 5 per group. Normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression with the 2–ΔΔCt method. (b–e) Th cell‐associated cytokine concentrations in serum. Interferon (IFN)‐γ, IL‐2, IL‐12 and IL‐18 for Th1 (b), IL‐4, IL‐5 and IL‐13 for Th2 (c), IL‐17A, IL‐22 and IL‐23 for Th17 (d), IL‐10 and transforming growth factor (TGF)‐β for Tregs (e). Dots represent individual animals. Horizontal bars refer to median values. Data obtained on day 5 post‐infection. Dotted lines represent lower enzyme‐linked immunosorbent assay (ELISA) detection limits. n.d. = not detected (detection limit 6·11 pg/ml); NI = not infected; Th = T helper. *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one to four experiments and representative of four independent experiments with at least five mice per group.
At the protein level, systemic production of typical Th1, Th2, Th17 and Treg cell‐related cytokines was examined. Serum levels of IFN‐γ, IL‐2, IL‐12p70 and IL‐18, all Th1‐associated or Th1‐stimulating cytokines, were elevated significantly in MCMV‐infected mice (Fig. 1b). With respect to Th2‐associated cytokines, IL‐4 was increased significantly in IFN‐γ KO mice, whereas levels of IL‐5 and IL‐13 remained unaltered post‐infection (Fig. 1c). The pattern of Th17‐associated cytokines was ambiguous. Serum IL‐17A was decreased in MCMV‐infected WT and IFN‐γ KO mice, while IL‐22 was increased. Levels of IL‐23 seemed reduced in infected IFN‐γ KO mice, whereas no clear difference was observed in WT mice. IL‐21 and GM‐CSF could not be detected (detection limit 8·81 and 15·6 pg/ml, respectively) (Fig. 1d and data not shown). Regarding Treg cell‐associated cytokines, IL‐10 was highly elevated post‐infection. The increase was particularly pronounced in infected IFN‐γ KO mice. In contrast, serum TGF‐β was decreased in infected mice, most significantly in WT mice (Fig. 1e), exhibiting a pattern analogous to the expression of FoxP3 in spleen and lymph nodes (Fig. 1a).
Taken together, the mRNA and protein data indicate that a Th1 response predominates both in WT and IFN‐γ KO mice with MCMV‐associated secondary HLH.
The regulatory T cell population is not reduced in MCMV‐induced secondary HLH
In murine primary HLH, hyperinflammation has been linked to an acquired Treg cell deficiency. Treg cells decline drastically during the fatal disease course, and as a consequence fail to suppress the aberrantly activated CD8+ T cells and concurrent immunopathology 24. Correspondingly, low Treg numbers were reported in untreated HLH patients 24. As the Treg population in experimental MCMV‐associated HLH seemed affected, i.e. decreased FoxP3 expression in spleen and lymph nodes, decreased secretion of TGF‐β, but highly increased serum IL‐10 (Fig. 1), this population was explored in further detail.
In WT and IFN‐γ KO BALB/c mice, the percentage of Treg cells present in lymph nodes appeared unaffected by the viral infection (Supporting information, Fig. S2a and S2d, left panel). In general, WT mice were leaning towards decreased Treg percentages, while IFN‐γ KO mice displayed a trend towards increased percentages; however, this was not significant in any experiment. A similar pattern was observed when the absolute Treg number was quantified in lymph nodes (Supporting information, Fig. S2a and S2d, middle panel). The quantity of FoxP3 protein per Treg cell reflects the cell's functional status and is considered a critical determinant of its suppressor function 27, 28. In MCMV‐infected WT and IFN‐γ KO mice, the FoxP3 levels per cell showed a tendency to decrease, although not significantly (Supporting information, Fig. S2a and S2d, right panel), indicating that Treg cell function was probably not hampered. MCMV infection induced the activation of Treg cells, as indicated by increased surface expression of the early activation marker CD69 (Supporting information, Fig. S2b and S2e) and stimulated their proliferation, evidenced by increased expression of the nuclear protein Ki‐67 (Supporting information, Fig. S2c and Fig. S2f), which is absent in resting [G(0)] cells 29.
Together, these data suggest that MCMV‐associated secondary HLH in WT and IFN‐γ KO mice does not result from a lack of Treg cell‐mediated control of inflammation.
CD4+ T cells are highly activated but dispensable in MCMV‐induced secondary HLH
As we reported previously, depletion experiments in the MCMV‐induced HLH model appointed no significant pathogenic role to CD8+ T cells, despite their hyperactivated phenotype 20. However, CD4+ T cells were also activated markedly post‐MCMV infection, demonstrated by increased expression of early activation marker CD69, intermediate–early activation marker CD25 (IL‐2 receptor α‐chain) and proliferation marker Ki‐67 (Fig. 2). The same pattern was observed in lymph nodes (Fig. 2), spleen, lung and blood (data not shown). Notably, CD4+ T cell activation was more pronounced in the absence of IFN‐γ, raising the question of whether this may account for increased disease severity and poor outcome in MCMV‐infected IFN‐γ KO mice compared to WT mice 20. To investigate the possible pathogenic role of CD4+ T cells in early development as well as subsequent progression of the HLH syndrome, an antibody depletion approach was chosen, administered either on day −1 prior to infection or on day 2 post‐infection, when the first symptoms had appeared. Complementarily, to decipher whether CD4+ T cells play a synergistic role with CD8+ T cells, CD4 and CD8 depletion were combined, revealing the overall role of T cells in the model.
Figure 2.
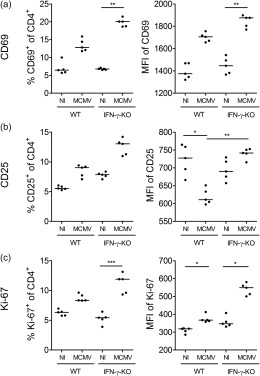
Activation status of CD4+ T cells in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) mice. Percentage of CD69+, CD25+ or Ki‐67+CD4+ T cells (a–c, left panel), gated as % CD69+, CD25+ or Ki‐67+ of live singlet CD4+ lymph node cells and their median fluorescence intensity (MFI) (a–c, right panel). Data obtained on day 5 post‐infection in the inguinal lymph nodes. Dots represent individual animals. Horizontal bars refer to median values. NI = not infected. *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment and representative of four to 10 independent experiments with at least five mice per group.
