

Abstract
Inactivation of the Adenomatous polyposis coli (APC) gene is an initiating and the most relevant event in most sporadic cases of colorectal cancer, providing a rationale for using Apc‐mutant mice as the disease model. Whereas carcinogenesis has been observed only at the organism level, the recent development of the organoid culture technique has enabled long‐term propagation of intestinal stem cells in a physiological setting, raising the possibility that organoids could serve as an alternative platform for modeling colon carcinogenesis. Indeed, it is demonstrated in the present study that lentivirus‐based RNAi‐mediated knockdown of Apc in intestinal organoids gave rise to subcutaneous tumors upon inoculation in immunodeficient mice. Reconstitution of common genetic aberrations in organoids resulted in development of various lesions, ranging from aberrant crypt foci to full‐blown cancer, recapitulating multi‐step colorectal tumorigenesis. Due to its simplicity and utility, similar organoid‐based approaches have been applied to both murine and human cells in many investigations, to gain mechanistic insight into tumorigenesis, to validate putative tumor suppressor genes or oncogenes, and to establish preclinical models for drug discovery. In this review article, we provide a multifaceted overview of these types of approaches that will likely accelerate and advance research on colon cancer.
Keywords: Apc, colon cancer, lentivirus, model, organoid
1. INTRODUCTION
Carcinoma arises from epithelial cells through accumulation of genetic aberrations1, 2 and the tissue‐specific microenvironment could further promote this process. More than 3 decades ago, rats served as a major animal model of human carcinogenesis, particularly for those types of carcinogenesis induced by chemical carcinogens.3, 4 For example, 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP), a food‐borne carcinogen in well‐cooked meat, induced tumor development in colon, prostate and mammary glands in rats,5 which was significantly promoted by a high‐fat diet,6 while it preferentially induced lymphoma in mice.7 Considering the sharp increase in deaths from these types of cancers, coinciding with the Westernization of diet in Japan,8 PhIP‐induced carcinogenesis models in rats, indeed, phenocopied many aspects of the pathogenesis of the human disease.
Ever since genetically engineered mice (GEM) became available as a result of gene‐targeting technology,9, 10 mice have been the gold standard in modeling cancer. GEM with conditional alleles could also be generated by using the Cre‐loxP system to achieve recombination in a tempo‐spatially controlled manner,11 paving the way for organ‐specific or adult‐onset carcinogenesis studies.12 Despite these benefits, critical drawbacks of GEM might include that its generation could take a long time and be laborious. This is particularly true if conditional gene targeting and multiple intercrossing are required. In addition, modifier genes could become problematic when intercrossing between different strains. Indeed, cooperation between Apc inactivation and Trp53 loss towards intestinal tumorigenesis became evident only in the C57BL/6J background after backcrossing for many generations.13, 14
Matrigel‐based organoid culture has recently emerged as a technique that essentially recapitulates tissue homeostasis in vitro exclusively with epithelial cells.15 It is likely that laminin, abundantly contained in Matrigel,16 and defined factors can reconstitute the intestinal stem cell niche. Both self‐renewal and differentiation of stem cells are maintained, thereby enabling infinite proliferation of stem cells without forced immortalization or transformation. By using this technique, we demonstrated that transformation of murine normal intestinal cells was feasible, through in vitro gene transduction and inoculation in nude mice.17 Importantly, induced carcinogenesis was highly concordant with earlier studies using GEM‐based in vivo models.18 Due to its simple and rapid nature, genetic engineering of organoids might be established as a next generation model of carcinogenesis. In this article, we illustrate the technical basics of this new approach, and review studies by others with similar techniques.
2. CONVENTIONAL MOUSE MODELS FOR INTESTINAL TUMOR DEVELOPMENT
In human colorectal cancer (CRC), mutations in the Adenomatous polyposis coli (APC) gene and the CTNNB1 gene encoding β‐catenin are found in > 80% and about 10%, respectively.19 Both mutations result in nuclear accumulation of β‐catenin, which cooperates with TCF4 to establish constitutive transcriptional activation of the Wnt pathway.20 Reflecting its critical role in tumor initiation, mouse models for intestinal carcinogenesis are based on the genetic aberrations in either gene.21, 22 For any candidate gene related to CRC, GEM are usually generated first and intercrossed with these models to evaluate the impact on tumor progression23, 24 by examining the changes in the incidence, size, multiplicity and histology of the tumors (Figure 1A).
Figure 1.
