

Abstract
Background
CD40 ligand (CD40L) is a thromboinflammatory molecule that predicts cardiovascular events. CD40L is a strong activator of nuclear factor kappa B (NF‐κB) in platelets that primes and enhances platelet activation in response to thrombotic stimuli. In addition to its classical receptor CD40, CD40L binds αIIbβ3, α5β1, and αMβ2 in various cell types. However, the function of the different CD40L receptors on platelets remains unexplored. The present study aims to identify the receptors of CD40L, involved in platelet NF‐κB activation, their downstream signaling and their implication in platelet aggregation.
Methods and Results
We showed that platelets express CD40, αIIbβ3, and α5β1 and release CD40L in response to sCD40L stimulation. sCD40L alone dose‐dependently induced platelet NF‐κB activation; this effect was absent in CD40−/− mouse platelets and inhibited by the CD40 blockade, but was unaffected by the αIIbβ3 or α5β1 blockade in human platelets. sCD40L/CD40 axis activates transforming growth factor‐β‐activated kinase 1 upstream of NF‐κB. In functional studies, sCD40L alone did not affect platelet aggregation but potentiated the aggregation response in the presence of suboptimal doses of thrombin; this effect was abolished by CD40, transforming growth factor‐β‐activated kinase 1, and NF‐κB inhibitors.
Conclusions
CD40L primes platelets via signaling pathways involving CD40/transforming growth factor‐β‐activated kinase 1/NF‐κB, which predisposes platelets to enhanced activation and aggregation in response to thrombotic stimuli.
Keywords: CD40/CD40L, NF/kB, platelet aggregation, TAK1, thrombosis
Subject Categories: Thrombosis, Coronary Artery Disease
Clinical Perspective
What Is New?
In platelets, activation of nuclear factor kappa B by sCD40L is CD40‐dependent.
sCD40L/CD40 axis activates TAK1 upstream of nuclear factor kappa B in platelets.
CD40L/CD40/TAK1/nuclear factor kappa B signaling primes platelets and potentiates aggregation in response to thrombotic stimuli.
What Are the Clinical Implications?
Targeting CD40L/CD40/TAK1/nuclear factor kappa B limits platelet priming and activation and may ultimately represent a therapeutic target in the treatment of thromboinflammatory diseases.
Introduction
Patients with inflammatory diseases exhibit higher than expected rates of ischemic cardiovascular events that are not attributable to common risk factors, but rather to inflammatory stimuli that trigger pathways contributing to the pathogenesis of cardiovascular diseases1, 2 In this regard, the thromboinflammatory mediator CD40L and its classical receptor CD40, molecules of critical importance in humoral immunity, have gained significant attention for their involvement in the pathophysiology of cardiovascular diseases and thrombotic events.3, 4, 5 Indeed, CD40L/CD40 interactions in different cell types regulate a plethora of inflammatory cascades (cytokine release, upregulation of adhesion molecules, and activation of leukocytes and platelets) at the forefront of cardiovascular alterations. Thus, CD40L/CD40 axis may represent a pivotal contributor to the establishment of accelerated vascular alterations in patients with high circulating levels of CD40L. Like most members of the tumor necrosis factor (TNF) family, CD40L can be detected in soluble form (sCD40L) in the blood circulation, whose principal source is activated platelets.6, 7, 8 Circulating levels of sCD40L in patients have now emerged as reliable indicators of cardiovascular risk, as there appears to be a significant correlation between levels of sCD40L and vascular complications such as atherosclerosis and acute coronary syndromes.9, 10 sCD40L activates platelets, as revealed by α‐ and dense‐granule release, and morphological changes typically associated with activation of αIIbβ3.11 In this regard, we have shown that enhanced levels of sCD40L exacerbate platelet activation and aggregation through the CD40/TRAF‐2/Rac‐1/p38 mitogen‐activated protein kinases (MAPK) signaling pathways.12 More recently, we have also shown that sCD40L triggers nuclear factor kappa B (NF‐κB) activation in platelets.13 Activation of NF‐κB by sCD40L is independent of p38 MAPK, which suggests that CD40L activates NF‐κB through a different pathway.