Following early (day −1) depletion of T cells, an adequate reduction of CD4+ and/or CD8+ T cell numbers was observed in lymph nodes (Fig. 3i,j) and in blood (data not shown). Nonetheless, neither depletion of CD4+ T cells alone (aCD4‐treated group) nor depletion of CD4+ and CD8+ T cells (aCD4 + 8‐treated group) was able to prevent HLH development. All treated mice continued to lose weight post‐infection, although the aCD4 + 8‐treated mice exhibited a lower percentage of total weight loss, suggesting a slightly milder disease course (Fig. 3a). Nevertheless, typical HLH‐like symptoms such as fever and cytopenia were unaffected by aCD4 or aCD4 + 8 treatment (Fig. 3b,c). Other features were attenuated, although not significantly, exclusively in aCD4 + 8‐treated mice. Hyperferritinaemia (Fig. 3d) and liver dysfunction, measured by the plasma level of liver enzymes (Fig. 3f), were tempered following co‐depletion of CD4+ and CD8+ T cells. Interestingly, elevation of plasma soluble CD25 (sCD25) was abolished completely following aCD4 or aCD4 + 8 treatment. When both CD4+ and CD8+ T cells were depleted, sCD25 levels dropped below those measured in naive mice (Fig. 3e), probably reflecting elimination of the cellular sources of sCD25. Conversely, aCD4 and aCD4 + 8 treatment resulted in increased activation of the residual CD4+ and CD8+ T cell population (Fig. 3g,h and data not shown), in line with previous findings following CD8+ T cell ablation in this model 20. Additionally, depletion increased splenic viral titres highly, particularly when CD4+ and CD8+ T cells were targeted together (Fig. 3k). The failure to control viral proliferation may explain, in part, the lack of efficacy of T cell depletion in the model. Additionally, when depleting antibodies were administered on day 2 post infection, so‐called ‘late’ T cell depletion, similar results were obtained (Supporting information, Fig. S3).
Figure 3.
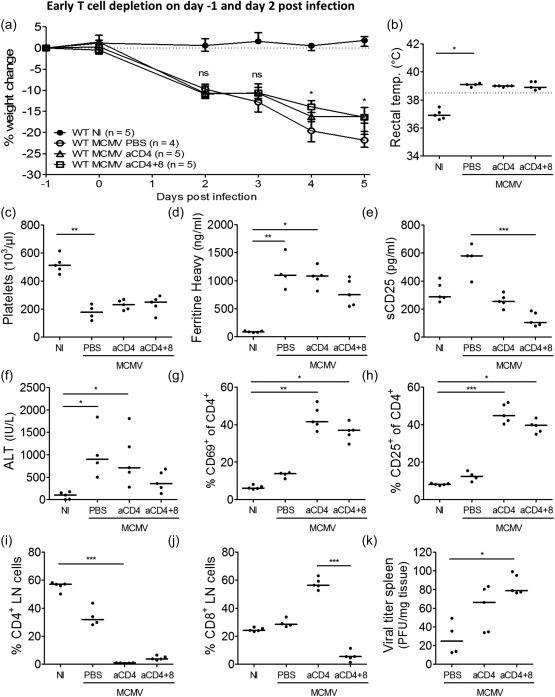
Early T cell depletion does not reduce disease severity in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) BALB/c mice. Phosphate‐buffered saline (PBS), anti‐CD4‐antibodies alone or combined with anti‐CD8‐antibodies were injected intraperitoneally on days −1 and 2 post‐infection (p.i.). (a) Percentage change in body weight relative from initial weight at day −1 p.i. Median with interquartile range of four to five mice per group. Weight change differed significantly between WT MCMV aCD4 + 8 and untreated WT MCMV on day 4 p.i. (*) and between WT MCMV aCD4 and untreated WT MCMV on day 5 p.i. (*). (b) Rectal body temperature (temp.) (°C) at day 2 p.i. Dotted line = 38·5°C. (c) Absolute platelet count in whole blood. (d) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one mouse. (e) Plasma concentration of sCD25 (pg/ml). (f) Plasma concentration of alanine transaminase (ALT) (IU/l). (g and h) Percentage of singlet, live lymph node‐derived CD4+ T cells positive for CD69 or CD25. (i,j) Percentage of CD4+ or CD8+ T cells in inguinal lymph nodes (LN), gated as CD4+ or CD8+ cells of live singlet cells. (k) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg tissue]. (c–k) Data obtained on day 5 p.i. (b–c,e–k) Dots represent individual animals. Horizontal bars refer to median values. aCD4 = anti‐CD4‐antibodies, aCD4 + 8 = anti‐CD4‐ and anti‐CD8 antibodies, NI = not infected; ns = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
In conclusion, neither early nor late ablation of CD4+ and/or CD8+ T cells was sufficient to inhibit development and propagation of the HLH‐like syndrome in MCMV‐infected mice, indicating that T cells play no major role in the pathogenesis of this model. They are, however, an essential defence against persistent viral infections.
Development of MCMV‐induced secondary HLH in SCID mice
As CD8+ and CD4+ T cells appeared to play a minor role in disease induction and propagation, we next investigated whether any adaptive lymphocytes were required for development of MCMV‐associated HLH. To this end, SCID mice, lacking functional B and T cells, were inoculated with MCMV. Disease developed more slowly in MCMV‐infected SCID mice, evidenced by more gradual progression of weight loss (Fig. 4a) and onset of hypothermia on day 9 post‐infection (Fig. 4c). Nonetheless, characteristic HLH‐like symptoms such as fever, mild lymphadenopathy, anaemia, haemophagocytosis and decreased NK cell numbers developed (Fig. 4b,d,f–h). Thrombocytopenia was absent (Fig. 4e). These data demonstrate that non‐lymphoid cells are sufficient to induce most features of HLH, suggesting an innate mechanism of pathogenesis.
Figure 4.
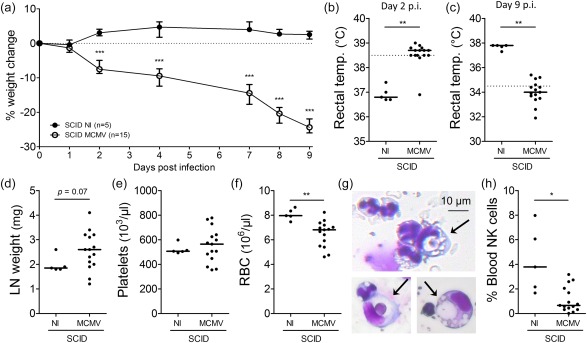
Severe combined immunodeficient (SCID) mice, lacking functional T and B cells, develop a haemophagocytic lymphohistiocytosis (HLH)‐like syndrome post‐infection with 5 × 103 plaque‐forming units (pfu) of mouse cytomegalovirus (MCMV). (a) Percentage change in body weight relative to initial weight at day 0 post‐infection (p.i.). Median with interquartile range of five to 15 mice per group. (b,c) Rectal body temperature (temp.) (°C) at days 2 and 9 p.i. Dotted line = 38·5°C (fever) or 34·5°C (end‐point as indication of mortality). (d) Weight of both inguinal lymph nodes (LN) (mg). (e,f) Absolute platelet and red blood cell count in whole blood. (g) Haemophagocytes detected in haematoxylin and eosin (H&E)‐stained cytospins from blood leucocytes of MCMV‐infected SCID mice. Arrows indicate engulfed cells. (h) Natural killer (NK) cell percentage in blood, gated as CD122+CD49b+ cells of CD3– propidium iodide (PI)– leucocytes. (b–f,h) Dots represent individual animals. Horizontal bars refer to median values. (c–h) Data were obtained on day 9 p.i. NI = not infected; *P < 0·05; **P < 0·01; ***P < 0·001; Mann–Whitney U‐test. Depicted data are from one experiment and representative of two experiments with at least five mice per group. [Colour figure can be viewed at wileyonlinelibrary.com]
CD25‐targeting therapy does not fully ameliorate disease in MCMV‐induced secondary HLH
As CD4+ and/or CD8+ T cell depletion had little effect on disease development, but favoured viral replication adversely, it appears that total T cell elimination is not a safe strategy in virus‐associated secondary HLH. A less drastic intervention would be to target aberrantly activated T cells specifically to halt hyperinflammation, while conserving part of the T cell population to respond to the viral threat. To this end, two clones of anti‐CD25 antibodies were used that either deplete CD25high‐expressing cells or block CD25 signalling, depriving target cells of the vital, stimulatory IL‐2 signal. In murine primary HLH, over‐expression of CD25 on CD8+ T cells was shown to reverse the IL‐2 consumption hierarchy, which is essential in regulating the magnitude of immune responses 24. This same observation was made in MCMV‐infected WT and IFN‐γ KO mice in lymph nodes (Fig. 5a,b), spleen and lung (data not shown). Hyperactivated CD8+ T cells increased their expression of CD25, surpassing that of Treg cells, thereby consuming the available IL‐2, escaping immunosurveillance and feeding inflammation 24. By targeting these CD25high‐expressing cells selectively, homeostasis could possibly be restored to limit immunopathology.