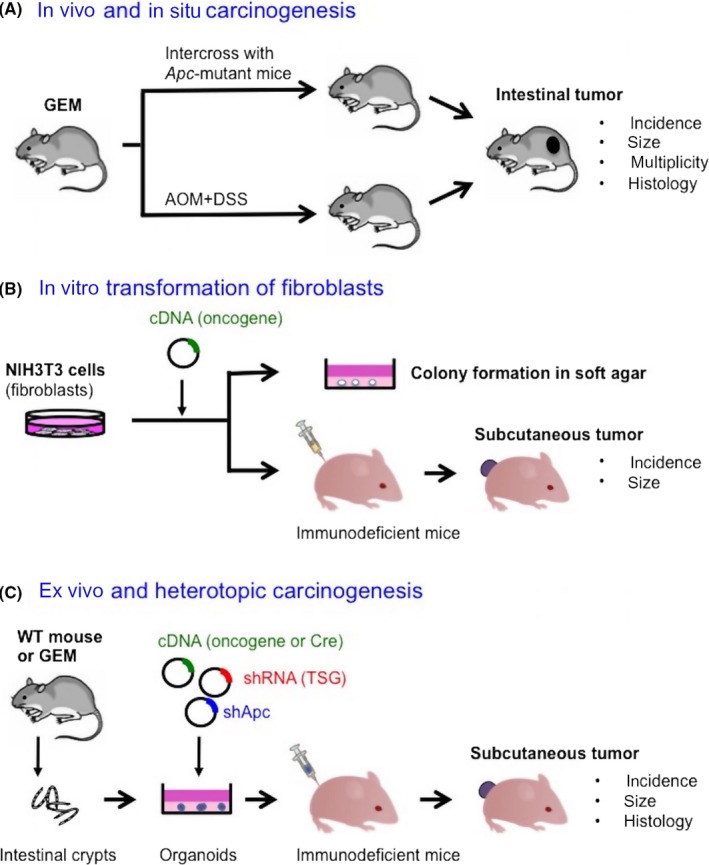
Models to validate tumorigenic potential of a candidate gene. A, In vivo mouse models for colon carcinogenesis. Genetically engineered mice (GEM) for a candidate gene is generated and intercrossed with Apc‐mutant mice or treated with azoxymethane (AOM)/dextran sulphate sodium (DSS) to monitor impact of the gene on tumor development or further progression. Representative readouts are shown. B, Cell‐based transformation assay. Candidate oncogenes are introduced into immortalized murine fibroblasts NIH3T3, followed by culture in soft agar or inoculation in the subcutis of nude mice. C, An ex vivo organoid‐based model. Primary organoid culture of intestinal cells is followed by lentiviral gene transduction. Organoids transduced with cDNA and/or shRNA are inoculated in immunodeficient mice to monitor tumor development
The APC gene is typically inactivated by truncating mutations.25 Apc Min/+ mice carrying the truncated allele were generated through N‐ethyl‐N‐nitrosourea (ENU)‐based mutagenesis.26 While homozygous ablation of Apc results in embryonic lethality, heterozygously mutant mice spontaneously develop numerous adenomatous polyps after the second hit in Apc,27, 28 resembling CRC‐prone hereditary syndrome familial adenomatous polyposis (FAP), except that tumors were predominantly observed in the small intestine.29, 30 Mice with a conditional allele of mutant Apc (eg, Apc flox/+) intercrossed with villin‐Cre mice also developed polyps preferentially in the small intestine.31 These observations strongly suggest that small intestinal cells are more susceptible to transformation by Apc inactivation than colonic cells in mice. To develop cancer in the colon, Apc flox/+ mice were intercrossed with transgenic mice that preferentially expressed Cre in the large intestine, such as Cdx2‐Cre31 or CAC‐Cre,32 or an adenovirus encoding Cre (adeno‐Cre) was directly infused into the rectum of Apc flox/flox.33 Colon carcinogenesis can be alternatively initiated by treatment with the carcinogen azoxymethane (AOM),34 which efficiently introduces an activating mutation in the Ctnnb1 gene in the murine colon,35 and promoted by dextran sulphate sodium (DSS) to induce colitis.36
3. AN ORGANOID‐BASED CARCINOGENESIS MODEL FOR MURINE INTESTINE
The NIH3T3‐based transformation assay in nude mice (Figure 1B) has contributed to the validation of oncogenic potential of many genes.37, 38 Given the development of the organoid culture for murine small intestinal cells,15 we reasoned that similar approaches with epithelial cells might become feasible. As a proof‐of‐concept experiment, knockdown of Apc and other tumor suppressor genes were achieved by lentiviral delivery of corresponding short‐hairpin RNA (shRNA), either alone or in combination, into primary intestinal organoids from wildtype mice with the C57BL/6J background (Figure 1C). Transduced organoids were inoculated in the subcutis of nude mice and monitored for tumor development. We here illustrate the key technical features of this model.17
3.1. Matrigel‐bilayer organoid culture for primary culture, passage and infection
Intestinal crypts or singly dissociated cells are usually resuspended in liquid Matrigel to form a dome‐like structure on a culture dish15 (Figure 2). In an effort to establish robust lentiviral infection in intestinal organoids, we found that single cells or organoids in Matrigel were resistant to lentiviral infection. However, they immediately died in the absence of Matrigel. To circumvent this issue, we plated the single cells on Matrigel and co‐incubated overnight with viral particles. After removing the virus and the floating dead cells the next day, the cells attaching to the Matrigel were covered with Matrigel. This simple procedure, referred to as Matrigel bilayer organoid culture (MBOC), achieved significantly high infection efficiency (approximately 90%) and robust propagation of transduced organonids39 (Figure 2). Consequently, we did not use the spin infection method, although widely used for hematological cells,40, 41 to avoid the risk of damaging naive single epithelial cells with several hours of centrifugation. We eventually adopted MBOC for the routine passage and primary culture as well, because it efficiently eliminated dead or differentiated cells that could digest Matrigel or exert toxic effects on viable cells, and robustly captured highly proliferative stem‐like cell populations on Matrigel, without conducting cell sorting by stem cell markers.