In nucleated cells, CD40L‐induced NF‐κB activation involves the phosphorylation of NF‐κB inhibitor κB protein (IκB), thus causing its proteasome‐mediated degradation. In response to stimuli, the NF‐κB dimers, in association with the inhibitory IκB subunit, are regulated by the IκB kinase (IKK). Upon phosphorylation by IKK, the IκB subunit is targeted for proteasome degradation, thereby releasing an active form of NF‐κB that translocates into the nucleus to transcript targeted genes.14 In anucleated platelets, phosphorylation of IκBα, which is indicative of NF‐κB activation, is also observed following platelet activation,15 and NF‐κB inhibitors lead to impairment of platelet function.16 Indeed, inhibition of platelet NF‐κB activation in response to sCD40L stimulation abolished the potentiation of platelet aggregation in response to suboptimal doses of agonists.13 This led us to hypothesize that CD40L/NF‐κB signaling in platelets plays a non‐genomic role as a primer that predisposes platelets to enhanced activation and aggregation and may represent an important target against atherothrombosis.
Nevertheless, the discovery of new CD40L receptors (αIIbβ3, α5β1, and αMβ2),17, 18, 19 in addition to its classical receptor CD40, adds complexity to the diverse interplays in which CD40L takes part in platelet function.5 Several studies showed that CD40L‐induced platelet activation passes through CD40,12, 20, 21, 22 while others examined the role of CD40L/αIIbβ3 interaction in platelet physiology;17, 23 one study reported a role for CD40L/α5β1 interactions in platelet activity.24 Despite these studies, the contribution of each of these different receptors in platelet activation and aggregation in response to CD40L, and in particular in the activation of the NF‐κB cascade, remains unexplored.
In search of the effector that leads to sCD40L‐induced NF‐κB activation, the transforming growth factor‐β‐activated kinase 1 (TAK1)25 appeared as a principal candidate. In fact, TAK1 has been shown to function upstream of IKKβ in response to various NF‐κB‐inducing stimuli.26, 27 Indeed, TAK1 is a major regulator of the inflammatory and immunity signaling pathways that can be activated through a variety of proinflammatory receptors, such as CD4028 Once activated, TAK1 leads to downstream activation of several pathways, including NF‐κB.26, 29 Although TAK1 is present in platelets30 its function in the CD40L/NF‐κB signaling remains unknown.
Thus, the present study was conducted to identify the CD40L receptors involved in platelet NF‐κB activation, their downstream signaling, and their impact on platelet aggregation. We found that sCD40L triggers activation of NF‐κB and primes platelets through the CD40 receptor and activation of TAK1, which suggests that CD40L primes platelets via signaling pathways involving CD40/TAK1/NF‐κB activation that predisposes platelets to enhanced activation and aggregation in response to thrombotic stimuli.
Methods
The data, analytic methods, and study materials will be made available, upon request from the corresponding author, to other researchers for the purpose of reproducing the results or replicating the procedure.
Preparation of Human Platelets
Venous blood was drawn from 30 healthy volunteers who had not taken medications known to interfere with platelet function for at least 3 weeks before the experiment. The protocol was approved by the Human Ethical Committee of the Montreal Heart Institute in accordance with the Declaration of Helsinki for experiments involving humans. Informed consent was obtained from all participants. Washed platelets were prepared as previously described12, 13 Briefly, platelet‐rich plasma was obtained by centrifugation of acid‐citrate‐dextrose, anticoagulated blood. Platelets were then pelleted from platelet‐rich plasma, to which 1 μg/mL of prostaglandin E1 (PGE1) was added, washed with HBSS‐Hank's sodium citrate buffer and finally resuspended in HBSS‐Hank's buffer, which contained 2 mmol/L MgCl2 and 2 mmol/L CaCl2. Platelets were adjusted to 250×106/mL and allowed to rest at 37°C for at least 30 minutes before further manipulation.
