

Abstract
Complement-mediated cytotoxicity may act as a selective pressure for tumor overexpression of complement regulators. We hypothesize that the same selective pressure could lead to complement alterations at the genetic level. We find that, when analyzed as a pathway, mutations in complement genes occur at a relatively high frequency and are associated with changes in overall survival across a number of cancer types. Analysis of pathways expressed in patients with complement mutations that are associated with poor overall survival reveals crosstalk between complement and hypoxia in colorectal cancer. The importance of this crosstalk is highlighted by two key findings: hypoxic signaling is increased in tumors harboring complement mutations, and hypoxic tumor cells are resistant to complement-mediated cytotoxicity due, in part, to hypoxia-induced expression of complement regulator CD55. The range of strategies employed by tumors to dysregulate the complement system testifies to the importance of this pathway in tumor progression.
Graphical Abstract
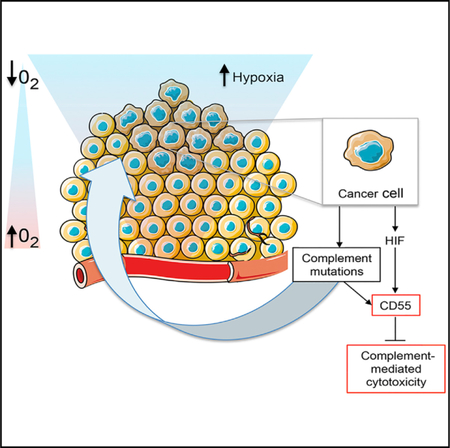
In Brief
Mutations in the complement system are prevalent across cancers. Olcina et al. find that colorectal cancers with complement component mutations are associated with increased hypoxic signaling and poor overall survival outcomes. Hypoxia-induced expression of complement regulator CD55 controls complement-mediated cytotoxicity.
INTRODUCTION
The complement system is composed of soluble and cell surface proteins (Reis et al., 2018). In the extracellular space, activation of complement components by proteolytic cleavage leads to the formation of downstream effectors that act as a first line of defense against pathogenic microorganisms or “altered” host cells (Ricklin et al., 2010). Since uncontrolled activation of the complement system is associated with cell lysis/cytotoxicity and can result in normal tissue toxicity, the pathway is tightly controlled by membrane-bound regulators such as CD35 (complement receptor type 1), CD46 (membrane cofactor protein), CD55 (complement decay accelerating factor), and CD59 (protectin) (Schmidt et al., 2016).
With respect to cancer, complement has been hypothesized to play a role in immunosurveillance through complement-mediated attack of incipient tumors (Pio et al., 2013). Indeed, the use of complement-evasion strategies such as increased tumor expression of complement regulators has been proposed as indirect evidence supporting an anti-tumor role for complement during tumorigenesis (Gorter and Meri, 1999). Elevated expression of complement regulators suggests a selective pressure in favor of protection against complement-mediated cytotoxicity (CMC) (Spiller et al., 2000). The detailed mechanisms underlying both global dysregulation of the complement system as well as altered tumor expression of specific complement regulators, however, have largely remained unexplored to date (Pio et al., 2013).
The existence of complement mutations across cancer types could provide additional evidence to support a role for complement in tumor progression. In cancer, the study of individual genes or even mutations in individual genes, has been suggested to be less important than the action of gene combinations (Leiserson et al., 2015). These gene combinations can work together to provide a survival advantage by converging on particular “molecular networks” (Hofree et al., 2013). As the complement system is a complex set of over 50 proteins converging on functional networks, interrogation of mutations across cancer types and within the whole pathway is likely to yield the most insights about the relevance of any potential alterations (Ricklin et al., 2010).
The Cancer Genome Atlas (TCGA) was developed with the aim to understand cancer by acquiring genome-wide information on thousands of patients (Hoadley et al., 2018). Exome sequencing efforts, including TCGA datasets, have also recently proved useful in finding patient-specific tumor-derived antigens (neoantigens) arising as a consequence of tumor-specific mutations (Rooney et al., 2015). Tumor neoantigens have gained increasing attention due to the realization that they likely contribute to the clinical activity observed for emerging immunotherapies. Neoantigen load is therefore considered both as a biomarker for cancer immunotherapies as well as a means to develop therapies that enhance neoantigen T cell reactivity (Schumacher and Schreiber, 2015).
By taking advantage of expanded TCGA datasets, we have uncovered previously unappreciated mutations in the complement system. These include a number of potential “driver” mutations as well as neoantigens derived from these mutations. Our analysis of pathways expressed in patients with complement mutations associated with poor prognostic outcomes reveals crosstalk between the complement system and hypoxia. In the context of cancer biology, hypoxia refers to the low oxygen tensions frequently found in solid tumors (Hammond et al., 2014). Hypoxia typically arises as a consequence of poor perfusion coupled with high tumor proliferation rates, leading to an imbalance in oxygen supply and demand (Bussink et al., 2003). Importantly, hypoxia correlates with negative patient prognosis and reduced efficacy of various forms of cancer treatment (Bussink et al., 2003). The negative prognostic outcome associated with hypoxia stems, at least in part, from hypoxia-induced pro-survival gene expression changes, including those driven by transcription factors known as hypoxia-inducible factors (HIFs) (Pugh and Ratcliffe, 2003). The importance of hypoxia-related gene expression changes is illustrated by the extensive interest in the development of hypoxia signatures derived from analyzing gene expression changes either in cancer cell lines exposed to normoxia and hypoxia in vitro and/or validated in clinical samples (Harris et al., 2015). These hypoxia signatures have been used to identify new hypoxia-induced genes and pathways associated with hypoxic signaling. Furthermore, these hypoxia signatures could be used clinically for patient stratification and to assess patient prognosis (Buffa et al., 2010; Chi et al., 2006; Harris et al., 2015; Winter et al., 2007). We have used the analysis of a number of hypoxia signatures to reveal associations between complement dysregulation and hypoxia in colorectal cancer. Importantly, those tumors with dysregulated complement through either protein or genetic alterations are associated with the worse prognostic outcome, highlighting the clinical relevance of these findings.
RESULTS
Complement Mutations Are Prevalent across Multiple Cancer Types
Using TCGA, we found that mutations and copy number alterations (CNAs) in complement system genes occurred in all tumor types queried (Figure 1A). We noted that the majority of cancer types not only presented dysregulation at the genetic level but also presented “multiple alterations” including mRNA expression changes (shown in gray in Figure 1A). We decided to assess the importance of these alterations with respect to changes known to have an impact on mRNA expression such as CNA and DNA methylation. We extracted information on correlation analysis between mRNA expression and CNA and mRNA expression and DNA methylation changes in complement genes in COADREAD (provided as an example in Tables S1A and S1B). These data indicate that CNAs of 7 complement genes are indeed correlated with mRNA expression changes (correlation coefficients ranging from 0.1767 to 0.4998). Furthermore, methylation changes of 40 complement genes are also significantly correlated with mRNA expression changes (correlation coefficients ranging from —0.5562 to 0.3180), suggesting that methylation and CNA could alter complement pathway activation through mechanisms independent of alterations in protein structure.
Figure 1. Complement System Mutations Are Prevalent across Multiple Cancer Types.

See also Figure S1.
(A) Frequency of mutations, deletions, amplifications, and multiple alterations in any complement gene are shown for all of 32 cancers analyzed by TCGA. Figure was adapted from http://www.cBioportal.org (Cerami et al., 2012; Gao et al., 2013). Cancer type abbreviations are found in STAR Methods.
(B) A modified KS test was performed on TCGA data to interrogate whether mutations in complement genes as a whole occur at a higher rate than mutations on any other gene in the genome. KS enrichment —log10 q-value for different cancer types is shown in the bar graph. The dotted line represents significance limit. The asterisk (*) represents significant at q < 0.1, —log10 q> 1.
(C) Graph shows Stouffer combined p value for all cancers analyzed by KS test as in Figure 1B above (referred to as PANCAN) and PANCAN without including SKCM.
(D) Table showing the complement genes shown to be significantly mutated by MutSig2CV analysis (q < 0.1) when cancers significant by KS test were queried.
To assess whether mutations (single-nucleotide variants [SNVs]) were occurring at a rate above background, we performed a modified Kolmogorov-Smirnov (KS) test on TCGA data across cancer types. KS test analysis revealed that mutations in complement system genes, as a group, occurred at a rate above background in 8 of the cancers analyzed (Figure 1B; Table S1C). A pan-cancer meta-analysis of the KS test results maintained significance even without melanoma, which had an individually high rate of complement mutations (Figure 1C; Table S1C).