Figure 2.
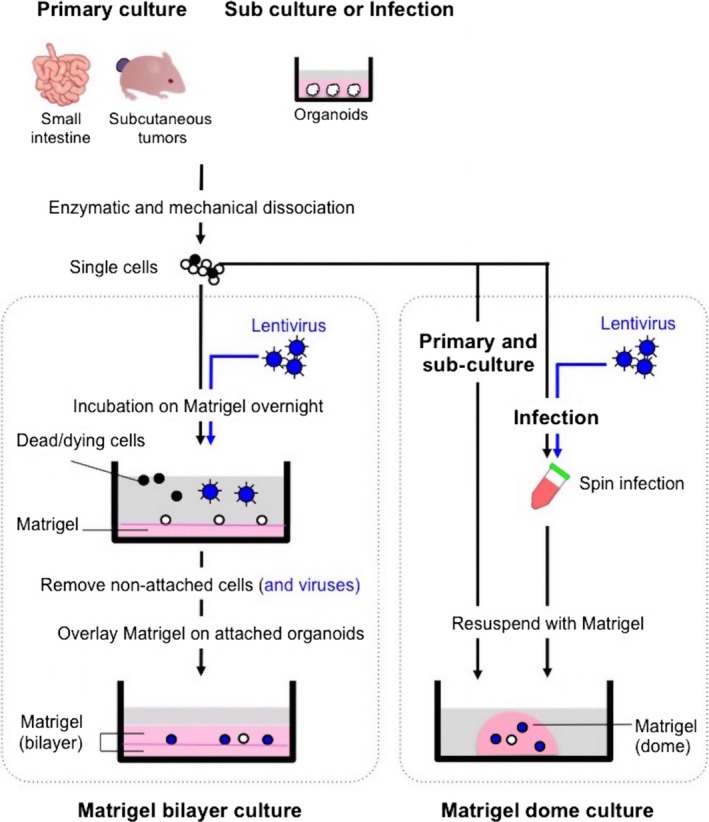
Matrigel bilayer culture for propagation and infection of organoids. Epithelial cells from normal small intestine and subcutaneous tumors can be propagated as organoids. Left panel: Matrigel bilayer culture. Singly dissociated cells are plated on Matrigel, and the cells attached on the lower layer of Matrigel are further covered with Matrigel the next day. By adopting Matrigel bilayer organoid culture, dead cells and tissue‐derived debris can be promptly eliminated from the culture, facilitating both primary‐ and sub‐culture and enabling efficient lentiviral infection. Open circles and closed circles depict viable single cells and dead cells, respectively. Lentivirally transduced cells are labeled in blue. Right panel: Matrigel dome culture. Cells are directly resuspended in Matrigel and poured on a dish to form a dome‐like structure. Spin infection is conducted in a tube
3.2. Re‐defining organoid‐derived tumors in the subcutis of nude mice
As readouts for the tumorigenicity in mice models, it is common to take the incidence, multiplicity, size and histology of the tumors into account.42 To evaluate the results from organoid‐derived carcinogenesis models, however, it was necessary to newly determine criteria for the diagnosis of “tumors.” Transduced intestinal organoids mixed with Matrigel formed subcutaneous nodules in nude mice, which fell into 4 categories based on their macroscopic and microscopic features. The former 2 were regarded as non‐tumors and the latter 2 as tumors (Figure 3).17
Figure 3.