Figure 5.
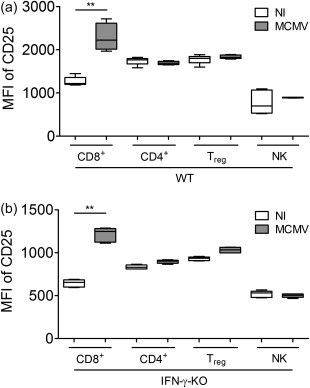
Reversal of the interleukin (IL)‐2 consumption hierarchy in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) mice. Median fluorescence intensity (MFI) of CD25 on CD25+CD8+, CD4+, regulatory T cells (Treg) and natural killer (NK) cells in inguinal lymph nodes in WT (a) and IFN‐γ KO mice (b). (a,b) Box‐plots represent median ±25% quartile, whiskers from minimum to maximum. White boxes, NI = not infected. Grey boxes, MCMV‐infected; five mice per group. Data obtained on day 5 post‐infection. **P < 0·01; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment and representative of three experiments with at least five mice per group.
Blocking (clone 7D4) or depleting (clone PC61) anti‐CD25‐antibodies (aCD25) were administered to WT mice on day 2 following MCMV infection. Depletion (clone PC61) was confirmed in lymph nodes (Fig. 6b) and lung (data not shown). Remarkably, ‘blocking’ clone 7D4 mediated a similar level of depletion (Fig. 6b). The antibodies did not improve weight loss post‐infection (Fig. 6a), nor altered the occurrence of lymphopenia, thrombocytopenia, anaemia or haemophagocytosis (Fig. 6c–f). As the treatment started on day 2 post‐infection, no effect could be observed on the degree of fever (data not shown). Plasma ferritin and sCD25 were not reduced by aCD25 treatment (Fig. 6g,h) and the drop in lung NK cells was not counteracted (Fig. 6i). As anticipated, specific targeting of CD25+ cells and not overall T cells preserved control over the viral infection, as viral titres did not differ between treated or untreated mice (Fig. 6k). Contrary to what was expected, however, aCD25 treatment was unable to normalize the IL‐2 consumption hierarchy (Fig. 6j), which could explain the lack of therapeutic efficacy.
Figure 6.
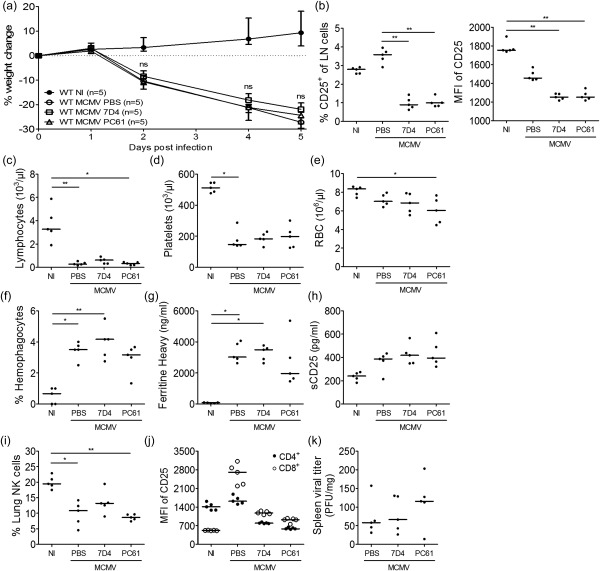
CD25‐targeting therapy does not ameliorate the haemophagocytic lymphohistiocytosis (HLH)‐like syndrome in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) BALB/c mice. Anti‐CD25‐antibodies (blocking clone 7D4 or depleting clone PC61) or phosphate‐buffered saline (PBS) were injected intraperitoneally on day 2 post‐infection (p.i.). (a) Percentage change in body weight relative to initial weight at day 0 p.i. Median with interquartile range of five mice per group. (b) Percentage CD25‐expressing cells of live singlet inguinal lymph node (LN) cells (left panel) and their median fluorescence intensity (MFI) of CD25. Clone 3C7 was used to detect remaining CD25 expression. (c–e) Absolute lymphocyte, platelet and red blood cell (RBC) count in whole blood. (f) Percentage haemophagocytes detected in cytospins of leucocytes derived from lung. Dots represent the average of triplicate counts of 100 cells from one mouse. (g) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one mouse. (h) Plasma concentration of sCD25 (pg/ml). (i) Natural killer (NK) cell percentage in lung, gated as CD122+CD49b+ cells of CD3– ZombieAqua– singlet cells. (j) MFI of CD25 on CD4+ and CD8+ T cells in the inguinal lymph nodes. Clone 3C7 was used to detect remaining CD25 expression. (k) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg tissue]. (b–k) Data obtained on day 5 p.i. (b–e, h–k). Dots represent individual animals. Horizontal bars refer to median values. NI = not infected; ns = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
Given that MCMV‐infected IFN‐γ KO mice showed higher expression levels of CD25 than WT mice (Fig. 2b), we also tested aCD25 treatment in IFN‐γ KO mice, as this might prove more effective. However, the treatment did not reduce weight loss post‐infection (Supporting information, Fig. S4a), nor repressed the development of pancytopenia, haemophagocytosis, hyperferritinaemia, elevated plasma sCD25, decreased NK cell numbers or splenomegaly (Supporting information, Fig S4c–i), an HLH symptom uniquely present in the absence of IFN‐γ 20. Analogous to the findings in WT mice, the IL‐2 consumption hierarchy was not restored by aCD25 treatment (Supporting information, Fig. S4k), despite adequate depletion of CD25+ cells in lymph nodes (Supporting information, Fig. S4b) and lungs (data not shown). Spleen viral titres remained unaffected (Supporting information, Fig. S4l).
In conclusion, blockade or depletion of CD25+ cells did not affect viral control negatively, but was insufficient to reinstate IL‐2 consumption homeostasis and had no therapeutic effect in MCMV‐induced secondary HLH.