In order to evaluate the potential functional impact of complement mutations, we extracted MutSig2CV analysis for cancer drivers on the significant tumor types by KS test analysis. Drivers promote tumor progression rather than just being a by-product of the high genomic instability found in tumors (Babur et al., 2015). Interestingly, we found that, across a number of cancer types, several complement mutations scored as significant (Figure 1D; Table S1D).
Predicted Driver Complement Mutations Give Rise to Neoantigens and Are Associated with Altered Immune Infiltration Profiles
Given the rising interest in the role of T cell-mediated responses mounted to tumor-derived antigens, we asked whether any of the mutations predicted to be drivers by MutSig2CV analysis could also give rise to neoantigens with sufficiently strong predicted binding affinities (<500 nM) by NetMHCpan (Schumacher and Schreiber, 2015). Interestingly, we found 43 out of the 68 peptides tested were found to be predicted true neoantigens (Table S1E). We collapsed mutations by amino acid change and counted them as a single event if they were predicted to bind the same allele and still found 64.10% predicted true neoantigens (Table S1E). To assess the relevance of the predicted binding affinities for complement mutation-derived neoantigens in comparison with frequently mutated cancer drivers, we analyzed an equivalent number of peptides derived from mutations in three driver genes in colorectal cancer as assessed by MutSig2CV (APC, TP53, and ARID1A). We found no predicted neoantigens from APC, TP53, and ARID1A with binding affinity <500 nM in colorectal cancer even when an expanded list of peptides (equivalent to the total number of complement mutation-derived peptides) was analyzed (Figure S1A; Tables S1F– S1H). The low binding affinity occurred despite the fact that the majority of peptides were actually predicted as true neoantigens. We further extended these analyses to include LDH1, the most significant MutSig2CV gene identified for lower-grade glioma (LGG) (another cancer type with MutSig2CV significant complement mutations). These analyses indicated that no significant predicted neoantigens (50–500 nM) were derived from LDH1 mutations (Figure S1B; Table S1I). Furthermore, average percentile binding affinity rank for complement mutation-derived neoantigens was much lower than that of top significant MutSig2CV genes (0.72 versus 44.45) (Tables S1E–S1I). These data suggest a selection for neoantigens with predicted binding affinities <500 nM derived from complement mutations.
Given the prevalence of complement mutation-derived neoantigens with high binding affinities, we asked whether any of the complement mutations predicted to be drivers would also be expected to result in changes in immune infiltrates. Interestingly, several of these mutations were associated with significant increased cytolytic activity, CD8+ T cells, and cytotoxic lymphocyte infiltration (Figures S1C–S1I).
Complement Mutations Are Associated with Significant Changes in Overall Survival
Most individual complement genes are mutated at rates below those of known canonical driver genes; thus, they may be part of the “long tail” of mutations (cancers being characterized by a large number of infrequently mutated genes and a small number of frequently mutated ones) (Ding et al., 2010). To investigate this possibility, a pathway-level analysis was performed at the “pan-cancer” level using Hotnet2 on TCGA and Genentech datasets (Seshagiri et al., 2012). This analysis confirmed that complement mutations occur at a global level, with 79.7% of datasets analyzed (including 34 different cancer types) containing at least one complement network and several datasets containing more than one mutated complement network (Figure 2A; Table S2A).
Figure 2. Complement System Mutations Are Associated with Changes in Overall Survival.

(A) Pie chart representing the number of significant (p < 0.05) Hotnet2 networks obtained per dataset queried. 79.7% of datasets have at least one network. An example of a network is shown to the right of the pie chart.
(B) Heatmap showing TCGA cancers with mutations in any gene in the subsets shown and their association with overall survival. q-value < 0.1.
(C) Kaplan-Meier (KM) curve for overall survival of COADREAD patients with mutations in any component gene versus patients without mutations in any of these genes (q = 0.00996).
(D) Heatmap showing TCGA cancers with amplifications in any gene in the subsets shown and their association with overall survival. q-value < 0.1.
(E) KM curve for overall survival of SKCM patients with amplifications in any component gene versus patients without mutations in any of these genes (q = 0.07377).
(F) Heatmap showing TCGA cancers with deletions in any gene in the subsets shown and their association with overall survival. qvalue < 0.1.
(G) KM curve for overall survival of LGG patients with deletions in any regulator gene versus patients without mutations in any of these genes (q = 0.07102).
These data support the hypothesis that mutations in complement genes may be cooperative in nature, with genes working within functional networks. We hypothesized that grouping mutations into subsets based on known gene function, and then performing downstream analysis, could provide information about the functional relevance of these mutations and their possible cooperation in cancer. We therefore decided to analyze the association between complement alterations within previously defined functional subgroups and overall survival (Table S2B) (Ricklin et al., 2010). Interestingly, when performing these analyses, we found that several groups of mutations and CNAs were associated with changes in overall survival outcome across different cancer types (Figures 2B–2G; Tables S2C–S2N). The association between component mutations in colorectal cancer and poor overall survival outcome is noteworthy (Figures 2B and2C). Components are the main “effectors” of the complement pathway (Ricklin et al., 2010). In addition, there is a strong association between poor overall survival and amplifications in components in skin cutaneous melanoma (SKCM) and deletions in regulators in LGG (Figures 2D–2G). Interestingly, mutations in components in SKCM as well as mutations in regulators in LGG are also associated with poor overall survival, suggesting that component mutations in SKCM could be gain of function while mutations in regulators in LGG could be loss of function mutations (Figure 2B). In addition, analyses were carried out on an individual gene basis (Tables S2I–S2N). For univariate analysis, several of these groups of genes contained only a few patients with alterations, resulting in an imbalance between the two compared arms. We therefore used the VALORATE algorithm rather than traditional log-rank to assess survival outcome p values (Tables S2U–S2W). Similarly to what we had observed from our multivariate analysis, we found that multiple complement gene mutations and CNA were significantly associated with poor overall survival outcomes (e.g., mutations in components in COADREAD, p value = 0.00273). Furthermore, we performed pan-cancer analyses to assess whether complement gene mutations and CNAs were associated with overall survival outcomes when all cancer types were analyzed together, or when grouping only epithelial cancers together (Tables S2O–S2T). Pan-cancer analyses were carried out either for the five gene subsets previously analyzed or on an individual gene basis. These analyses showed significant correlations with overall survival for certain groups of genes (e.g., protease and receptor mutations, q < 0.05), as well as for individual genes (e.g., mutations and deletions in C1QA, p < 0.05), even when performing stage, age, and cancer-type corrections (Tables S2O–S2T). Overall, these analyses demonstrate that certain complement mutations and CNAs (either analyzed at the individual gene level or in gene subsets) are associated with changes in overall survival, both in specific cancer types as well as when analyzed at the pan-cancer level.
Complement Mutations Are Associated with Hypoxia Signatures in Colorectal Cancer
To gain further insights into processes that could be occurring in patients with complement mutations and may account for the differences in survival, we investigated which genes were differentially expressed in patients with complement mutations and in patients without. To do so, we performed gene set enrichment analysis (GSEA) and examined pathways associated with malignant progression. We found that a recently described hypoxia-related signature (referred to as hypoxia signature) was ranked as the most differentially expressed pathway between these two groups of patients (Figure 3A; Tables S3A and S3C) (Li et al., 2014). “Graft vs Host Disease” and “Antigen Presentation,” both KEGG pathways containing genes previously associated with complement activation, were the other two pathways ranked within the top three (Figures S2A and S2B; Table S3A) (Thornton et al., 1994; Zhang et al., 2016).
Figure 3. Hypoxia Inhibits CMC in Colorectal Cancer.

See also Figure S2.
(A) GSEA plot for “Hypoxia_Li2014” in COADREAD patients with any complement mutation.
(B) GSEA plot for “Hypoxia_Li2014” in COADREAD patients with component mutations.
(C) GSEA plot for “Hypoxia_Buffa2010” in COADREAD patients with component mutations.
(D) GSEA plot for “Hypoxia_Winter2007 upregulated” in COADREAD patients with component mutations.
(E) GSEA plot for “Hypoxia_Winter2007 from literature” in COADREAD patients with component mutations.