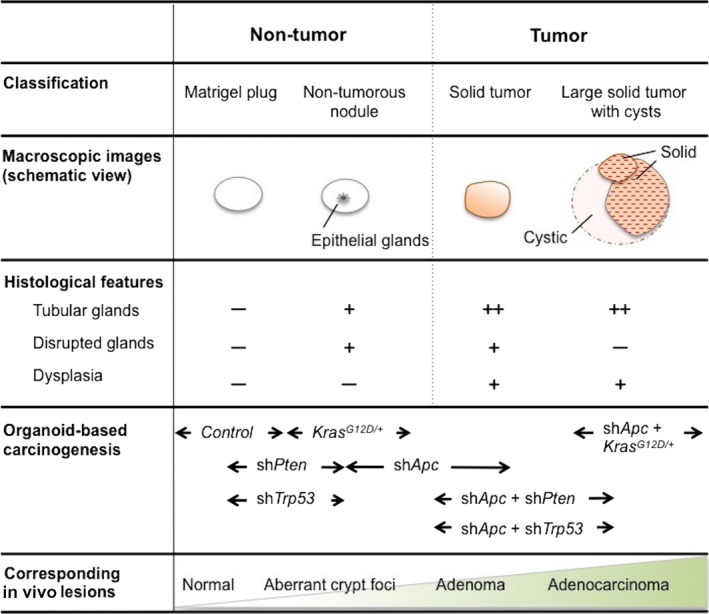
Classification of subcutaneous nodules developed in nude mice. Tumors and non‐tumors developed in the subcutis of nude mice are classified based on their macroscopic and microscopic features. The results from organoid‐based carcinogenesis in murine intestinal cells (Ref. 17) are mapped in the lower row
3.2.1. Matrigel plug
Unless Apc is silenced in inoculated organoids, no nodule can be detected, yet flat white semi‐transparent materials occasionally remained in the injection sites. Microscopically, no epithelial cells were observed, while fibroblasts, immune cells or calcified remains of organoids could be detected. These materials without viable epithelial glands are defined as Matrigel plugs.
3.2.2. Non‐tumorous nodule
Matrigel plug‐like nodules could contain a tiny population of epithelial glands. Histologically, a few non‐dysplastic tubular glands are lined up in a monolayer. Despite the presence of epithelial glands in the subcutis, these lesions were classified as non‐tumorous nodules, as bona fide tumors would have potently outgrown Matrigel. No viable organoids were, in fact, recovered from these nodules, denying the presence of tumor‐initiating cells. It is possible that organoids acquired a growth advantage as a result of random genomic integration of lentiviral vectors.43
Organoids expressing Kras G12D can be generated by delivery of Cre into organoids from conditional knock‐in mice heterozygous for the LSL‐KrasG12D allele.44 Upon inoculation, a few enlarged glands could be transiently formed in non‐tumorous nodules, but eventually disappeared to form Matrigel plugs,17 consistent with the low tumorigenic potential of Kras G12D alone in vivo.45, 46 Similar histology and disappearance have been observed in chemically‐induced aberrant crypt foci (ACF), putative early lesions in colon carcinogenesis.47 Whereas dysplastic ACF represent a pre‐neoplastic lesion,48, 49 hyperplastic ACF are associated with Kras mutation but do not progress into tumors.50, 51 Thus, hyperplastic ACF might also be present, at least temporarily, in non‐tumorous nodules.
3.2.3. Solid tumor
A solid tumor is defined as a palpable round‐shaped nodule. It typically has a similar color to white muscle, and consists of tubular glands accompanied by stromal infiltration. Co‐injected Matrigel is no longer observed, indicating that the tumor outgrew and substituted Matrigel with newly developed tumor stroma. In approximately 70% of the cases tested, organoids transduced with shRNA against Apc (shApc) developed small but solid tumors over several weeks, while no tumor developed without Apc knockdown. With the co‐introduction of shPten or shTrp53, the development rate for solid tumors reached 100% and the size became larger, consistent with earlier in vivo studies.14, 52 Interestingly, shApc‐driven tumors contained prominent mucus pools in the stroma, due to the intra‐tumoral disruption of glands. Similarly, simultaneous augmentation of proliferation and apoptosis has been observed upon acute inactivation of Apc in the murine intestine.53 As it proved to be mediated by the major Wnt‐target gene myc,54 these observations might be a reflection of myc's dual roles.
3.2.4. Large solid tumor with cysts
LSL‐KrasG12D organoids co‐infected with lentiviruses encoding Cre and shApc rapidly gave rise to significantly large tumors with both cystic and solid components, consistent with a strong synergy between Kras mutation and Apc inactivation in vivo.45, 46 Reflecting active angiogenesis, the surface of these tumors tended to be red and contained a huge amount of serous or hematogenous fluid inside cysts. Histologically, tumor glands were densely packed, while disrupted glands were rarely observed, suggestive of anti‐apoptotic effects by Kras G12D. Our experience in organoid‐based tumorigenesis for the lung,55 pancreas (Matsuura et al, submitted) and hepato‐biliary tract (Ochiai et al, submitted) suggests that emergence of a cystic component is closely associated with Kras G12D. Accordingly, future classification of the nodules might as well include “cyst” in the category of non‐tumorous nodules.