Neutrophils are highly increased but play no role in development of MCMV‐induced secondary HLH
As adaptive immunity T and B cells appeared to play a limited role in the pathogenesis of murine virus‐induced secondary HLH, we turned our focus towards the innate immune compartment. During active disease, neutrophil numbers were highly increased in blood and tissue (Fig. 7a–c), both in infected WT and IFN‐γ KO mice. Neutrophils are not only an important first line of defence against invading pathogens, initiating the earliest phases of inflammation, they are equally known to cause abundant organ damage in several autoimmune and autoinflammatory diseases, among others by the formation of neutrophil extracellular traps 30. Considering their striking expansion following MCMV infection, these deleterious effects may contribute to the aberrant immune responses and tissue damage observed in the mouse model of secondary HLH. To investigate this hypothesis, two monoclonal antibodies targeting neutrophils (anti‐Gr1 and anti‐Ly6G) were administered to the mice on day −1 prior to MCMV infection, in order to prevent the development of HLH. Preventive treatment with anti‐Gr1 could not reduce neutrophil numbers adequately (data not shown), while anti‐Ly6G administration resulted in a pronounced depletion of neutrophils in blood as well as in lung tissue (Fig. 7b,c). Nonetheless, the absence of neutrophils did not alter the development or severity of the HLH‐like syndrome in MCMV‐infected mice. Weight loss, fever, lymphopenia, thrombocytopenia, anaemia, hyperferritinaemia and hepatitis were undiminished in neutrophil‐depleted mice (Fig. 7d–h), indicating that the pathogenesis of murine virus‐associated secondary HLH is independent of this cell type.
Figure 7.
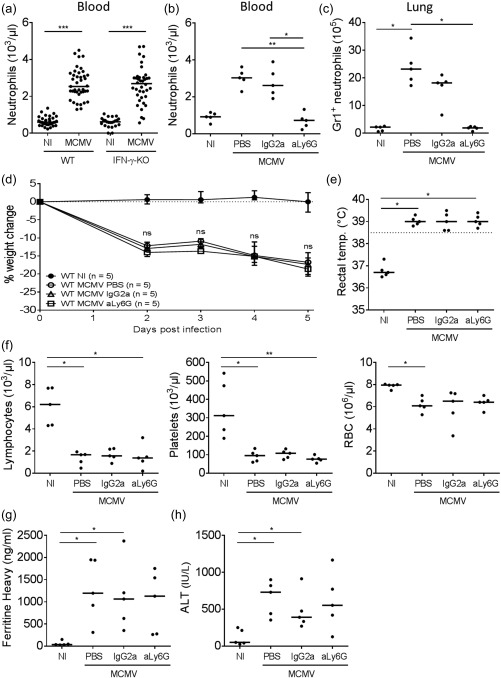
Depletion of lymphocyte antigen 6 complex locus G6D (Ly6G)‐expressing neutrophils does not ameliorate the haemophagocytic lymphohistiocytosis (HLH)‐like syndrome in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) mice. Anti‐Ly6G‐antibodies (clone 1A8) or immunoglobulin (Ig)G2a isotype control (clone 2A3) were injected intraperitoneally on day −1 prior to infection. (a) Absolute neutrophil count in whole blood in the HLH model, in WT and interferon (IFN)‐γ knock‐out (KO) mice, 5 days post‐infection (p.i.). (b) Absolute neutrophil count in whole blood of WT mice, after anti‐Ly6G‐mediated depletion, day 5 post‐infection (p.i.) (c) Absolute count of Gr1‐expressing neutrophils in lung, gated as Gr1+ cells of live (ZombieAqua–) singlet lung cells, day 5 p.i. (d) Percentage change in body weight relative from initial body weight at day 0. Median with interquartile range of five mice per group. (e) Rectal body temperature (temp.) (°C) at day 2 p.i. Dotted line = 38·5°C. (f) Absolute lymphocyte, platelet and red blood cell (RBC) count in whole blood, day 5 p.i. (g) Plasma concentration of the ferritin heavy chain (ng/ml), day 5 p.i. Dots represent the average of two dilutions for one single mouse. (h) Plasma concentration of alanine transaminase (ALT) (IU/L), day 5 p.i. (a–c,e,f,h) Dots represent individual animals. Horizontal bars refer to medians. aLy6G = depleting monoclonal antibody; IgG2a = isotype control; NI = not infected; PBS = no antibody, only saline treatment; ns = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
Discussion
Animal models of HLH have contributed greatly to our current knowledge of the mechanisms driving disease pathogenesis and have paved the way towards targeted therapy 31, 32. However, as the diversity of these animal models is great, given the heterogeneous nature of the underlying aetiologies of HLH, models of primary and secondary HLH often report conflicting data. This allows us to hypothesize that subtypes of HLH may be distinguished from other subtypes by a divergent pathogenesis 33. In particular, differences in the role of specific T cell subsets requires further exploration. Primary HLH models are unanimous in that CD8+ T cells are a crucial pathogenic factor, whereas CD4+ T cells are dispensable 8, 10, 11. However, in murine secondary HLH, both CD8+ and CD4+ T cells appear redundant 12, 14, 15. In the current study, Th cell profiles were examined during murine cytomegalovirus (MCMV)‐induced secondary HLH, including assessment of the regulatory T cell compartment. The CD4+ and CD8+ T cell dependency of the model was studied, as well as the requirement for adaptive immunity and the importance of IL‐2 consumption homeostasis, dictated by gradations in the expression of CD25 on activated T cells. CD25‐targeting therapy was evaluated for its efficacy in virus‐associated secondary HLH. Lastly, the focus was turned towards the innate immune compartment, observing striking increases in neutrophil numbers during active HLH‐like disease. Despite their abundance, targeted depletion experiments revealed no crucial role for these polymorphonuclear cells in the pathogenesis of the model.
Based on the expression of Th cell lineage‐defining transcription factors and the serum concentration of Th cell‐associated cytokines, the immune response in MCMV‐associated secondary HLH appeared to be predominantly Th1‐orientated. This Th1 signature parallels observations in HLH patients. Compared to healthy controls, levels of IFN‐γ and IL‐12, important Th1‐related cytokines, are often highly elevated in primary and secondary HLH, whereas levels of IL‐4, a Th2‐associated cytokine, are reported as undetectable, normal, decreased or only slightly increased 34, 35, 36, 37, signifying domination of Th1 over Th2 cytokines. Conversely, IL‐10, an anti‐inflammatory cytokine produced by Tregs which antagonizes Th1 cytokines, was also highly increased post‐infection. This was remarkable considering the reduced serum levels of TGF‐β, another important Treg cytokine, and concurrent decreased transcription of FoxP3 in lymph nodes and spleen of infected animals, indicating a decline in Treg numbers or activity 27, 28. However, flow cytometry did not reveal a pronounced drop in Treg cell percentage or absolute numbers, and Treg cell activation and proliferation markers were increased significantly post‐infection, suggesting that Treg cells are not defective in MCMV‐induced secondary HLH. In contrast, Treg dysfunction may be inherent in primary HLH, as the cells use granule‐mediated cytotoxicity to kill activated, autologous cells when dampening immune responses 38, 39. In addition to this inherent defect, primary HLH may result from an acquired Treg defect. During active murine primary HLH, the Treg cell compartment was found to be deprived of stimulatory IL‐2 signals, causing a drastic decline in Treg numbers and affecting their immunoregulatory function 24. Nonetheless, the serum IL‐10 concentration is elevated highly in most primary HLH models 8, 11, 40, 41. Correspondingly, augmented serum IL‐10 is reported frequently in HLH patients 34, 35, 36, 37, although disease may be associated with a marked reduction of Treg cells 24. Therefore, rather than originating from Treg cells, the serum IL‐10 present in HLH patients and mice probably emanates from macrophages, actively performing haemophagocytosis 42, or even from NK 41 or CD8+ T cells 43.