(F) Graph represents the % of CMC/total lysis in CT26 cells exposed to normoxia (21% O2) or hypoxia (1% O2) for 24 hr. % CMC/total lysis was assessed by calculating calcein release/total lysis following treatment with either normal mouse serum or heat-inactivated normal mouse serum (as control). * = p value < 0.05, unpaired t test. Error bars represent the SEM for a representative experiment. n = 3.
Intrigued by the fact that a hypoxia signature appeared within the list of pathways differentially expressed in these patients, we asked whether other hypoxia signatures were also significantly overrepresented in the patients with complement mutations. Overall, hypoxia gene sets as a whole were enriched in patients with complement mutations (p value = 0.0005, GSEA-squared), with two of the four additional hypoxia signatures nominally enriched (p < 0.05) in these two groups of patients (Figures S2C and S2D; Tables S3A, S3D, and S3E). An additional signature containing genes known to be downregulated in hypoxia as anticipated was not enriched in these patients (Figure S2E; Tables S3A and S3F). We then refined the analysis by asking whether hypoxia signatures were also enriched specifically in patients with component mutations in colorectal cancer, where a strong association between these mutations and poor overall survival had been observed. Overall, hypoxia signatures as a whole were enriched in patients with component mutations (p value = 0.0008, GSEA-squared) with four of five signatures being nominally enriched (p < 0.05) (Figures 3B–3E; Tables S3B–S3E and S3G–S3I). Once again, the “Hypoxia_Winter2007_downregulated” signature was not enriched in these patients (Figure S2F; Table S3B). To assess the specificity of the association between hypoxia and complement mutations in colorectal cancer, we asked whether component mutations were also associated with hypoxia signatures in SKCM, another cancer type where this group of mutations was associated with poor outcome. Interestingly, no significant association was found between component mutations in SKCM and hypoxia signatures in this group (Figure S2G; Table S3J). To extend these analyses, we also asked whether hypoxia signatures would be enriched in patients with mutations in a different group of complement genes, the regulators, also associated with poor overall survival in LGG, and once again observed no significant association (Figure S2H; Table S3K). These data suggest certain specificity in the association between component mutations and hypoxia in colorectal cancer, and therefore we decided to focus on colorectal cancer for our subsequent experiments. Furthermore, these data suggest that colorectal cancer patients with component mutations have tumors associated with high expression of hypoxia signatures, and therefore may have more hypoxic tumors, providing one potential explanation for the poor overall survival observed in this patient population.
Hypoxia Inhibits CMC in Colorectal Cancer
Since the terminal consequence of complement activation is CMC, we asked whether there were any differences in susceptibility to CMC in colorectal cancer cells exposed to hypoxia. We first compared CMC between normal human colon cells and colorectal cancer cells, and (as has been previously reported) we observed that colorectal cancer cells were more resistant to CMC than normal colon cells (Figure S2I) (Gorter and Meri, 1999). Interestingly, when assessing cytotoxicity levels in hypoxia, we found that both human and murine colorectal cancer cells exposed to hypoxia were less susceptible to CMC than normoxic colorectal cancer cells (Figures 3F and S2J). Notably, the murine colorectal cancer cells tested, CT26, have a mutation in central complement component C3 (pV254I). The human colorectal cancer cells tested, HCT116 also harbor a C3 mutation, but in this case the C3 mutation is silent, and therefore should not alter amino acid sequence. These data suggest that the crosstalk between hypoxia and complement occurs in two directions in colorectal cancer. On one hand, complement component mutations are highly associated with hypoxia signatures and therefore hypoxia-induced gene expression changes. On the other, we note that hypoxia itself can also regulate the terminal consequence of complement system activation by making cancer cells (including those with complement component mutations) more resistant to CMC.
Hypoxia-Induced Expression of Complement Regulator CD55 Contributes to Inhibition of CMC
Membrane-bound regulators tightly control CMC (Schmidt et al., 2016). Intrigued by the increased resistance to CMC observed in colorectal cancer cells exposed to hypoxia, we asked whether hypoxia-induced expression of complement regulators could account for this effect. To address this hypothesis, we first asked which membrane-bound complement regulator (CD35, CD46, CD55, or CD59) was differentially expressed at the mRNA level between colorectal cancer and normal tissue using the Oncomine database. We found that CD46 and CD55 were significantly overexpressed in colorectal cancer compared to normal tissue and CD55 showed the highest increase in fold change (fold change, 3.438) (Figure 4A). We asked whether increased mRNA levels of either of these regulators was associated with a hypoxia signature (same as was used in Figure 3A) and found that there was a positive and significant correlation between CD55 mRNA expression and the hypoxia signature tested in colorectal cancer TCGA patient samples (Figure 4B). In contrast, a slight negative association was observed between CD46 expression and the hypoxia signature tested (Figure S3A). Since CD35 has been shown to be induced following hypoxia/reoxygenation, we also performed in silico analysis to assess the potential hypoxia inducibility of CD35 in colorectal cancer (Collard et al., 1999). We found that CD35 mRNA expression is correlated with the hypoxia signature tested (Figure S3B). However, since mRNA expression changes of CD35 were not markedly increased in colorectal cancers, we did not experimentally pursue the hypoxia inducibility of CD35 further. Notably, CD55 mRNA expression in colorectal cancer correlates with poor patient prognosis (Figure 4C). We therefore decided to focus on CD55 and tested whether mRNA or protein levels of CD55 increased under hypoxic conditions in colorectal cancer cells. Interestingly, we found that mRNA and protein expression of CD55 was increased under hypoxic conditions (1% O2) in several colorectal cancer cells tested in vitro (Figures 4D–4F and S3C). Furthermore, in agreement with our in silico analysis, CD46 mRNA expression levels were not significantly increased in hypoxia, but instead mRNA levels trended toward a decrease (Figure S3D). GLUT1 mRNA expression was assessed in these experiments as a control for exposure to hypoxic conditions (Figures S3E and S3F). Interestingly, CD55 expression changes appeared to be oxygen dependent, since exposure of cells to reoxygenation (21% O2), following a period of hypoxia, reduced CD55 mRNA and protein levels back to normoxic baseline (Figures 4D and4E). We assessed the HIF dependence of CD55 gene expression using both short hairpin RNA (shRNA)- and small interfering RNA (siRNA)-mediated knockdown of HIF members. These data suggested that both HIF1α and HIF2α contribute to the regulation of CD55 under hypoxic conditions (Figures S3G and S3H). Since the greatest decrease in CD55 expression was observed following depletion of the dimerization HIFa partner, HIF1β, these data indicate that the hypoxia-inducible expression of CD55 is likely dependent on both HIF1α and HIF2α (since loss of HIF1β leads to loss of HIF1α- and HIF2α-dependent transcriptional activation) (Pugh and Ratcliffe, 2003).
Figure 4. Hypoxia-Induced Expression of Complement Regulator CD55 Contributes to Inhibition of CMC.

See also Figure S3.
(A) mRNA expression fold changes (FC) and p values for complement regulators CD35, CD46, CD55, and CD59 in different colorectal cancer/adenocarcinoma versus normal tissue pairs. Data were acquired from https://www.oncomine.org/resource/login.html (GSE20916) (Skrzypczak et al., 2010).
(B) Relative expression of CD55 (log10 conversion) in COADREAD patients is shown against hypoxia signature expression (log10 conversion) (Li et al., 2014). Two-tailed p value is shown for the Pearson r (correlation coefficient).
(C) KM curve for colorectal cancer patients with high (red) or low (blue) CD55 mRNA expression levels is shown. This analysis was based on the PrognoScan database (http://dna00.bio.kyutech.ac.jp/PrognoScan/) using the publicly available Gene Expression Omnibus data (https://www.ncbi.nlm.nih.gov/geo) with the accession number GSE14333 (Freeman et al., 2012; Smith et al., 2010). p = 0.011327.
(D) mRNA expression of CD55/18S is shown. qPCR was carried out following exposure of HCT116 cells to 0, 6, or 24 hr of hypoxia (1% O2) or 24 hr of hypoxia followed by 1 hr reoxygenation (21% O2). ** = p value < 0.01, 1-way ANOVA with Tukey’s multiple comparison test. Error bars represent the SEM for a representative experiment. n = 3.
(E) HCT116 cells were treated with 0, 6, or 24 hr of hypoxia (1% O2) or 24 hr of hypoxia followed by 1 hr reoxygenation (21% O2). Western blotting (WB) was carried with the antibodies indicated. β-actin, loading control. n = 3.
(F) CT26 cells were treated with 0 or 24 hr of hypoxia (1% O2). WB was carried with the antibodies indicated. β-actin, loading control. n = 3.