4. OTHER ORGANOID‐BASED CARCINOGENESIS MODELS FOR THE INTESTINE AND COLON
Following our previous study,17 several studies reported modeling colon carcinogenesis with similar approaches.56, 57, 58, 59, 60 Although based on analogous concepts, they varied in many respects, including the methods for cell culture, genetic engineering and inoculation, as well as which organs or species were used. These studies are listed in Table 1.
Table 1.
A list of representative studies on modeling colon carcinogenesis using organoid‐based approaches
| Organoids | Genetic alterations | Inoculation | Diagnosis | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Organ | Type | 3‐D culture | Genotypes | Transduction | shRNA/cDNA/Gene editing | Mouse | Site | ||
| Mouse | SI | Normal | Matrigel (bilayer) | WT | Lentivirus | shApc, shTrp53, shPten | Nude | Subcutis | Adenoma‐adenocarcinoma | Onuma et al17 |
| Kras LSL‐G12D/+ | shApc, Cre | Aberrant crypt foci‐adenocarcinoma | ||||||||
| Trp53 −/− | shApc, shPten | Adenoma‐adenocarcinoma | ||||||||
| Pten flox/flox ;Villin‐Cre | shApc, shTrp53 | Adenoma‐adenocarcinoma | ||||||||
| Mouse | SI | Normal | Collagen (air‐liquid interface) | Apc flox/flox ; Villin‐Cre ER | Tamoxifen +retrovirus | shTrp53, shSmad4, Kras G12D | (in vitro) | Tubular adenomatous polyp‐adenocarcinoma | Li et al56 | |
| LI | shTrp53, shSmad4, Kras G12D | Minimal dysplasia‐adenocarcinoma | ||||||||
| Apc flox/flox ; Trp53 flox/flox ; Kras LSL‐G12D/+ | Adenovirus | Cre | High‐grade focal dysplasia | |||||||
| Apc flox/flox ; Villin‐Cre ER | Tamoxifen +retrovirus | shTrp53, shSmad4, Kras G12D | NSG | Subcutis | Adenocarcinoma | |||||
| Human | SI | Normal | Matrigel (dome) | WT | CRISPR/Cas9 (lipofection) | APC, TP53, KRAS G12D | NSG | Subcutis | Adenoma | Drost et al57 |
| APC, TP53, KRAS G12V, SMAD4 | Invasive carcinoma | |||||||||
| LI | APC, TP53, KRAS G12D | Well differentiated carcinoma | ||||||||
| APC, TP53, KRAS G12V, SMAD4 | Poorly differentiated carcinoma | |||||||||
| Human | LI | Normal | Matrigel (dome) | WT | CRISPR/Cas9 (electroporation) | APC | NOG | Kidney subcapsule | None | Matano et al58 |
| APC, TP53, KRAS G12V, SMAD4, PIK3CA E545K | Adenocarcinoma | |||||||||
| Lentivirus | CTNNB1 S33Y, KRAS G12V, PIK3CA E545K, shTP53, shSMAD4 | |||||||||
| Adenoma | ND | CRISPR/Cas9 (electroporation) | ー | None | ||||||
| TP53, KRAS G12V, SMAD4, PIK3CAE 545K | Spleen | Adenocarcinoma with liver metastasis | ||||||||
| Mouse | LI | Normal | Matrigel (dome) | shApc | Doxycycline+adenovirus | Cre | Nude | Colon (+DSS) | Benign tubular adenomas | O'Rourke et al60 |
| shApc; Kras LSL‐G12D ; p53 R127H/− | Doxycycline +adenovirus | Cre | Colon (+DSS) | Carcinoma with submucosal invasion | ||||||
| Kras LSL‐G12D ; p53 flox/flox | CRISPR/Cas9 (lipofection) | Cre, APC | C57BL/6J | Colon (+DSS), spleen, tail vein | High‐grade adenocarcinoma(liver, lung metastasis) | |||||
| shApc; Kras LSL‐G12D ; p53 R127H/− ; shSmad4 | Doxycycline +adenovirus | Cre | Spleen | Liver metastasis | ||||||
| Human | CRC | Tumor (T3) | ND | ー | ー | NSG | Colon (+DSS) | Liver metastasis | ||
| Mouse | LI | Adenoma | Matrigel (dome) | Apc min/+ ; Kras LSL‐G12D/+ ; Villin Cre ; Lgr5 DTR/eGFP | CRISPR/Cas9 (lipofection) | Trp53, Smad4 | NSG | Colon | Tumor development and liver metastasis | De Sousa e Melo et al59 |
| Portal vein | Liver metastasis | |||||||||
CRC, colorectal cancer; DSS, dextran sulfate sodium; LI, large intestine; NSG/NOG, NOD scid gamma mouse (severe immunodeficient mouse); SI, small intestine; WT; wildtype.