As we could not provide evidence previously for a role of CD8+ T cells in the pathogenesis of MCMV‐associated secondary HLH 20, data contrasting the findings in primary HLH mouse models 8, 10, 11, the present study focused on the CD4+ T cell population and the overall role of T and B lymphocytes. Remarkably, although the activation pattern of CD4+ T cells correlated closely to disease severity in WT and IFN‐γ KO mice, CD4+ T cell depletion did not affect disease development or progression. In addition, combined targeting of CD4+ and CD8+ T cells indicated that T cells were not required for disease pathogenesis, while functional T and B cells were dispensable to induce HLH‐like disease in MCMV‐infected SCID mice. These observations are in line with other models of secondary HLH, reporting lymphocyte‐independent mechanisms of action. In a CpG‐induced model of macrophage activation syndrome (MAS), mice lacking T, B and NK T cells could still develop HLH 12. Similarly, recombination activating gene 1 (Rag1) deficiency could not ameliorate HLH‐like disease in HIF1α‐transgenic mice 15, and another humanized transgenic mouse model of MAS did not respond to ablation of T and/or B cells 14. In patients, it was published recently that the degree of CD8+ and CD4+ T cell activation could be a distinguishing feature between primary and secondary HLH, which may be used for rapid diagnostic purposes. T cell populations were activated markedly and effector‐differentiated in primary HLH, while this signature was mainly absent in patients with secondary HLH. T cell activation in virus‐associated secondary HLH was situated between the spectrum from non‐virus‐associated secondary HLH to primary HLH 21, 22. Thus, in contrast with primary HLH, T cells do not appear to play a similar, dominant pathogenic role in secondary HLH, demonstrating that disease is not driven necessarily by over‐activation of the adaptive immune system. One hypothesis speculates that over‐stimulation of innate immune pathways, via excessive Toll‐like receptor triggering 12, 42, 44 or hypersecretion of myeloid‐related cytokines 14, may underlie secondary HLH. In the humanized mouse model of MAS, anti‐CD33‐mediated elimination of myeloid cells reverted disease completely 14. These observations are clinically relevant, as there is evidence of patients who develop HLH independently of T cells or in the absence of T cell activation 21. Primary immunodeficient patients, including SCID patients, may present with HLH despite severe T cell deficiencies 23. In these patients T cell‐targeting therapy is contraindicated, as innate mechanisms are probably driving pathogenesis. Similarly, a subgroup of HLH patients was reported recently to carry gain‐of‐function mutations in NLRC4, which cause spontaneous inflammasome activation and excessive IL‐18 production, driving systemic macrophage activation and HLH flares 45. Targeted inhibition of IL‐18 could control the HLH syndrome successfully in one intriguing case report 46, demonstrating that innate immune defects can underlie HLH. Notably, serum levels of IL‐18 were also increased highly in the MCMV‐induced mouse model of secondary HLH.
As depletion of CD4+ and CD8+ T cells increased viral replication dramatically in the model, we hypothesized that selective targeting of hyperactivated, CD25high‐expressing T cells might be more suited to break hyperinflammation and restore immune homeostasis, while maintaining sufficient control over the viral infection by preserving part of the mature T cell population. In this attempt, a blocking clone (7D4, IgM antibody, operating predominantly via the complement pathway) and a depleting clone (PC61, IgG antibody, functioning via antibody‐dependent cell‐mediated cytotoxicity) were used 47. To specifically target T cells in HLH is not a new idea. Daclizumab, a CD25‐blocking antibody similar to clone 7D4 used in the present study, has been administered successfully in three cases of paediatric and adult HLH, either as monotherapy or in combination with other immunosuppressants 48, 49, 50. Additionally, daclizumab was shown to boost NK cell function in patients with multiple sclerosis, which could be of importance in HLH patients harbouring acquired NK cell defects. However in our model, no clear therapeutic effects of CD25 depletion or blockade were observed. This lack of effect could be associated to either the general lack of T cell dependency of our model or to a failure of the treatment to restore disruptions in the IL‐2 consumption hierarchy. First, the success of CD25‐targeting therapy in human HLH relies most probably on the elimination of activated T cells and a correction of elevated sCD25 serum levels. Thus, failure of the treatment in MCMV‐associated secondary HLH may be associated with the finding that, in contrast to primary HLH models, there is no evidence for a pathogenic role of T cells. Possible application of anti‐CD25 antibodies in the treatment of HLH should therefore be investigated further in animal models of primary, T cell‐dependent HLH. Secondly, the failure of anti‐CD25 therapy in our model could be explained by a lack of efficacy on the IL‐2 consumption hierarchy. Expression levels of CD25, the IL‐2 receptor α‐chain, determine the intake of IL‐2 by immune cells. During homeostasis, CD25 expression is highest on Treg cells, which suppress aberrant immune activation. When responding to an infection, CD25 expression on CD8+ T cells increases transiently, redirecting IL‐2 consumption away from Treg cells and initiating a temporary contraction of the Treg population, allowing effector CD8‐mediated immune responses to expand. When the pathogen has been eradicated, the CD25 expression hierarchy is restored to its initial state. In murine primary HLH, however, reversion of the IL‐2 consumption hierarchy was reported to persist. Decreased IL‐2 production, increased levels of competitive sCD25 and heightened consumption of IL‐2 by CD25high‐expressing CD8+ T cells resulted in an IL‐2‐limiting environment, depriving Treg cells of crucial survival stimuli. The reversed CD25 hierarchy instead nourished activation of pathogenic CD8+ T cells, stimulating immunopathology 24. Although CD8+ T cells are not pathogenic players in MCMV‐induced secondary HLH 20, a similarly inverted IL‐2 consumption hierarchy was observed which could not be overturned by anti‐CD25 antibodies.
