
Figure 4. Proposed models for loss-of-function phenotype of disease-predisposing variant PTPN22-R620W in TCR signaling.
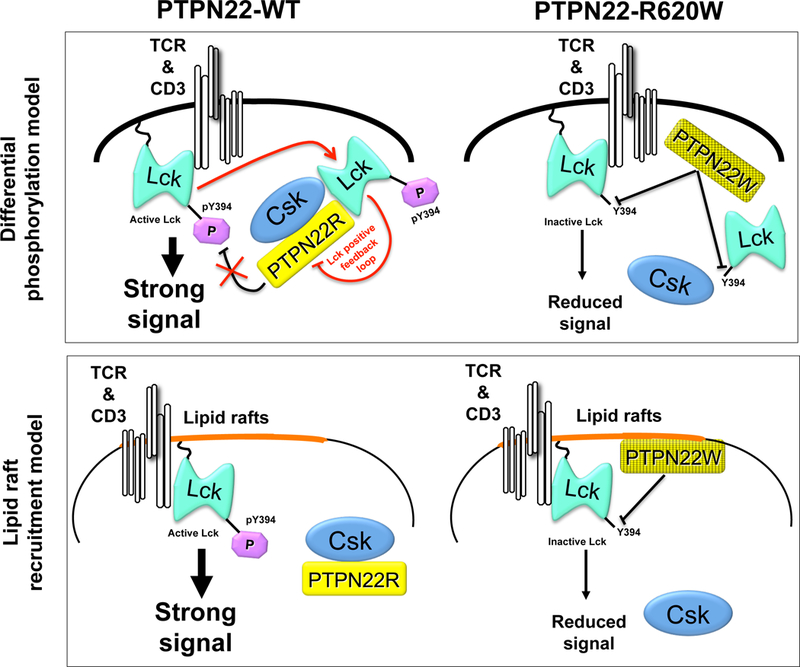
(A) In the initial model(18, 36, 39) the interaction between PTPN22-WT and Csk leads to a synergistic reciprocal potentiation of negative regulation of TCR signaling. The synergism is disrupted by the R620W variation that results in increased phosphorylation of Y394 and decreased phosphorylation of Y505. Interaction with Csk might enhance the function of PTPN22/Ptpn22, however evidence of a reciprocal action of PTPN22/Ptpn22 on Csk is lacking. (B) In the isoform model(17), PTPN22.6-WT, a catalytically-inactive isoform binds to Csk and acts as a dominant negative isoform by inhibiting the action of active PTPN22-WT. The autoimmune-predisposing SNP leads to the appearance of a variant PTPN22.6-R620W isoform. PTPN22.6-R620W displays reduced binding to Csk. However, PTPN22.6-R620W causes enhanced TCR signaling through dominant interference with the negative regulatory effects of PTPN22. As a consequence, Lck may undergo increased phosphorylation of Y394, and downstream TCR signals are enhanced. It is still unclear whether PTPN22.6-R620W inhibits the action of PTPN22-WT and PTPN22-R620W to the same extent. (C) In the degradation model(47), the half-life of PTPN22 is controlled by calpain binding which directs PTPN22 to proteasomal degradation. Csk competes with calpain for binding to PTPN22; thus PTPN22-R620W undergoes increased binding to calpain and proteasomal degradation. Lck is activated by increased phosphorylation of Y394, and downstream TCR signals are enhanced.