4.1. Liquid‐air interface culture‐based heterotopic carcinogenesis in nude mice
Minced tissue fragments were embedded in collagen gel, set in a culture insert, and exposed to serum‐containing media on the bottom and to free air on the top.61 This setting, referred to as liquid‐air interface culture, enables the generation of gradients in concentration of nutrients and oxygen, regenerating tissue structure in a configuration close to the intestinal mucosa. As both epithelial cells and stromal cells are maintained in culture, histological analysis can be directly conducted. Gene transduction was basically conducted in an inducible manner with tamoxifen or acute introduction of Cre. Notably, delivery of Cre worked efficiently for adenovirus, but not for retrovirus, through brief exposure (approximately 30 minutes) to dissociated cells. Then, direct microinjection of the viral particles encoding shRNA or cDNA into the organoids was conducted, which successfully validated the pro‐tumorigenic effects by 11p15.5 amplicon in Apc‐null colon organoids.56
4.2. Colon organoid‐based orthotopic carcinogenesis models in severe immunodeficient or syngenic mice
Colon organoids isolated from GEM harboring doxycycline (Dox)‐inducible shApc were treated with Dox to knockdown Apc. To generate a niche for engraftment in the colon of host mice, mucosal damage had been induced in advance through oral administration of DSS. This procedure enabled development of benign adenomas in nude mice by Apc knockdown alone.60 When further interbred with Kras G12D/+ ; Trp53 f/f mice, their colon organoids, infected with adenovirus‐Cre and treated with Dox to achieve triple mutations, gave rise to invasive carcinoma. Intriguingly, similar results were obtained for orthotopical inoculation in syngenic C57BL/6J mice, demonstrating significant augmentation of tumor engraftment by DSS. Moreover, metastases to the liver and the lung were also observed following injection to the spleen and tail vein, respectively.60 In another study, organoids derived from colon adenoma in Apc min/+ ; Kras G12D/+ mice were subject to further deletion of Trp53 and Smad4 through genome editing with CRSIPR/Cas9. Orthotopic inoculation of these quadruple mutant organoids in severe immunodeficient NSG/NOG mice resulted in adenocarcinoma development with spontaneous liver metastasis, which was inhibited by ablation of Lgr5+ cells, demonstrating critical roles of cancer stem cells in establishing metastasis.59 These studies highlight the potential of the organoid‐based adoptive transplant approaches in extending the research field, from tumorigenesis to metastatic progression and tumor immunity.
4.3. Human organoid‐based carcinogenesis models in severe immunodeficient mice
Organoid culture is also feasible for human intestinal cells.62 Normal small intestinal cells and colon cells were genetically engineered by CRISPR/Cas9 to reconstitute triple mutations in APC, TP53 and KRAS, followed by depletion of factors or addition of inhibitors to enrich cells that acquired independency of the stem cell niches through introduced mutations.57 Engineered organoids from intestine and colon developed adenomas and well‐differentiated adenocarcinomas, respectively. Additional inactivation of SMAD4 resulted in development of invasive carcinomas and poorly differentiated adenocarcinomas, respectively. In contrast, patient‐derived colon adenoma organoids or APC inactivation in the colon organoids were not tumorigenic in the kidney subcapsule of NSG mice. These results suggest that APC inactivation alone in human cells is not sufficient to induce neoplastic changes even in NSG/NOG mice. Not surprisingly, CRISPR/Cas9‐mediated introduction of mutations in TP53, KRAS and PIK3CA cooperated with APC inactivation to generate full‐blown tumors and metastatic tumors from normal organoids and adenomas, respectively.58 Taken together, these observations strongly suggest that human colon organoids can develop adenocarcinomas if only multiple mutations are introduced but not develop premalignant lesions, including ACF or adenomas.
5. CONCLUSION
Each organoid‐based colon carcinogenesis model overviewed herein has unique features. Accordingly, researchers will want to select the right model for their research, depending on the aim of their study, as well as taking into consideration that the time and cost that can be afforded. For example, models with murine colon organoids largely depend on the use of GEM with multiple mutations and place more emphasis on generating tumors or phenomena in the advanced stages.57, 58, 60 Although multiple intercrossing is required, these models might be most suitable for studies on tumor progression and metastasis and for preclinical trials. However, models with murine small intestinal organoids could detect genetic cooperation towards tumorigenesis with the highest sensitivity as preneoplastic lesions or tumors. Hence, validation of candidate genes, whether identified by a murine forward genetic screen63 or in human CRC samples,19 is warranted.
Whereas organoids from inbred young laboratory mice of the C57BL/6 strain might be regarded as “clean” in terms of the homogenous nature of genomes and epigenomes, human organoids may not be ideal in this regard because they might harbor genetic polymorphisms and accumulate mutations from environmental carcinogens and aging. Nonetheless, it is noteworthy that they for the first time enabled human models of carcinogenesis, which will provide valuable information not only for basic science but also for future practice of personalized medicine.