In conclusion, the presented data indicate that adaptive immune cells, and in particular T cells, were dispensable for the development of MCMV‐associated secondary HLH, although they played a major part in anti‐viral defences by controlling the viral proliferation rate. These findings highlight an important difference between primary and secondary HLH. Both animal models and human data indicate that adaptive lymphoid cells may be of minor importance in secondary HLH when compared to primary HLH. These insights appear to divide the spectrum of HLH into T cell‐driven disease and haemophagocytic syndromes occurring in the absence of measurable T cell activation, and carry important therapeutic implications. Aggressive T cell‐targeting drugs may only be indicated in patients with true ‘lymphocytic’ histiocytosis 21. To assess the involvement of other cell populations in the development of ‘non‐lymphocytic’ haemophagocytic histiocytosis, we focused here upon possible pathogenic contributions of neutrophils within the innate immune compartment. Neutrophil counts were increased highly during active disease, but preventive depletion of this cell type could not ameliorate any HLH‐like symptoms in the mouse model. Thus, other innate immune cells may be involved in the pathogenesis of ‘non‐lymphocytic’ haemophagocytic histiocytosis. Further research into this HLH subtype should focus upon the myeloid compartment, including monocytes and macrophages, but also upon different subsets of innate lymphoid cells including, but not limited to, NK cells and γδ‐T cells. Furthermore, attention should be devoted to non‐haematopoietic cells such as fibroblasts, epithelial and endothelial cells, as they could play a thus far under‐appreciated role in cytokine storm development, as addressed recently in MAS patients by our research group 51, but also by others in severe influenza infections 52, 53, 54. Fibroblasts and endothelial cells can contribute additionally to elevations in serum sCD25, attributed previously to excessive T cell activation in HLH, but shown recently to correlate poorly with T cell activation markers in patients with secondary HLH 21.
Disclosure
The authors declare no commercial or financial conflicts of interest.
Author contributions
E. B. was responsible for the design and concept of the study, acquisition, analysis and interpretation of data and writing the manuscript. M. I., T. M., J. V. and N. B. contributed to the acquisition and analysis and interpretation of data. L. B. produced the depleting antibodies used in the study. C. W., R. S., G. A. and P. M. were responsible for the design and concept of the study, interpretation of the data and revision of the manuscript. All authors read and approved the final manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1. Schematic representation of the murine model of virus‐associated secondary haemophagocytic lymphohistiocytosis (HLH). The clinical and laboratory features of this model were described recently 20. HLH‐like disease is present in wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) BALB/c mice post‐intraperitoneal (i.p.) infection with 5 × 10³ plaque‐forming units (pfu) of mouse cytomegalovirus (MCMV). Symptoms are analysed 5 days post‐infection. *Fever, pancytopenia, haemophagocytosis, hyperferritinaemia and elevated soluble CD25 levels; **lymphadenopathy, liver dysfunction and decreased natural killer (NK) cell numbers; ***in addition to WT BALB/c mice, also splenomegaly, coagulopathy and decreased NK cell cytotoxicity; ↑ = more pronounced compared to WT BALB/c.
Fig. S2. Number and activation status of regulatory T cells in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) mice. Percentage and absolute number of regulatory T cells (Treg) in WT BALB/c mice (a, left and centre panel) and IFN‐γ KO mice (d, left and centre panel), gated as % forkhead box protein 3 (FoxP3)+CD25+CD4+ of live singlet lymph node cells. Median fluorescence intensity (MFI) of FoxP3 per Treg cell in WT BALB/c mice (a, right panel) and IFN‐γ KO mice (d, right panel). Percentage of CD69+ or Ki‐67+ Treg cells in WT BALB/c mice (b,c, left panel) and IFN‐γ KO mice (e,f, left panel) and their median fluorescence intensity (MFI) of CD69 or Ki‐67 (b,c,e,f, right panel). All data were obtained on day 5 post‐infection in the inguinal lymph nodes. Dots represent individual animals. Horizontal bars refer to median group values. NI = not infected; * P < 0·05; **P < 0·01; Mann–Whitney U‐test. Depicted data are from one experiment and representative of three to five independent experiments with at least five mice per experimental group.
Fig. S3. Late T cell depletion does not reduce disease severity in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) BALB/c mice. Phosphate‐buffered saline (PBS), anti‐CD4‐antibodies alone or combined with anti‐CD8‐antibodies, were injected intraperitoneally on day 2 post infection (p.i.). (a) Percentage change in body weight relative from initial weight at day 0 p.i. Median with interquartile range of five mice per group. (b) Rectal body temperature (temp.) (°C) at day 2 p.i. Dotted line = 38·5°C. (c) Absolute platelet count in whole blood. (d) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one mouse. (e) Plasma concentration of sCD25 (pg/ml). (f) Plasma concentration of alanine transaminase (ALT) (IU/l). (g,h) Percentage of singlet, live lymph node‐derived CD4+ T cells positive for CD69 or CD25. (i,j) Percentage of CD4+ or CD8+ T cells in inguinal lymph nodes (LN), gated as CD4+ or CD8+ cells of live singlet cells. (k) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg tissue]. (c–k) Data obtained on day 5 p.i. (b,c,e–k) Dots represent individual animals. Horizontal bars refer to median values. aCD4 = anti‐CD4‐antibodies; aCD4 + 8 = anti‐CD4‐ and anti‐CD8‐antibodies; NI = not infected; n.s. = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
Fig. S4. Blocking CD25 signalling or depleting CD25‐expressing cells does not ameliorate the haemophagocytic lymphohistiocytosis (HLH)‐like syndrome in mouse cytomegalovirus (MCMV)‐infected interferon (IFN)‐γ knock‐out (KO) mice. Anti‐CD25‐antibodies (blocking clone 7D4 or depleting clone PC61) were injected intraperitoneally on day 2 post‐infection (p.i.). (a) Percentage change in body weight relative from initial body weight at day 0 p.i. Median with interquartile range of five mice per group. (b) Percentage CD25‐expressing cells of all live singlet inguinal lymph node (LN) cells (left panel) and their median fluorescence intensity (MFI) of CD25. Clone 3C7 was used to detect remaining CD25 expression. (c–e) Absolute lymphocyte, platelet and red blood cell (RBC) count in whole blood. (f) Percentage haemophagocytes detected in cytospins of white blood cells derived from lung. Dots represent the average of triplicate counts of 100 cells from an individual mouse. (g) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one single mouse. (h) Plasma concentration of sCD25 (pg/ml). (i) Percentage of natural killer (NK) cells in lung, gated as CD122+ CD49b+ cells of CD3– ZombieAqua– cells. (j) Absolute spleen weight (g). (k) Median fluorescence intensity (MFI) of CD25 on CD4+ and CD8+ T cells in the inguinal lymph nodes. Clone 3C7 was used to detect remaining CD25 expression. (l) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg spleen tissue]. (b–l) Data obtained on day 5 p.i. (b–e,h–l) Dots represent individual animals. Horizontal bars refer to medians. 7D4 = blocking clone; NI = not infected; phosphate‐buffered saline (PBS) = no antibody treatment; PC61 = depleting clone; n.s. = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
Acknowledgements
The current study was supported by grants from the Agency for Innovation by Science and Technology (IWT), the Research Foundation Flanders (FWO), the Regional Government of Flanders (GOA and C1 program) and the Interuniversity Attraction Poles (IAP). E. B. received an IWT fellowship and J. V. an SB fellowship from FWO.