Preparation of Murine Platelets
The handling and care of mice complied with the guidelines established by the Animal Care and Ethical Committee of the centre hospitalier de l'Université de Montréal (CHUM) research center in agreement with the Canadian Council on Animal Care guidelines. Age‐ and sex‐matched 18 wild‐type and 18 CD40−/− mice, both on C57BLK/J6 background, were purchased from the Jackson Laboratory and housed under pathogen‐free conditions. Briefly12 mice (3–4 months; 20–30 g) were anesthetized using an intraperitoneal injection of a mixture of 75 mg/kg ketamine (vetalar) and 0.5 mg/kg medetomidine (Domitor, Pfizer). Blood was collected from the ventricular puncture into syringes containing one twentieth of the blood volume of heparin as an anticoagulant. For each experiment, we combined the blood collected from 3 mice (750–1000 μL/mouse), which was diluted (1:1) using a modified Tyrode buffer containing 0.1 μg/mL PGE1 and centrifuged. Washed platelets were then prepared from platelet‐rich plasma and resuspended in a modified Tyrode buffer to yield a final concentration of 250×106/mL.
Determination of Platelet CD40L Receptors
The known CD40L receptors in various cell types are CD40, αIIbβ3, α5β1, and αMβ2. Their expressions on human platelets were first measured by flow cytometry. Isolated platelets were fixed with 1% paraformaldehyde and were then washed and stained with saturating concentrations of primary antibody against CD40, α5, β1, αIIb, β3, αM, and β2 integrins (R&D systems) for 30 minutes at 4°C or their isotype‐matched control immunoglobulin Gs. Platelets were washed 3 times with PBS/0.2% Fetal Bovine Serum (FBS), and Alexa 488 coupled secondary antibody was then added and incubated for 30 minutes at 4°C. Samples were analyzed (20 000 events) on a FACSCalibur flow cytometer (Becton Dickinson), and platelets were gated by their characteristic forward and side scatter properties, as described previously12
The presence of the different CD40L receptors in platelets was also measured by Western blot (WB) of platelet lysates. Proteins were resolved in 10% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% non‐fat dry milk for 1 hour, washed 3) times with Tris‐buffered saline (TBS) (150 mmol/L NaCl, 20 mmol/L Tris, pH 7.4, 0.1% Tween‐20) and incubated with appropriate primary antibody overnight at 4°C. Following the washing steps, membranes were labeled with horseradish peroxidase‐conjugated secondary antibodies for 1 hour; washing and bound peroxidase activity was detected using enhanced chemiluminescence (PerkinElmer Life Sciences).
Measurement of Platelet CD40L
The presence of CD40L in platelet lysates was determined by WB, and its expression on the platelet surface was analyzed by flow cytometry. The concentration of sCD40L secreted by platelets in a resting state and after stimulation with sCD40L was determined by using an ELISA kit (R&D system) that recognizes the sCD40L backbone.