Taken together, organoid‐based carcinogenesis models are promising tools in cancer research, which would likely substitute and complement in vivo mouse models to various degrees. Therefore, efforts toward establishing similar models for other types of cancer driven by different genetic aberrations are warranted.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Maru Y, Onuma K, Ochiai M, Imai T, Hippo Y. Shortcuts to intestinal carcinogenesis by genetic engineering in organoids. Cancer Sci. 2019;110:858–866. 10.1111/cas.13938
REFERENCES
- 1. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759‐767. [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maronpot RR, Nyska A, Foreman JE, Ramot Y. The legacy of the F344 rat as a cancer bioassay model (a retrospective summary of three common F344 rat neoplasms). Crit Rev Toxicol. 2016;46:641‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kemp CJ. Animal models of chemical carcinogenesis: driving breakthroughs in cancer research for 100 years. Cold Spring Harb Protoc. 2015;2015:865‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito N, Hasegawa R, Sano M, et al. A new colon and mammary carcinogen in cooked food, 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP). Carcinogenesis. 1991;12:1503‐1506. [DOI] [PubMed] [Google Scholar]
- 6. Ochiai M, Nakagama H, Watanabe M, Ishiguro Y, Sugimura T, Nagao M. Efficient method for rapid induction of aberrant crypt foci in rats with 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine. Jpn J Cancer Res. 1996;87:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ochiai M, Imai H, Sugimura T, Nagao M, Nakagama H. Induction of intestinal tumors and lymphomas in C57BL/6N mice by a food‐borne carcinogen, 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine. Jpn J Cancer Res. 2002;93:478‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuriki K, Tajima K. The increasing incidence of colorectal cancer and the preventive strategy in Japan. Asian Pac J Cancer Prev. 2006;7:495‐501. [PubMed] [Google Scholar]
- 9. Lampreht Tratar U, Horvat S, Cemazar M. Transgenic mouse models in cancer research. Front Oncol. 2018;8:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty‐first century. Nat Rev Genet. 2005;6:507‐512. [DOI] [PubMed] [Google Scholar]
- 11. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99‐109. [PubMed] [Google Scholar]
- 12. Deng CX. Conditional knockout mouse models of cancer. Cold Spring Harb Protoc. 2014;2014:1217‐1233. [DOI] [PubMed] [Google Scholar]
- 13. Clarke AR, Cummings MC, Harrison DJ. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene. 1995;11:1913‐1920. [PubMed] [Google Scholar]
- 14. Halberg RB, Katzung DS, Hoff PD, et al. Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc Natl Acad Sci USA. 2000;97:3461‐3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262‐265. [DOI] [PubMed] [Google Scholar]
- 16. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010;10:1886‐1890. [DOI] [PubMed] [Google Scholar]
- 17. Onuma K, Ochiai M, Orihashi K, et al. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc Natl Acad Sci USA. 2013;110:11127‐11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackstadt R, Sansom OJ. Mouse models of intestinal cancer. J Pathol. 2016;238:141‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC‐/‐ colon carcinoma. Science. 1997;275:1784‐1787. [DOI] [PubMed] [Google Scholar]
- 21. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570‐579. [DOI] [PubMed] [Google Scholar]
- 22. Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta‐catenin gene. EMBO J. 1999;18:5931‐5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Higurashi T, Endo H, Uchiyama T, et al. Conditional knockout of the leptin receptor in the colonic epithelium revealed the local effects of leptin receptor signaling in the progression of colonic tumors in mice. Carcinogenesis. 2014;35:2134‐2141. [DOI] [PubMed] [Google Scholar]
- 24. Musteanu M, Blaas L, Mair M, et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology. 2010;138:1003‐1011. e1‐5. [DOI] [PubMed] [Google Scholar]
- 25. Zhang L, Shay JW. Multiple roles of APC and its therapeutic implications in colorectal cancer. J Natl Cancer Inst. 2017;109 10.1093/jnci/djw332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322‐324. [DOI] [PubMed] [Google Scholar]
- 27. Fodde R, Edelmann W, Yang K, et al. A targeted chain‐termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA. 1994;91:8969‐8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482‐4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, de Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathology. 2018;50:49‐59. [DOI] [PubMed] [Google Scholar]
- 30. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589‐600. [DOI] [PubMed] [Google Scholar]
- 31. Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma‐carcinoma progression based on somatic Apc inactivation. Can Res. 2007;67:9721‐9730. [DOI] [PubMed] [Google Scholar]