References
- 1. Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol 2013; 161:609–22. [DOI] [PubMed] [Google Scholar]
- 2. Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: update on diagnostic challenges and therapeutic options. Br J Haematol 2016; 174:175–87. [DOI] [PubMed] [Google Scholar]
- 3. de Saint Basile G, Sepulveda FE, Maschalidi S, Fischer A. Cytotoxic granule secretion by lymphocytes and its link to immune homeostasis. F1000Res 2015; 4:930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med 2014; 5:69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis 2000; 6:601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis 2007; 7:814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramos‐Casals M, Brito‐Zerón P, López‐Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet 2014; 383:1503–16. [DOI] [PubMed] [Google Scholar]
- 8. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004; 104:735–43. [DOI] [PubMed] [Google Scholar]
- 9. Billiau AD, Roskams T, Van Damme‐Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN‐gamma‐producing lymphocytes and IL‐6‐ and TNF‐alpha‐producing macrophages. Blood 2005; 105:1648–51. [DOI] [PubMed] [Google Scholar]
- 10. Kögl T, Müller J, Jessen B et al Hemophagocytic lymphohistiocytosis in syntaxin‐11‐deficient mice: T‐cell exhaustion limits fatal disease. Blood 2013; 121:604–13. [DOI] [PubMed] [Google Scholar]
- 11. Chen M, Felix K, Wang J. Critical role for perforin and Fas‐dependent killing of dendritic cells in the control of inflammation. Blood 2012; 119:127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Behrens EM, Canna SW, Slade K et al Repeated TLR9 stimulation results in macrophage activation syndrome‐like disease in mice. J Clin Invest 2011; 121:2264–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, Behrens EM. Interferon‐γ mediates anemia but is dispensable for fulminant toll‐like receptor 9‐induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum 2013; 65:1764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wunderlich M, Stockman C, Devarajan M et al A xenograft model of macrophage activation syndrome amenable to anti‐CD33 and anti‐IL‐6R treatment. JCI Insight 2016; 1:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang R, Hayashi Y, Yan X et al HIF1A is a critical downstream mediator for hemophagocytic lymphohistiocytosis. Haematologica 2017; 102:1956–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tesi B, Sieni E, Neves C et al Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN‐γ receptor deficiency. J Allergy Clin Immunol 2015; 135:1638–41. [DOI] [PubMed] [Google Scholar]
- 17. Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol 2014; 155:118–25. [DOI] [PubMed] [Google Scholar]
- 18. Burns C, Cheung A, Stark Z et al A novel presentation of homozygous loss‐of‐function STAT‐1 mutation in an infant with hyperinflammation – a case report and review of the literature. J Allergy Clin Immunol Pract 2016; 4:777–9. [DOI] [PubMed] [Google Scholar]
- 19. Staines‐Boone AT, Deswarte C, Venegas Montoya E et al Multifocal recurrent osteomyelitis and hemophagocytic lymphohistiocytosis in a boy with partial dominant IFN‐γR1 deficiency: case report and review of the literature. Front Pediatr 2017; 5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brisse E, Imbrechts M, Put K et al Mouse cytomegalovirus infection in BALB/c mice resembles virus‐associated secondary hemophagocytic lymphohistiocytosis and shows a pathogenesis distinct from primary hemophagocytic lymphohistiocytosis. J Immunol 2016; 196:3124–34. [DOI] [PubMed] [Google Scholar]
- 21. Ammann S, Lehmberg K, Stadt U et al Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T‐cell activation, differentiation and repertoire. Eur J Immunol 2017; 47:364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsh RA. Diagnostic dilemmas in HLH: can T‐cell phenotyping help?. Eur J Immunol 2017; 47:240–3. [DOI] [PubMed] [Google Scholar]
- 23. Bode SFN, Ammann S, Al‐Herz W et al The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica 2015; 100:978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Humblet‐Baron S, Franckaert D, Dooley J et al IL‐2 consumption by highly activated CD8 T cells induces regulatory T‐cell dysfunction in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol 2016; 138:200–9.e8. [DOI] [PubMed] [Google Scholar]
- 25. Santambrogio P, Cozzi A, Levi S et al Functional and immunological analysis of recombinant mouse H‐ and L‐ferritins from Escherichia coli . Protein Expr Purif 2000; 19:212–8. [DOI] [PubMed] [Google Scholar]
- 26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 27. Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol 2009; 182:148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev 2011; 241:260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scholzen T, Gerdes J. The Ki‐67 protein: from the known and the unknown. J Cell Physiol 2000; 182:311–22. [DOI] [PubMed] [Google Scholar]
- 30. Delgado‐Rizo V, Martínez‐Guzmán MA, Iñiguez‐Gutierrez L, García‐Orozco A, Alvarado‐Navarro A, Fafutis‐Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol 2017; 8:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strippoli R, Caiello I, De Benedetti F. Reaching the threshold: a multilayer pathogenesis of macrophage activation syndrome. J Rheumatol 2013; 40:761–7. [DOI] [PubMed] [Google Scholar]
- 32. Brisse E, Wouters CH, Matthys P. Hemophagocytic lymphohistiocytosis (HLH): a heterogeneous spectrum of cytokine‐driven immune disorders. Cytokine Growth Factor Rev 2015; 26:263–80. [DOI] [PubMed] [Google Scholar]
- 33. Canna SW, Behrens EM. Not all hemophagocytes are created equally: appreciating the heterogeneity of the hemophagocytic syndromes. Curr Opin Rheumatol 2012; 24:113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Osugi Y, Hara J, Tagawa S et al Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997; 89:4100–3. [PubMed] [Google Scholar]
- 35. Tang Y, Xu X, Song H et al Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. Br J Haematol 2008; 143:84–91. [DOI] [PubMed] [Google Scholar]
- 36. Xu X‐J, Tang Y‐M, Song H et al Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr 2012; 160:984–90.e1. [DOI] [PubMed] [Google Scholar]
- 37. Chen Y, Wang Z, Luo Z, Zhao N, Yang S, Tang Y. Comparison of Th1/Th2 cytokine profiles between primary and secondary haemophagocytic lymphohistiocytosis. Ital J Pediatr 2016; 42:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004; 21:589–601. [DOI] [PubMed] [Google Scholar]
- 39. Verbsky JW, Grossman WJ. Hemophagocytic lymphohistiocytosis: diagnosis, pathophysiology, treatment, and future perspectives. Ann Med 2006; 38:20–31. [DOI] [PubMed] [Google Scholar]
- 40. Pachlopnik Schmid J, Ho C‐H, Diana J et al A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH). Eur J Immunol 2008; 38:3219–25. [DOI] [PubMed] [Google Scholar]
- 41. Lee S‐H, Kim K‐S, Fodil‐Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL‐10 production, and CD8 T cell regulation during viral infection. J Exp Med 2009; 206:2235–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohyagi H, Onai N, Sato T et al Monocyte‐derived dendritic cells perform hemophagocytosis to fine‐tune excessive immune responses. Immunity 2013; 39:584–98. [DOI] [PubMed] [Google Scholar]