(G) Graph shows the % CMC/total lysis in HCT116 cells treated in 21% O2 (normoxia) and either CD55 blocking antibody or IgG control (for the last hour of treatment). % CMC/total lysis was assessed by calculating calcein release/total lysis (and total cell number) following treatment with either normal human serum or heat-inactivated normal human serum. **** = p value < 0.0001, unpaired t test, two-tailed. Error bars represent the SEM for a representative experiment. n = 3.
(H) Graph shows the % CMC/total lysis in HCT116 cells treated with 24 hr of 1% O2 (hypoxia) and either CD55 blocking antibody or IgG control (for the last hour of treatment). % CMC/total lysis was assessed by calculating calcein release/total lysis (and total cell number) following treatment with either normal human serum or heat-inactivated normal human serum. **** = p value < 0.0001, unpaired t test, two-tailed. Error bars represent the SEM for a representative experiment. n = 3
(I) Graph shows the % CMC/total lysis in CT26 cells treated in 21% O2 (normoxia) and either CD55 blocking antibody or IgG control (for the last hour of treatment). % CMC/total lysis was assessed by calculating calcein release/total lysis following treatment with either normal human serum or heat-inactivated normal human serum. * = p value < 0.05, unpaired t test. Error bars represent the SEM for a representative experiment. n = 3.
(J) Graph shows the % CMC/total lysis in CT26 cells treated with 24 hr of 1% O2 (hypoxia) and either CD55 blocking antibody or IgG control (for the last hour of treatment). % CMC/total lysis was assessed by calculating calcein release/total lysis following treatment with either normal human serum or heat-inactivated normal human serum. **** = p value < 0.0001, unpaired t test, two-tailed. Error bars represent the SEM for a representative experiment. n = 3.
Finally, we asked whether the increase in CD55 expression in colorectal cancer cells could account for the increased resistance to CMC observed in hypoxia. To do so, we assessed CMC in HCT116 and CT26 cells treated with a CD55 blocking antibody or IgG control, and exposed to either normoxia (Figures 4G and4I) or hypoxia (Figures 4H and4J). Cells in normoxia showed increased levels of cytotoxicity compared to cells treated with hypoxia. Furthermore, blocking CD55 antibody treatment increased susceptibility to CMC in both normoxia and hypoxia (Figures 4G–4J). We further confirmed these data by using C5b-9 staining as a marker for formation of the membrane attack complex (and therefore a surrogate for CMC). Decreased C5b-9 staining was observed in cells exposed to hypoxia compared to normoxia (Figure S3I). Furthermore, treatment with CD55 antibody increased levels of C5b-9 in both normoxic and hypoxic conditions (Figure S3I). These data lead us to conclude that blocking increased expression of CD55 could sensitize cells to CMC.
DISCUSSION
Our data indicate that looking for mutations in whole pathways or within functional subgroups may aid in the identification of genetic alterations that predict overall survival outcomes. This approach was helpful in uncovering a pathway that while having clear associations with cancer progression has not been previously appreciated as significantly mutated in cancer, and may have otherwise received little attention due to the perceived “low” frequency of mutations in individual genes. Despite the relatively low frequency of individual mutations (typically <10%), certain complement genes are significant when analyzed by MutSig2CV. Pathway-level mutational analysis revealed that complement pathway mutations are widespread across many cancer types. Additionally, KS test analysis revealed that mutations in complement genes, as a group, occur at a rate above background in a number of the cancers analyzed. However, it should be noted that complement mutations in cancer types below the significance threshold set in the KS test may still be relevant. This is due to the fact that specific groups of mutations in these cancer types could still be clinically significant if not analyzed as a block.
Interestingly, we report that analysis of peptides derived from predicted driver complement mutations suggests that a high proportion of these mutations (over 60%) can give rise to neoantigens that could potentially be used to enhance T cell-mediated reactivity. Indeed, analysis of immune infiltration differences between patients with and without CD55 mutations (one of the mutations giving rise to neoantigens) correlates with altered immune infiltrates including increased cytotoxic T cell responses. It would be interesting to test these further to assess their full potential in therapeutic strategies such as cancer vaccines. Predicted neoantigens have typically been reported in passenger mutations occurring at very low frequencies (around 0.2%–2%), suggesting that any therapeutic use of these mutations would have to be on a patient-by-patient basis (Schumacher and Schreiber, 2015). Several of the complement mutation-derived neoantigens we report here occur at far higher frequencies and therefore could present an opportunity for targeting these mutations in a much wider population of patients.
A closer look at the CD55 mutations identified in colorectal cancer suggests that these mutations occur across different short consensus repeat (SCR) domains, suggesting that they are likely to be loss of function (Vogelstein et al., 2013). This would be in line with CD55 polymorphisms in other disease states, which often result in reduced CD55 protein expression or function (Ozen et al., 2017). However, to our knowledge, none of the CD55 mutations we report here overlap with variants identified in other diseases such as Cromer Inab phenotype (lack of all Cromer complex blood group antigens) or even those variants that have been associated with lung cancer or gastric cancer risk (Ozen et al., 2017; Zhang et al., 2017). Interestingly, Lukacik et al. (2004) showed that missense mutation L205F (present in a colon adenocarcinoma patient) results in 50% reduced CD55 function compared to wild-type CD55. We note, however, that CD55 mutations are not associated with changes in overall survival in colorectal cancer patients.
In contrast, we report that high CD55 mRNA expression is significantly associated with decreased disease-free survival in colorectal cancer. Our results further suggest that hypoxia-induced CD55 expression can act as a barrier to maximal CMC. We should note, however, that we only investigated the effects of hypoxia on CMC in colorectal cancer cells and appreciate that the effects of hypoxia on CMC in other cell types could be different (Okroj et al., 2009). The effectiveness of monoclonal antibody-based therapies, including immunotherapies, has been suggested to rely on CMC for maximal efficacy in certain tumor types (Derer et al., 2014). Targeting CD55 in this context could enhance the efficacy of certain therapeutic strategies. Even though CD55 is ubiquitously expressed in most human cells, colorectal cancers express CD55 at much higher levels than normal colorectal tissue, providing the basis for a therapeutic window (Gorter and Meri, 1999). In line with previous reports, we find that hypoxia-induced expression of CD55 is HIF1α dependent, although we also find HIF2α-dependent regulation of CD55 under hypoxic conditions (Louis et al., 2005). While we describe the direct contribution of hypoxia to CD55 regulation, we acknowledge that other factors present in the tumor microenvironment may also regulate CD55. In fact, angiogenic factors, including hypoxia-inducible vascular endothelial growth factor, have been shown to regulate CD55 expression on endothelial cells (Mason et al., 2001). Furthermore, cyclooxygenase-derived prostaglandin E2, interleukin-1β, tumor necrosis factor a, and lipopolysaccharide, which have all been shown to regulate CD55, can in turn also increase HIF1α stabilization/transcriptional activity (Frede et al., 2006; Jung et al., 2003; Liu et al., 2002). These findings highlight the intricate crosstalk that exists between hypoxia and other known regulators of CD55.
Furthermore, previous reports have described roles for hypoxia in regulating complement proteins in different disease contexts, including inflammatory bowel conditions and ischemia reperfusion injury (Collard et al., 1999; Louis et al., 2005; Olcina et al., 2018). Reports on ischemia/reperfusion injury may provide insights into the role of complement in hypoxic tumor regions, given the similarities that exist between ischemia/reperfusion and certain tumor contexts. For instance, the interruption of blood flow experienced during an ischemic event can induce molecular changes similar to those occurring in tumors with temporarily occluded blood vessels (Gorsuch et al., 2012; Hammond et al., 2014). These molecular changes include a decrease in pH, a factor that should also be considered in the context of alterations in complement regulation (Fishelson et al., 1987). Furthermore, temporarily occluded blood vessels that can give rise to hypoxic regions in tumors may reopen, resulting in reoxygenation events reminiscent of those occurring in ischemia/reperfusion injuries (Gorsuch et al., 2012). Nevertheless, it is important to note that differences also exist in the downstream consequence of hypoxia/reoxygenation in ischemia/reperfusion injury and cancer. A notable difference relates to the apoptotic response mounted following hypoxia/reoxygenation in these two contexts. Apoptosis following ischemia/reperfusion injury can lead to permanent tissue damage (Gorsuch et al., 2012). However, while apoptosis programs may also be triggered in hypoxic tumors (especially in a p53 wild-type tumor), it is common for tumors to evade such apoptosis through mechanisms including p53 mutations (Graeber et al., 1996). Our data suggest that hypoxia may likewise provide a selective pressure for cancer cells to evade complement-mediated attack. These findings highlight the importance of the tumor microenvironment in shaping the cancer mutational landscape.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Monica Olcina (molcina@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and treatments
HCT116 (male adult human epithelial colorectal cancer), cells originally purchased from ATCC (CCL-247TM) were a kind gift from Ester Hammond (Oxford, UK). Cells were grown in DMEM with 10% FBS, in a standard humidified incubator at 37 °C and 5% CO2. FHC (fetal human colon, 13 weeks gestation, sex information unknown) cells were purchased from ATCC (CRL-1831) and grown as per ATCC instructions. CT26 (mouse colon carcinoma) cells were a kind gift from Matt Bogyo’s labortory (Stanford, USA) and were grown in RPMI with 10% FBS, in a standard humidified incubator at 37 °C and 5% CO2. CT26 is a N-nitroso-N-methylurethane (NNMU)-induced undifferentiated colon carcinoma cell line (sex information unknown) induced in BALB/c mice, (Corbett et al., 1975; Wang et al., 1995). All cell lines were routinely tested for mycoplasma and found to be negative.