Platelet NF‐κB and TAK1 Activation Assay
Following stimuli that solicit the NF‐κB pathway, the IκB inhibitory subunit of the NF‐κB complex is phosphorylated and degraded, thus allowing the p65 subunit to be phosphorylated and activated. As described previously13 we used primary antibodies against phospho‐IκBα serine32/36, phospho‐TAK1 threonine184/187, phospho‐NF‐κB p65 serine536, TAK1, NF‐κB p65, and β‐actin (Cell Signaling Technology). Isolated platelets were suspended in Hanks' balanced Salt Solution (HBSS) complete medium at 109 platelets/mL and were rested at 37°C for 30 minutes. Resting platelets (control) and platelets stimulated with sCD40L for 5 minutes at 37°C were prepared. To elucidate the involvement of each CD40L receptor in NF‐κB activation, ReoPro (abciximab, 10 μg/mL, Janssen/Lilly)17, 31 and JBS5 (10 μg/mL, calbiochem)24 were used to block αIIbβ3 and α5β1, respectively. To block the CD40 receptor, a direct CD40 blocker could not be used because the available anti‐CD40 monoclonal antibody has agonistic features32, 33 Therefore, we acquired the Fab‐fragment 5C8 (10 μg/mL), an anti‐CD40L antagonist that specifically blocks the binding site of CD40L that is recognized by CD40, thereby inhibiting CD40/CD40L binding.18, 34 The corresponding immunoglobulin G isoforms for each antibody were used as a control. BAY 11‐7082 (10 μmol/L, selective IκBα inhibitor; Sigma‐Aldrich)13 5Z‐7‐Oxozeaenol (ZOL, 1–1000 nmol/L, a selective TAK1 inhibitor; Sigma‐Aldrich)35, 36 and Takinib (0.01–100 μmol/L, a selective TAK1 inhibitor, MedChemExpress)37 were used to study TAK1 activation by sCD40L. Platelets were then lysed by adding hot SDS‐blue containing β‐mercaptoethanol and protease inhibitors and the lysates immunoblotted, as described previously.12, 13
Platelets isolated from wild‐type or CD40−/− mice were suspended in a modified Tyrode buffer at 109 platelets/mL and rested at 37°C for 30 minutes. They were then stimulated with mouse sCD40L (R&D System) to evaluate NF‐κB complex activation. Lysates from mouse platelets were resolved in 10% SDS–PAGE and assessed for phospho (p) p‐IκBα and p‐P65. β‐actin blots were generated from stripped membranes of p‐IκB and p‐P65 blots.
Measurement of Platelet Aggregation
We monitored aggregation of washed human platelets on a 4‐channel optical aggregometer (Chronolog Corp) under shear (1000 rpm) at 37°C. Platelet suspensions were preincubated with increasing concentrations (10–1000 ng/mL) of sCD40L (R&D systems) at 37°C, for 5, 15, or 30 minutes. Platelet suspensions were also pretreated with several inhibitors: 5C8 (10 μg/mL), JBS5 (10 μg/mL), BAY 11‐7082 (10 μmol/L), ZOL (10, 50, and 100 nmol/L), or Takinib (1, 5, and 10 μmol/L), for 10 minutes followed by a treatment of platelets with sCD40L (1000 ng/mL) for 30 minutes. Platelet aggregation was then triggered by a suboptimal dose of α‐thrombin (Sigma‐Aldrich) approximating 0.025±0.01 U/mL that induces no >30% aggregation, as observed in the dose‐response curve of platelet aggregation in response to thrombin ranging from 0.0125 to 0.1 U/mL (Figure S1). Traces were recorded until stabilization of platelet aggregation was reached12, 13
Statistical Analysis
Statistical analysis was performed using IBM SPSS statistics 25. Results are presented as mean±SEM. Statistical comparisons were done using a paired t test or a 1‐way ANOVA followed by a Dunnett's test for comparison against a single group. In human platelets, each experiment (n) represents data obtained from 1 donor blood; whereas in mouse, each experiment (n) represents data obtained from platelets isolated from combined 3 mouse blood. The specific statistical tests used, the mean of data, the number of experiments, and the P values are specified in the figure legends. A P<0.05 was considered statistically significant.
Results
Detection of the CD40L System in Platelets
First, we aimed to identify the different platelet CD40L receptors. This was done by analyzing, using flow cytometry and WB, the different subunits of the known receptors for CD40L (CD40, αIIbβ3, α5β1, and αMβ2) on various cell types. As shown in Figure 1A, we detected by flow cytometry the presence of CD40, αIIbβ3, and α5β1 on platelets; however, αMβ2, whose expression is restricted to leukocytes, was absent. This was confirmed by immunoblotting of the different subunits in platelet lysates (Figure 1B).
Figure 1.