- 43. Rood JE, Canna SW, Weaver LK, Tobias JW, Behrens EM. IL‐10 distinguishes a unique population of activated, effector‐like CD8 + T cells in murine acute liver inflammation. J Leukoc Biol 2017; 101:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Strippoli R, Carvello F, Scianaro R et al Amplification of the response to Toll‐like receptor ligands by prolonged exposure to interleukin‐6 in mice: implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum 2012; 64:1680–8. [DOI] [PubMed] [Google Scholar]
- 45. Canna SW, de Jesus AA, Gouni S et al An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014; 46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canna SW, Girard C, Malle L et al Life‐threatening NLRC4‐associated hyperinflammation successfully treated with Interleukin‐18 inhibition. J Allergy Clin Immunol 2016; 139:1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stephens LA, Anderton SM. Comment on ‘cutting edge: anti‐CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells’. J Immunol 2006; 177:2036. [DOI] [PubMed] [Google Scholar]
- 48. Tomaske M, Amon O, Bosk A, Handgretinger R, Schneider EM, Niethammer D. Alpha‐CD25 antibody treatment in a child with hemophagocytic lymphohistiocytosis. Med Pediatr Oncol 2002; 38:141–2. [DOI] [PubMed] [Google Scholar]
- 49. Olin RL, Nichols KE, Naghashpour M et al Successful use of the anti‐CD25 antibody daclizumab in an adult patient with hemophagocytic lymphohistiocytosis. Am J Hematol 2008; 83:747–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lackner H, Urban C, Sovinz P, Benesch M, Moser A, Schwinger W. Hemophagocytic lymphohistiocytosis as severe adverse event of antineoplastic treatment in children. Haematologica 2008; 93:291–4. [DOI] [PubMed] [Google Scholar]
- 51. Put K, Avau A, Brisse E et al Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin‐18 and interferon‐γ. Rheumatology 2015; 54:1507–17. [DOI] [PubMed] [Google Scholar]
- 52. Teijaro JR, Walsh KB, Cahalan S et al Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011; 146:980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sanders CJ, Doherty PC, Thomas PG. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res 2011; 343:13–21. [DOI] [PubMed] [Google Scholar]
- 54. Oslund KL, Baumgarth N. Influenza‐induced innate immunity: regulators of viral replication, respiratory tract pathology and adaptive immunity. Future Virol 2011; 6:951–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1. Schematic representation of the murine model of virus‐associated secondary haemophagocytic lymphohistiocytosis (HLH). The clinical and laboratory features of this model were described recently 20. HLH‐like disease is present in wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) BALB/c mice post‐intraperitoneal (i.p.) infection with 5 × 10³ plaque‐forming units (pfu) of mouse cytomegalovirus (MCMV). Symptoms are analysed 5 days post‐infection. *Fever, pancytopenia, haemophagocytosis, hyperferritinaemia and elevated soluble CD25 levels; **lymphadenopathy, liver dysfunction and decreased natural killer (NK) cell numbers; ***in addition to WT BALB/c mice, also splenomegaly, coagulopathy and decreased NK cell cytotoxicity; ↑ = more pronounced compared to WT BALB/c.
Fig. S2. Number and activation status of regulatory T cells in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) and interferon (IFN)‐γ knock‐out (KO) mice. Percentage and absolute number of regulatory T cells (Treg) in WT BALB/c mice (a, left and centre panel) and IFN‐γ KO mice (d, left and centre panel), gated as % forkhead box protein 3 (FoxP3)+CD25+CD4+ of live singlet lymph node cells. Median fluorescence intensity (MFI) of FoxP3 per Treg cell in WT BALB/c mice (a, right panel) and IFN‐γ KO mice (d, right panel). Percentage of CD69+ or Ki‐67+ Treg cells in WT BALB/c mice (b,c, left panel) and IFN‐γ KO mice (e,f, left panel) and their median fluorescence intensity (MFI) of CD69 or Ki‐67 (b,c,e,f, right panel). All data were obtained on day 5 post‐infection in the inguinal lymph nodes. Dots represent individual animals. Horizontal bars refer to median group values. NI = not infected; * P < 0·05; **P < 0·01; Mann–Whitney U‐test. Depicted data are from one experiment and representative of three to five independent experiments with at least five mice per experimental group.
Fig. S3. Late T cell depletion does not reduce disease severity in mouse cytomegalovirus (MCMV)‐infected wild‐type (WT) BALB/c mice. Phosphate‐buffered saline (PBS), anti‐CD4‐antibodies alone or combined with anti‐CD8‐antibodies, were injected intraperitoneally on day 2 post infection (p.i.). (a) Percentage change in body weight relative from initial weight at day 0 p.i. Median with interquartile range of five mice per group. (b) Rectal body temperature (temp.) (°C) at day 2 p.i. Dotted line = 38·5°C. (c) Absolute platelet count in whole blood. (d) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one mouse. (e) Plasma concentration of sCD25 (pg/ml). (f) Plasma concentration of alanine transaminase (ALT) (IU/l). (g,h) Percentage of singlet, live lymph node‐derived CD4+ T cells positive for CD69 or CD25. (i,j) Percentage of CD4+ or CD8+ T cells in inguinal lymph nodes (LN), gated as CD4+ or CD8+ cells of live singlet cells. (k) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg tissue]. (c–k) Data obtained on day 5 p.i. (b,c,e–k) Dots represent individual animals. Horizontal bars refer to median values. aCD4 = anti‐CD4‐antibodies; aCD4 + 8 = anti‐CD4‐ and anti‐CD8‐antibodies; NI = not infected; n.s. = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; ***P < 0·001; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.
Fig. S4. Blocking CD25 signalling or depleting CD25‐expressing cells does not ameliorate the haemophagocytic lymphohistiocytosis (HLH)‐like syndrome in mouse cytomegalovirus (MCMV)‐infected interferon (IFN)‐γ knock‐out (KO) mice. Anti‐CD25‐antibodies (blocking clone 7D4 or depleting clone PC61) were injected intraperitoneally on day 2 post‐infection (p.i.). (a) Percentage change in body weight relative from initial body weight at day 0 p.i. Median with interquartile range of five mice per group. (b) Percentage CD25‐expressing cells of all live singlet inguinal lymph node (LN) cells (left panel) and their median fluorescence intensity (MFI) of CD25. Clone 3C7 was used to detect remaining CD25 expression. (c–e) Absolute lymphocyte, platelet and red blood cell (RBC) count in whole blood. (f) Percentage haemophagocytes detected in cytospins of white blood cells derived from lung. Dots represent the average of triplicate counts of 100 cells from an individual mouse. (g) Plasma concentration of the ferritin heavy chain (ng/ml). Dots represent the average of two dilutions for one single mouse. (h) Plasma concentration of sCD25 (pg/ml). (i) Percentage of natural killer (NK) cells in lung, gated as CD122+ CD49b+ cells of CD3– ZombieAqua– cells. (j) Absolute spleen weight (g). (k) Median fluorescence intensity (MFI) of CD25 on CD4+ and CD8+ T cells in the inguinal lymph nodes. Clone 3C7 was used to detect remaining CD25 expression. (l) Titre of infectious virus in spleen [plaque‐forming units (pfu) per mg spleen tissue]. (b–l) Data obtained on day 5 p.i. (b–e,h–l) Dots represent individual animals. Horizontal bars refer to medians. 7D4 = blocking clone; NI = not infected; phosphate‐buffered saline (PBS) = no antibody treatment; PC61 = depleting clone; n.s. = not significant (P ≥ 0·05); *P < 0·05; **P < 0·01; Kruskal–Wallis with Dunn's post‐test. Depicted data are from one experiment.