METHOD DETAILS
Hypoxia treatment
Hypoxic treatments at 1% O2 were carried out in an In Vivo2 400 (Ruskinn) (LaGory et al., 2015). Unless the experiment involved periods of reoxygenation cells were harvested inside the chamber with equilibrated solutions.
Immunoblotting
Cells were lysed in UTB (9 M urea, 75 mM Tris-HCl pH 7.5 and 0.15 M β-mercaptoethanol) and sonicated briefly before quantification. Antibodies used were anti-human mouse CD55 (BioRad, concentration # MCA914: 1:500), anti-mouse rat CD55 (R&D # MAB5376, concentration: 1:500) β-actin (Sigma # A5441, concentration: 1:5000), H3 (Abcam, #ab1791, concentration: 1:1000), HIF1α (BD-Biosciences, #610959, concentration: 1:500), HIF2α (Novus, # NB100–122, concentration: 1:500), ARNT (Novus, # NB100–124, concentration: 1:500), CD59 (Santa Cruz, concentration # sc-133171, 1:500). The BioRad Chemidoc XRS system was used. In each case experiments were carried out in triplicate and a representative blot is shown unless otherwise stated.
Immunofluorescence
5 × 104 HCT116 cells were seeded on coverslip slides. Treated cells were washed in PBS before being fixed in 4% paraformaldehyde (Santa Cruz, # sc-281692) for 15 minutes at room temperature. Cells were then washed once in PBS and incubated with 1% PBS-Triton X-100 (PBS-T) for 10 minutes at room temperature. Blocking was then carried out for 1 hour in 0.5–1 mL 2% (w/v) BSA in 0.1% PBS-T. Following a wash in ice-cold 0.25% PBS-T, slides were incubated in primary C5b-9 antibody (Abcam, cat # 55811, concentration: 1:1000), diluted in 2%(w/v) BSA-PBS-T 0.1% for 1 hour at 37○C in a humidified chamber. Following three washes in ice-cold 0.25% PBS-T, slides were incubated in Alexa Fluor A459 (Invitrogen #A21207, concentration: 1:250) for 1 hour at 37○C in a humidified chamber in the dark. Slides were mounted with ProLong™ Gold Antifade mountant with DAPI (Thermofisher, P36935) following two further washes in ice-cold 0.25% PBS-T and a final wash in PBS (Olcina et al., 2013). Cells were visualized using a DSM6000 or Dmi8 (Leica) microscope. Experiments were carried out in triplicate and a representative set of images are shown.
qRT-PCR
RNA was extracted using TRIzol (Invitrogen/Life Technologies, # 15596018). iScript cDNA synthesis kit (Bio-Rad, # 1708891) was used to reverse transcribe cDNA from total RNA according to manufacturer’s instructions. Relative mRNA levels were calculated using the standard curve methodology using a 7900HT Fast Real-Time PCR System. In each case experiments were carried out in triplicate and data for a representative experiment is shown unless otherwise stated. Primers used: 18S F: TAGAGGGACA AGTGGCGTTC, 18S R: CGGACATCTAAGGGCATCAC. CD55 F: CCACAAAAACCACCACACC, CD55 R: GCCCAGATAGAAGACG GGTAGTA, GLUT1 F: ATACTCATGACCATCGCGCTAG GLUT1 R: AAAGAAGGCCACAAAGCCAAAG, CD46 F: TGCTGCTCCAGA GTGTAAAGTG, CD46 R: CGCTGCCATCGAGGTAAA.
CMC assay
CMC was assessed as previously described with minor modifications (Ajona et al., 2007). Full details of the protocol followed in this manuscript are described here. Cells were trypsinized and washed in PBS. Cells were resuspended in calcein buffer containing 2 μM calcein AM (Trevigen, cat # 4892–010-K). Cells were loaded with calcein for 1 hour at 37°C and then washed once with calcein buffer before addition of complement-containing pooled serum or heat-inactivated serum (Innovative Research Inc, Cat # IPLA-SER for human serum and IGMSSER for mouse serum). Heat-inactivated serum was treated by incubation at 60°C for 30 minutes and was used as a control in order to calculate specific CMC. In experiments requiring use of blocking antibodies, CD55 antibody (BRIC 110, American Research Products # 08–9402-02 for human cells or Biolegend CD55 antibody #131802, for mouse cells) and IgG control (mouse IgG, Santa Cruz # sc-2025 for human cells or Biolegend Armenian hamster # 400902, IgG for mouse cells) treatment was performed 1 hour before addition of serum (Ajona et al., 2007). Supernatant calcein release was assessed by measuring fluorescence at 485 nm excitation and 520 nm emission, using a Synergy H1-mono plate reader (Biotek) following 1 hour incubation at 37○C with either normal or heat-inactivated serum. The remaining pellet was lysed with 0.1% triton-X and also read by measuring fluorescence at 485 nm excitation and 520 nm emission. Specific CMC was calculated by subtracting calcein release/total lysis from heat-inactivated serum treated cells from calcein release/total lysis from normal serum treated cells as follows: [Reading from calcein release in cells treated with normal serum/total lysis (calcein release in normal serum treated cells+ calcein release from 0.1% triton treated pellet)] – [Reading from calcein release in cells treated with heat-inactivated normal serum/total lysis (calcein release in heat-inactivated normal serum treated cells + calcein release from 0.1% triton treated pellet)]. In each case experiments were carried out in triplicate and data for a representative experiment is shown unless otherwise stated. For experiments involving hypoxia treatment, all steps (prior to measurement of calcein release by fluorescence reading) were carried out in the hypoxia chamber In Vivo2 400 (Ruskinn).
siRNA transfection
HIF1α (L-004018–00). HIF2α (L-004814–00) and ARNT pool (L-007207–00) (Dharmacon) or non-targeting RNAi negative control (Scramble, D-001810–10) (Dharmacon) were transfected into HCT116 cells using Lipofectamine RNAiMax transfection reagent (Invitrogen, #13778075) at a final concentration of 50 nM; according to the manufacturers’ instructions. Cells were harvested 72 hours post-transfection.
Lentiviral transduction
shRNAs targeting HIF1α, HIF2α, or non-targeting control (Scramble, RHS6848) were used. HCT116 cells were transduced with shRNA lentiviruses as previously described (LaGory et al., 2015). Full details of the protocol followed in this manuscript are described here. Inititally, shRNAs were transfected into 293T cells using Lipofectamine 2000 transfection reagent (Invitrogen, #11668027) according to the manufacturers’ instructions for virus production. 24 hours post-transfection, medium was replaced with fresh medium. 24 hours later, HCT116 cells were transduced with each virus on two consecutive days to produce stable Scramble (Scr), HIF1α and HIF2α knockdown cells. 24 hours after the last infection cells were washed extensively and grown in antibiotic selection medium. All procedures were carried out in compliance with BSL-2 protocol standards.
KS statistic of mutations
To determine the enrichment of complement mutations in each TCGA cancer, a modified Kolmogorov-Smirnov (KS) test using the method of GSEA (kt.test2; https://github.com/franapoli/signed-ks-test) was performed on “mutation ranks.” For each patient in a cohort the mutation rank of a gene was determined from total mutation number divided by gene coding length which was then put into a rank ordered list. Complement mutations as a group were compared to the background of all other coding genes. Coding sequence length was obtained from the Consensus CDS database (Pruitt et al., 2009). q-values were computed across all cancers using the method of Benjamani-Hochberg. The pan-cancer meta-analysis p value was calculated using Stouffers method.