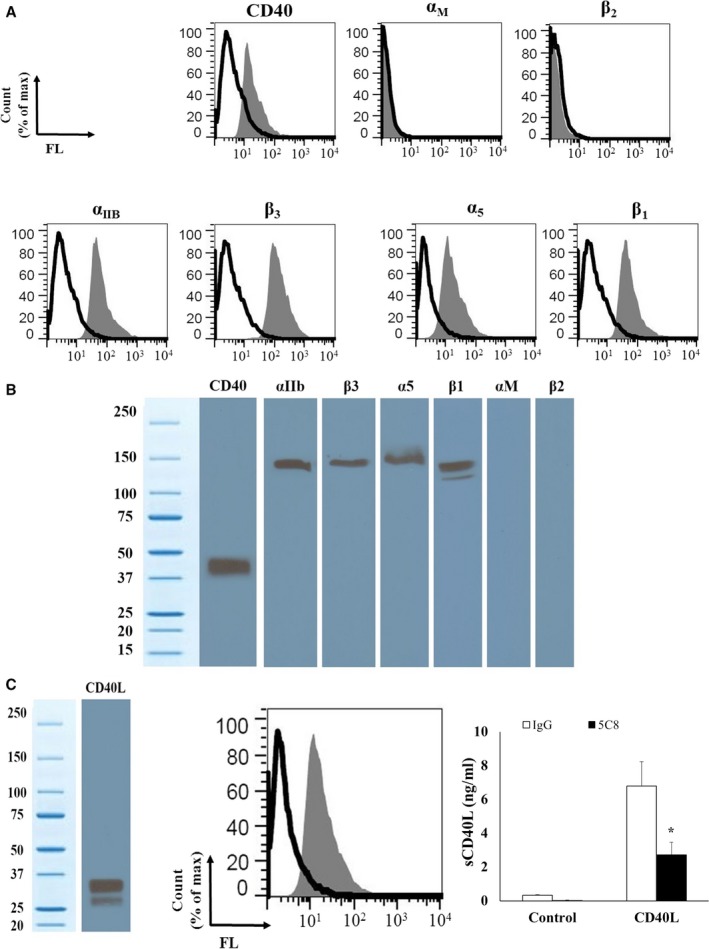
Human platelets express 3 receptors for CD40L: CD40, αIIbβ3, and α5β1. A, CD40L receptor expression was evaluated by flow cytometry. Isolated platelets were stained with primary antibodies against CD40, α5, β1, αIIb, β3, αM, and β2 integrins (grey curves), for 30 minutes at 4°C or their isotype‐matched control immunoglobulin Gs (black curves). The graphs of the number of platelets were normalized to the mode to depict the data regarding “% of max.” The % of max denotes the number of cells in each bin (the numerical ranges for the parameter on the x‐axis) divided by the number of cells in the bin that contains the largest number of cells). B, The presence of CD40L receptors in human platelets was also determined by WB. Platelet lysates were resolved in 10% SDS–PAGE and assessed for CD40, α5, β1, αIIb, β3, αM, and β2 integrins. C, Human platelets express and release sCD40L. Left: Platelet lysates were resolved in 10% SDS–PAGE and assessed for CD40L by WB. Center: Isolated platelets were analyzed by flow cytometry using primary antibody against CD40L (grey curve) and its corresponding isotype‐matched control immunoglobulin G (black curve). Right: sCD40L secretion profile of platelets after stimulation with exogenous sCD40L (1 ng/mL), as analyzed by ELISA. Also showing, the effect of CD40L/CD40 blockade using 5C8 on sCD40L release in CD40L‐stimulated platelets (n=3, mean±SEM). *P<0.01 (Paired t test; 5C8 vs its immunoglobulin G control). FL indicates Forward light scatter.