- 32. Xue Y, Johnson R, Desmet M, Snyder PW, Fleet JC. Generation of a transgenic mouse for colorectal cancer research with intestinal cre expression limited to the large intestine. Mol Cancer Res. 2010;8:1095‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shibata H, Toyama K, Shioya H, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120‐123. [DOI] [PubMed] [Google Scholar]
- 34. Ochiai M, Ubagai T, Kawamori T, Imai H, Sugimura T, Nakagama H. High susceptibility of Scid mice to colon carcinogenesis induced by azoxymethane indicates a possible caretaker role for DNA‐dependent protein kinase. Carcinogenesis. 2001;22:1551‐1555. [DOI] [PubMed] [Google Scholar]
- 35. Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the beta‐catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis. 2000;21:1117‐1120. [PubMed] [Google Scholar]
- 36. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation‐related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59:1427‐1434. [DOI] [PubMed] [Google Scholar]
- 38. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448:561‐566. [DOI] [PubMed] [Google Scholar]
- 39. Maru Y, Orihashi K, Hippo Y. Lentivirus‐based stable gene delivery into intestinal organoids. Methods Mol Biol. 2016;1422:13‐21. [DOI] [PubMed] [Google Scholar]
- 40. Kotani H, Newton PB 3rd, Zhang S, et al. Improved methods of retroviral vector transduction and production for gene therapy. Hum Gene Ther. 1994;5:19‐28. [DOI] [PubMed] [Google Scholar]
- 41. Biagi E, Bambacioni F, Gaipa G, et al. Efficient lentiviral transduction of primary human acute myelogenous and lymphoblastic leukemia cells. Haematologica. 2001;86:13‐16. [PubMed] [Google Scholar]
- 42. Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Biffi A, Bartolomae CC, Cesana D, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332‐5339. [DOI] [PubMed] [Google Scholar]
- 44. Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K‐ras. Genes Dev. 2001;15:3243‐3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Janssen KP, Alberici P, Fsihi H, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096‐1109. [DOI] [PubMed] [Google Scholar]
- 46. Sansom OJ, Meniel V, Wilkins JA, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K‐ras oncogene in vivo. Proc Natl Acad Sci USA. 2006;103:14122‐14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McLellan EA, Medline A, Bird RP. Sequential analyses of the growth and morphological characteristics of aberrant crypt foci: putative preneoplastic lesions. Can Res. 1991;51:5270‐5274. [PubMed] [Google Scholar]
- 48. Ochiai M, Hippo Y, Izumiya M, Watanabe M, Nakagama H. Newly defined aberrant crypt foci as a marker for dysplasia in the rat colon. Cancer Sci. 2014;105:943‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ochiai M, Watanabe M, Nakanishi M, Taguchi A, Sugimura T, Nakagama H. Differential staining of dysplastic aberrant crypt foci in the colon facilitates prediction of carcinogenic potentials of chemicals in rats. Cancer Lett. 2005;220:67‐74. [DOI] [PubMed] [Google Scholar]
- 50. Calcagno SR, Li S, Colon M, et al. Oncogenic K‐ras promotes early carcinogenesis in the mouse proximal colon. Int J Cancer. 2008;122:2462‐2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Papanikolaou A, Wang QS, Papanikolaou D, Whiteley HE, Rosenberg DW. Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane‐induced colon carcinogenesis. Carcinogenesis. 2000;21:1567‐1572. [PubMed] [Google Scholar]
- 52. Marsh V, Winton DJ, Williams GT, et al. Epithelial Pten is dispensable for intestinal homeostasis but suppresses adenoma development and progression after Apc mutation. Nat Genet. 2008;40:1436‐1444. [DOI] [PubMed] [Google Scholar]
- 53. Sansom OJ, Reed KR, Hayes AJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676‐679. [DOI] [PubMed] [Google Scholar]
- 55. Sato T, Morita M, Tanaka R, et al. Ex vivo model of non‐small cell lung cancer using mouse lung epithelial cells. Oncol Lett. 2017;14:6863‐6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li X, Nadauld L, Ootani A, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20:769‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Drost J, van Jaarsveld RH, Ponsioen B, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43‐47. [DOI] [PubMed] [Google Scholar]
- 58. Matano M, Date S, Shimokawa M, et al. Modeling colorectal cancer using CRISPR‐Cas9‐mediated engineering of human intestinal organoids. Nat Med. 2015;21:256‐262. [DOI] [PubMed] [Google Scholar]
- 59. de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature. 2017;543:676‐680. [DOI] [PubMed] [Google Scholar]
- 60. O'Rourke KP, Loizou E, Livshits G, et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat Biotechnol. 2017;35:577‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt‐dependent stem cell niche. Nat Med. 2009;15:701‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sato T, Stange DE, Ferrante M, et al. Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762‐1772. [DOI] [PubMed] [Google Scholar]
- 63. Starr TK, Allaei R, Silverstein KA, et al. A transposon‐based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747‐1750. [DOI] [PMC free article] [PubMed] [Google Scholar]