Cancer type abbreviations
ACC = Adrenocortical carcinoma, BLCA = Bladder urothelial carcinoma, BRCA = Breast invasive carcinoma, CESC = Cervical squamous cell carcinoma and endocervial adenocarcinoma, CHOL = Cholangiocarcinoma, COADREAD = Colorectal adenocarcinoma, DLBC = Lymphoid neoplasm diffuse large B cell lymphoma, ESCA = Esophageal carcinoma, GBM = Glioblastoma multiforme, HNSC = Head and Neck squamous cell carcinoma, KICH = Kidney Chromophobe, KIRC = Kidney renal clear cell carcinoma, KIRP = Kidney renal papillary cell carcinoma, LAML = Acute myeloid leukemia, LGG = Brain lower grade glioma, LIHC = Liver hepatocellular carcinoma, LUAD = Lung adenocarcinoma, LUSC = Lung squamous cell carcinoma. MESO = Mesothelioma, OV = Ovarian serous cystadenocarcinoma, PAAD = Pancreatic adenocarcinoma, PCPG = Pheochromocytoma and paraganglioma, PRAD = Prostate adenocarcinoma, SARC = Sarcoma, SKCM = Skin cutaneous melanoma, STAD = Stomach adenocarcinoma, TGCT = Testicular germ cell tumors, THCA = Thyroid carcinoma, THYM = Thymoma UCEC = Uterine corpus endometrial carcinoma, UCS = Uterine carcinosarcoma, UVM = Uveal melanoma.
TCGA RNA-sequencing (RNA-seq) analysis
TCGA raw counts were acquired from the TCGA data portal using TCGA-Assembler. Counts were rounded then inputted into DESEQ2 to perform differential expression analysis using default size factor normalization (Love et al., 2014). The Independent filtering and Cooks Cutoff flags were set to FALSE. Benjamini–Hochberg method was used for multiple hypothesis testing.
To examine tumor-associated HIF-activity (referred to as hypoxia signature in Figure 4), raw data for each sequenced gene were rescaled to set the median equal to 1, and HIF-activity was quantified by averaging the normalized expression of 44 target genes, associated with HIF activity (encoding ADM, IGFBP3, EDN2, PFKFB4, FLT1, TFR2, BNIP3L, TGFA, BNIP3, PGK1, EGLN1, LDHA, EGLN3, CP, TGFB3, PFKFB3, HK1, TFRC, EDN1, CDKN1A, CA9, HMOX1, SERPINE1, LOX, NDRG1, CA12, PDK1, VEGFA, ERO1L, RORA, P4HA1, MXI1, SLC2A1(GLUT1), STC2, MIF, DDIT4, ENO1, CXCR4, PLOD1, P4HA2, GAPDH, PGAM1, TMEM45A and PIM1) (Li et al., 2014). Log10 conversion of the hypoxia signature was plotted against Log10 conversion of raw data for CD55, CD46 and CD35 (also rescaled to set the median equal to 1). Two-tailed p value is shown for each Pearson r (correlation coefficient). Raw RNA-seq data were downloaded from the TCGA project (accessed through cBioportal: http://www.cbioportal.org/) on February 15th 2016).
GSEA
Gene set enrichment analysis was performed using COADREAD, SKCM or LGG expression data, where groups were defined by complement mutational status. For each comparison DESEQ2 was run on RNaseq data using the mutation stratification with default settings except cooksCutoff = F and independentFiltering = F. Gene set enrichment was run with the preRanked setting using the “stat” column from DESEQ as ranking with the associated hypoxia-related signature using the JAVA based GSEA tool with default settings (Buffa et al., 2010; Eustace et al., 2013; Li et al., 2014; Winter et al., 2007). Full gene lists for each hypoxia-related signature used are given in Tables S3C–S3I).
To capture if the hypoxia-related signatures were, as a group, more enriched in complement gene mutated samples than expected by chance we performed “GSEA-squared.” To capture a biological background, GSEA was first performed with the C2 Kegg database with the hypoxia-related signatures appended as individual gene sets (KEGG+Hypoxia). The input of the second round was a ranked list consisting of the gene sets of KEGG+Hypoxia ranked by their NES scores. A modified Kolmogorov-Smirnov test that uses the method of GSEA was then used to derive a p value for the enrichment of the Hypoxia sets on the KEGG gene set background (kt.test2; https://github.com/franapoli/signed-ks-test).
MutSig2CV v3.1
MutSig2CV v3.1 data was obtained from firebrowse.org (Accessed 26.1.2016). MutSig is used to identify genes that are mutated at higher rates than would randomly be expected to occur by chance (Lawrence et al., 2013). MutSig accounts for the background mutational rate for each gene by taking into account any process leading to positive selection of mutations (for example, frequency of non-synonymous versus silent mutations, hotspots of mutation clusters, or being enriched in evolutionary conserved regions). As such it can give an indication of whether mutations are likely “passengers” or “drivers.” “Drivers” would be expected to be selected for due to their direct effects on tumor progression as opposed to be indirectly selected for due to co-occurrence with other genetic alterations (Babur et al., 2015). Significance levels (p values) are determined by testing whether the observed mutations significantly exceeded the expected counts based on the background model. False-discovery rates (q-values) are also calculated and genes with q < 0.1 are reported as significantly mutated (full list in Table S1D).
Neoantigen Prediction
The list of variants predicted as “cancer drivers” by Mutsig2CV was used for neoantigen prediction (following removal of synonymous mutations), Genes, mutations, neoantigens and predicted binding affinities (IC50) are shown in Tables S1E–S1I). The term neoantigen refers to tumor specific DNA alterations that give rise to novel peptide sequences, usually entirely absent from the normal human genome (Schumacher and Schreiber, 2015). Of note, each mutation gave rise to multiple peptides and from those, neoantigens in turn were predicted to bind to one or multiple alleles. To prevent overestimation by counting multiple neoantigens from a single amino-acid changing mutation binding to the same allele, we collapsed mutations by amino acid change and counted them as a single event if they were predicted to bind the same allele.
The list of HLA alleles in the whole TCGA cohort was a kind gift from Michael Rooney and these alleles were reduced to the most common 20 alleles. For an improved coverage of common HLA alleles, the missing most common alleles from the US population (as reported by http://www.allelefrequencies.net) was also added into the list resulting in a total of 24 alleles. The list of variants and the list of HLA alleles was fed into the NetMHCpan (v3) via topiary tool (https://github.com/openvax/topiary) to calculate predicted binding affinities of predicted true neoantigens (Nielsen and Andreatta, 2016).
Copy number and expression correlation data
Copy number variation and mRNA expression correlation data for COADREAD was downloaded from Firebrowse.org (accessed August 15th 2018). Data generated through the pipeline was downloaded and used to assess correlations between copy number and expression data. The original downloaded report contained “the calculated correlation coefficients based on measurements of genomic copy number (log2) values” and the level of expression of the corresponding gene for each patient as described in firebrowse.org. “Gene level (TCGA Level III) expression data and copy number data of the corresponding loci derived by using the CNTools package of Bioconductor were used for the calculations. Pearson correlation coefficients were calculated for each pair of genes shared by the two data sets across all the samples that were common. The input file “*.medianexp.txt” is generated in the pipeline mRNA_Preprocess_Median in the stddata run.” Once data for all copy number and gene expression data was downloaded, data on all complement genes shown in Table S2B was queried. Significant genes are shown in Table S1A.
DNA methylation and expression correlation data
DNA methylation and mRNA expression correlation data for COADREAD was downloaded from firebrowse.org (accessed August 15th 2018). The data was generated as described in firebrowse.org: “Level 3 methylation and gene expression arrays were paired on the basis of Entrez Gene ID concordance.” Spearman correlation was calculated to assess the association between DNA methylation and the level of expression of the corresponding genes, with the top 25 correlated methylation probes per gene presented. Platforms used: Methylation Array Platforms: Illumina Infinium HumanMethylation27, Illumina Infinium HumanMethylation450, Illumina DNA Methylation OMA002, Illumina DNA Methylation OMA003. Gene Expression Platforms: Agilent 244K Gene Expression G4502A-07–1, Agilent 244K Gene Expression G4502A-07–2, Agilent 244K Gene Expression. Once data for all copy number and gene expression data was downloaded, data on all complement genes shown in Table S2B was queried. Significant genes are shown in Table S1B.
Hotnet2
Hotnet2 addresses the “long-tail” phenomenon by discovering significant alterations in pathways or networks of genes, rather than singularly mutated genes (Leiserson et al., 2015). TCGA data used in Hotnet2 was converted from “.maf” files from Firebrowse.org. Data from the Genetech/gRed (colorectal cancer) dataset generated in (Seshagiri et al., 2012) was also used for hotnet2 analysis. FHCRC and PRMET datasets (from (Kumar et al., 2016), (Robinson et al., 2015) respectively) were downloaded from cbioportal. org using the cgdsr package. ‘NEPC & variants’ dataset was obtained from (Beltran et al., 2016). “Silent” and “RNA” mutations were filtered out from the list of patient mutations. Hotnet2 determines a heat score for each gene in a given cancer (Leiserson et al., 2015). Heat scores are calculated using an insulated heat diffusion process, determined by the significance and number of times the gene was mutated. 1000 random permutations of the Human Protein Reference Database (HPRD) were used to test against subnetworks identified by the Hotnet2 algorithm. Once the randomly permuted networks were made, Hotnet2 was run using default parameters, except the minimum subnetwork size was changed from 2 genes to 3 genes.
Clinical association analysis
Alterations were only evaluated if they occurred in at least 3 samples. Somatic mutations, CNA and survival information, from the public TCGA data portal (https://tcga-data.nci.nih.gov/docs/publications/tcga/) for individual cancers were first analyzed using a univariate model on the alteration status using the logrank test with the survival package in R (variable: logrank.pval;Tables S2U–S2W). Unlike clinical studies, analysis of patient populations based on genomic data such as the TCGA often have unbalanced patient populations based on dichotomies generated from individual genomic features. In the context of these “unbalanced arms” the asymptotically normal assumption of the traditional log-rank test is not met and often gives misleading false positives (Vandin et al., 2015). To correct for this problem we used VALORATE, an algorithm that creates an accurate approximation of the p value using the conditional distributions of the co-occurrences between events and mutations (Treviño and Tamez-Pena, 2017). The VALORATE method was recently shown to provide a more accurate assessment of survival statistics in imbalanced populations such as those found in genetic studies (Treviño and Tamez-Pena, 2017). The VALORATE algorithm is additionally useful in cases where the wald test does not converge (variable: valorate.pval; Tables S2U–S2W). Analysis of all complement genes across 23 tumor types was performed. Additionally 6 different metagene signatures based on a previous functional classification of complement genes (All genes, Components, Pattern Recognition, Proteases, Receptors, Regulators) were analyzed (Ricklin et al., 2010).
Individual cancers were additionally analyzed using a multivariate wald test using the coxph module in R on samples with clinical annotation. In this multivariate model clinical covariates of stage, age, and gender were included when available (variable: not corrected for load.wald.pval; Tables S2D, S2F, S2H, S2J, S2L, and S2N). An additional covariate of mutational load which is the sum of all non-silent mutations in a cancer sample was used (variable: corrected.for.load.wal.pval; Tables S2C, S2E, S2G, S2I, S2K, and S2M) to control for genomic instability and as a proxy for MSI (Chalmers et al., 2017). Data for correlations between associations and overall survival where q < 0.1 are reported as dark red squares in the heatmap in Figure 2. These data represent Benjamani Hochberg corrected Log q-values of the model including age, stage, gender, and in the cases of COADREAD, ESCA, STAD and UCEC also mutational load correction (Cortes-Ciriano et al., 2017).
Similarly, analyses were also performed at the pan-cancer level. In the pan-cancer analysis the multivariable cox regression was performed either controlling for type (variable: corrected.for.type.wald.pval; Tables S2O–S2T) or controlling for type, age and gender (variable: corrected.for.type.age.gender.wald.pval; Tables S2O–S2T). The improvement LRT was performed on these two models.
Immune infiltration analysis
For each cancer, samples with and without mutation were compared for enrichment of immune infiltration signatures using RNaseq data. Single sample GSEA (ssGSEA) was performed with immune signatures derived from (MCP-counter) (Becht et al., 2016). ssGSEA scores were compared between mutant and non mutant groups using a Student’s t test to find significant differences in specific immune infiltration signals.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for the quantification of experimental data was performed with GraphPad Prism Software version 7.0c. Three independent biological replicates were carried out for all experiments unless otherwise stated in the figure legend and “n” denotes the number of independent biological replicates performed. The statistical tests used and the levels of significance are stated in the figure legend for each figure. Information describing how data was analyzed and quantified can also be found in the relevant “Method Details” section for each method.
Supplementary Material
Table 1. KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human CD55 | BioRad, | Cat# MCA914; RRID:AB_321792 |
| Rat polyclonal CD55 | R&D | Cat# MAB5376; RRID:AB_10640505 |
| Mouse monoclonal β-actin | Sigma | Cat# A5441; RRID:AB_476744 |
| Rabbit polyclonal H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Mouse monoclonal HIF1α | BD-Biosciences | Cat# 610959; RRID:AB_398272 |
| Rabbit HIF2α | Novus | Cat# NB100–122; RRID:AB_10002593 |
| ARNT | Novus | Cat# NBP1–02476; RRID:AB_1520290 |
| Mouse CD59 | Santa Cruz | Cat# sc-133171; RRID:AB_2244558 |
| Anti-human CD55 (blocking) antibody (BRIC 110) | American Research Products | Cat# 08–9402-02; RRID:AB_1540745 |
| Anti-mouse CD55 (blocking) antibody | Biolegend | Cat# 131802; RRID:AB_1279269 |
| Armenian hamster anti-mouse IgG antibody | Biolegend | Cat# 400902 |
| Mouse anti-human IgG control | Santa Cruz | Cat# sc-2025; RRID:AB_737182 |
| Alexa Fluor A459 | Invitrogen | Cat# A21207; RRID:AB_141637 |
| Critical Commercial Assays | ||
| Calcein AM Cell Viability Assay | R&D | 4892–010-K |
| Deposited Data | ||
| Computational raw and analyzed data (including p values, q-values, correlation coefficients, predicted IC50 values,Hotnet2 networks and GSEA data) | This paper | Tables S1A–S1I, S2A–S2W, and S3A–S3K |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247TM |
| CT26 | Laboratory of Matt Bogyo (Stanford, USA) | RRID:CVCL_7256 |
| FHC | ATCC | CRL-1831 |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810–10 |
| SMARTpool: ON-TARGETplus HIF1A siRNA | Dharmacon | L-004018–00 |
| SMARTpool: ON-TARGETplus EPAS1(HIF2A) siRNA | Dharmacon | L-004814–00 |
| SMARTpool: ON-TARGETplus ARNT (HIF1β) siRNA | Dharmacon | L-007207–00 |
| Recombinant DNA | ||
| shRNA control: TRC1/1.5-pLKO.1 CCGGCAACAAGATGAAGA GCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT | Open biosystems | RHS6848 |
| shRNA HIF1α: pSIREN GTCTAGAGATGCAGCAAGA | (LaGory et al., 2015) | N/A |
| shRNA HIF2α: TRC1/1.5-pLKO.1TATGTCCTGTTAGCTCCACCT | Open biosystems | N/A |
| Software and Algorithms | ||
| DESeq2 | (Love et al., 2014) | RRID: SCR_015687 |
| GSEA | RRID:SCR_003199 | |
| MutSig2CV v3.1 | (Lawrence et al., 2013). | Firebrowse.org |
| NetMHCpan (v3) | (Nielsen and Andreatta, 2016). | http://www.cbs.dtu.dk/services/NetMHCpan/ |
| Hotnet2 | (Leiserson et al., 2015). | http://compbio.cs.brown.edu/projects/hotnet2/ |
| VALORATE | (Treviño and Tamez-Pena,2017). | N/A |
| MCP-counter | (Becht et al., 2016) | http://cit.ligue-cancer.net/language/en/mcp-counter/ |
| SSGSEA | (Hänzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| GraphPad Prism version 7.0c | GraphPad Software | N/A |
Highlights.
Complement mutations occur at a significantly higher rate than background mutations
Complement component mutations are associated with poor overall survival
Tumors with complement component mutations harbor increased hypoxic signaling
Hypoxic colorectal cancer cells are resistant to complement-mediated cytotoxicity
ACKNOWLEDGMENTS
This work was supported by NIH grants CA67166, CA197713, and CA198291, the Silicon Valley Foundation, the Sydney Frank Foundation, and the Kimmelman Fund (A.J.G.). M.M.O. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. R.K.K. was supported by the Stanford ChEM-H Undergraduate Scholars Program. T.G.G. is supported by the NCI/NIH (P01 CA168585) and an American Cancer Society Research Scholar Award (RSG-12-257-01-TBE). We would like to thank Stavros Melemenidis for his contribution to the graphical abstract.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ajona D, Hsu Y-F, Corrales L, Montuenga LM, and Pio R (2007). Downregulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J. Immunol 178, 5991–5998. [DOI] [PubMed] [Google Scholar]
- Babur Ö, Gönen M, Aksoy BA, Schultz N, Ciriello G, Sander C, and Demir E (2015). Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol 16, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Saute` s-Fridman C, Fridman WH, and de Reynie` s A (2016). Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 17, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffa FM, Harris AL, West CM, and Miller CJ (2010). Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 102, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussink J, Kaanders JHAM, and van der Kogel AJ (2003). Tumor hypoxia at the micro-regional level: clinical relevance and predictive value of exogenous and endogenous hypoxic cell markers. Radiother. Oncol 67, 3–15. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, et al. (2017). Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi J-T, Wang Z, Nuyten DSA, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Børresen-Dale A-L, et al. (2006). Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med 3, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collard CD, Bukusoglu C, Agah A, Colgan SP, Reenstra WR, Morgan BP, and Stahl GL (1999). Hypoxia-induced expression of complement receptor type 1 (CR1, CD35) in human vascular endothelial cells. Am. J. Physiol 276, C450–C458. [DOI] [PubMed] [Google Scholar]
- Corbett TH, Griswold DP Jr., Roberts BJ, Peckham JC, and Schabel FM Jr. (1975). Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 35, 2434–2439. [PubMed] [Google Scholar]
- Cortes-Ciriano I, Lee S, Park WY, Kim TM, and Park PJ (2017). A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun 8, 15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derer S, Beurskens FJ, Rosner T, Peipp M, and Valerius T (2014). Complement in antibody-based tumor therapy. Crit. Rev. Immunol 34, 199–214. [DOI] [PubMed] [Google Scholar]
- Ding L, Wendl MC, Koboldt DC, and Mardis ER (2010). Analysis of nextgeneration genomic data in cancer: accomplishments and challenges. Hum. Mol. Genet 19 (R2), R188–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace A, Mani N, Span PN, Irlam JJ, Taylor J, Betts GNJ, Denley H, Miller CJ, Homer JJ, Rojas AM, et al. (2013). A 26-gene hypoxia signature predicts benefit from hypoxia-modifying therapy in laryngeal cancer but not bladder cancer. Clin. Cancer Res 19, 4879–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishelson Z, Horstmann RD, and Mu€ller-Eberhard HJ (1987). Regulation of the alternative pathway of complement by pH. J. Immunol 138, 3392–3395. [PubMed] [Google Scholar]
- Frede S, Stockmann C, Freitag P, and Fandrey J (2006). Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem. J 396, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG, and Beauchamp RD (2012). Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 142, 562–571.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch WB, Chrysanthou E, Schwaeble WJ, and Stahl GL (2012). The complement system in ischemia-reperfusion injuries. Immunobiology 217, 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter A, and Meri S (1999). Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol. Today 20, 576–582. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, and Giaccia AJ (1996). Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379, 88–91. [DOI] [PubMed] [Google Scholar]
- Hammond EM, Asselin MC, Forster D, O’Connor JPB, Senra JM, and Williams KJ (2014). The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin. Oncol. (R. Coll. Radiol.) 26, 277–288. [DOI] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BHL, Barberis A, West CML, and Buffa FM (2015). Gene expression signatures as biomarkers of tumour hypoxia. Clin. Oncol. (R. Coll. Radiol.) 27, 547–560. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018). Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofree M, Shen JP, Carter H, Gross A, and Ideker T (2013). Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Isaacs JS, Lee S, Trepel J, Liu Z-G, and Neckers L (2003). Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem. J 370, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. (2016). Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, Scott DA, Richardson AD, and Giaccia AJ (2015). Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep 12, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Siva-chenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. (2015). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet 47, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TPF, Keith B, Nissim I, and Simon MC (2014). Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Kirschenbaum A, Lu M, Yao S, Dosoretz A, Holland JF, and Levine AC (2002). Prostaglandin E2 induces hypoxia-inducible factor-1alpha stabilization and nuclear localization in a human prostate cancer cell line. J. Biol. Chem 277, 50081–50086. [DOI] [PubMed] [Google Scholar]
- Louis NA, Hamilton KE, Kong T, and Colgan SP (2005). HIF-dependent induction of apical CD55 coordinates epithelial clearance of neutrophils. FASEB J 19, 950–959. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacik P, Roversi P, White J, Esser D, Smith GP, Billington J, Williams PA, Rudd PM, Wormald MR, Harvey DJ, et al. (2004). Complement regulation at the molecular level: the structure of decay-accelerating factor. Proc. Natl. Acad. Sci. USA 101, 1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JC, Lidington EA, Yarwood H, Lublin DM, and Haskard DO (2001). Induction of endothelial cell decay-accelerating factor by vascular endothelial growth factor: a mechanism for cytoprotection against complement-mediated injury during inflammatory angiogenesis. Arthritis Rheum 44, 138–150. [DOI] [PubMed] [Google Scholar]
- Nielsen M, and Andreatta M (2016). NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okroj M, Corrales L, Stokowska A, Pio R, and Blom AM (2009). Hypoxia increases susceptibility of non-small cell lung cancer cells to complement attack. Cancer Immunol. Immunother 58, 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcina MM, Foskolou IP, Anbalagan S, Senra JM, Pires IM, Jiang Y, Ryan AJ, and Hammond EM (2013). Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol. Cell 52, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcina MM, Kim RK, Melemenidis S, Graves EE, and Giaccia AJ (2018). The tumour microenvironment links complement system dysregulation and hypoxic signalling. Br. J. Radiol Published online May 15, 2018 10.1259/bjr.20180069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen A, Comrie WA, Ardy RC, Domı´nguez Conde C, Dalgic B, Beser ÖF, Morawski AR, Karakoc-Aydiner E, Tutar E, Baris S, et al. (2017). CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. N. Engl. J. Med 377, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pio R, Ajona D, and Lambris JD (2013). Complement inhibition in cancer therapy. Semin. Immunol 25, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, et al. (2009). The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19, 1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, and Ratcliffe PJ (2003). Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med 9, 677–684. [DOI] [PubMed] [Google Scholar]
- Reis ES, Mastellos DC, Ricklin D, Mantovani A, and Lambris JD (2018). Complement in cancer: untangling an intricate relationship. Nat. Rev. Immunol 18, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, and Lambris JD (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol 11, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CQ, Lambris JD, and Ricklin D (2016). Protection of host cells by complement regulators. Immunol. Rev 274, 152–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. [DOI] [PubMed] [Google Scholar]
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. (2012). Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, and Ostrowski J (2010). Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One 5, e13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, Lu P, Johnson JC, Schmidt C, Bailey CE, et al. (2010). Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 138, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller OB, Criado-Garcı´a O, Rodrı´guez De Co´ rdoba S, and Morgan BP (2000). Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin. Exp. Immunol 121, 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BP, Vĕtvicka V, and Ross GD (1994). Natural antibody and complement-mediated antigen processing and presentation by B lymphocytes. J. Immunol 152, 1727–1737. [PubMed] [Google Scholar]
- Treviño V, and Tamez-Pena J (2017). VALORATE: fast and accurate log-rank test in balanced and unbalanced comparisons of survival curves and cancer genomics. Bioinformatics 33, 1900–1901. [DOI] [PubMed] [Google Scholar]
- Vandin F, Papoutsaki A, Raphael BJ, and Upfal E (2015). Accurate computation of survival statistics in genome-wide studies. PLoS Comput. Biol 11, e1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, and Restifo NP (1995). Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J. Immunol 154, 4685–4692. [PMC free article] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, et al. (2007). Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67, 3441–3449. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chu J, Yu J, and Wei W (2016). Cellular and molecular mechanisms in graft-versus-host disease. J. Leukoc. Biol 99, 279–287. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang Z, Cao L, Lin J, Yang Z, and Zhang X (2017). A common CD55 rs2564978 variant is associated with the susceptibility of non-small cell lung cancer. Oncotarget 8, 6216–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.