We also confirmed the presence of the CD40L protein in platelet lysates through flow cytometry and WB (Figure 1C). Moreover, we demonstrated its release by treating platelets with sCD40L. The platelet release of sCD40L was abolished by the blockade of CD40L/CD40 axis (Figure 1C) using 5C8, a monoclonal antibody that binds with high affinity to CD40L and neutralizes its function by specifically blocking its binding site with CD4034
sCD40L Activates Platelet NF‐κB Via CD40
We have previously shown that NF‐κB is required for sCD40L‐induced platelet activation and potentiation of platelet aggregation independently of p38‐MAPK activation.13 Having shown that platelets express different receptors for CD40L, we aimed to determine the CD40L receptors involved in NF‐κB activation. To this end, we first confirmed the activation of NF‐κB by sCD40L by assessing IκBα phosphorylation, which leads to the degradation and release of the active form of NF‐κB, and p65 phosphorylation, a subunit of NF‐κB. Our results showed that sCD40L induces a dose‐dependent phosphorylation of IκBα (Figure 2A) and p65 (Figure 2B). To confirm the specific action of sCD40L on platelet NF‐κB activation, its action was abolished by a blocking monoclonal CD40L antibody (TNFSF5)38 (Figure 2). Moreover, sCD40L alone primes platelets via either NF‐κB or p38 MAPK, independently of the presence of agonists such as thrombin or collagen, which were without any effects on NF‐κB or p38 MAPK activation (Figure S2).
Figure 2.
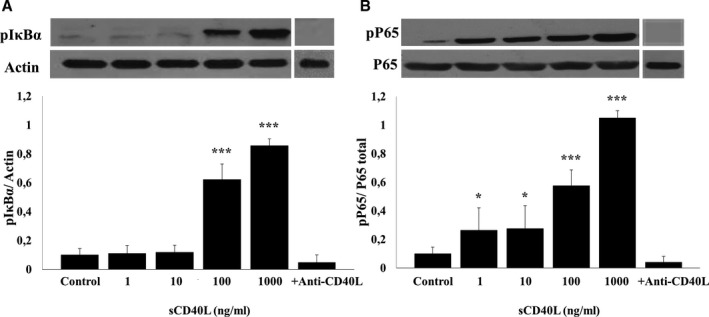
sCD40L phosphorylates IκBα and P65 in human platelets. Washed human platelets (1000×106/mL) were treated with different concentrations of sCD40L (1, 10, 100, and 1000 ng/mL) for 5 minutes at 37°C. Platelets were also pretreated with anti‐CD40L and stimulated with sCD40L (1000 ng/mL). Platelet lysates were resolved in 10% SDS–PAGE and assessed for (A) pIκBα and (B) pP65. Actin blot is from stripped membranes of pIκBα blot; and P65 blot is from stripped membranes of pP65 blot. Blots are representative of 4 independent experiments. Histograms represent the mean of data, expressed in optical density (n=4, mean±SEM). *P<0.05; † P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs control).
To determine the CD40L receptors involved in platelet NF‐κB activation, we used specific blocking monoclonal antibodies to antagonize each receptor: ReoPro for αIIbβ3,17 JBS5 for α5β124 and 5C8 for CD40L/CD40 binding34 As shown in Figure 3, blockade of αIIbβ3 (Figure 3A) and α5β1 (Figure 3B) receptors on platelets with their corresponding antagonists has no influence on IκB and p65 phosphorylation in response to sCD40L. In contrast, phosphorylation of both IκB and p65 (Figure 3C) were abolished by 5C8, which binds with high affinity to CD40L and neutralizes its function by specifically blocking its binding site with CD40.34 We confirmed the unique contribution of CD40 to platelet NF‐κB activation in response to sCD40L using a genomic approach in CD40‐deficient mouse platelets. Indeed, we demonstrated an increase in IκBα (Figure 4A) and p65 phosphorylation (Figure 4B) in msCD40L‐treated, wild‐type mouse platelets; this effect was absent in CD40−/− mouse platelets. These results clearly showed that the CD40L receptor responsible for NF‐κB activation in platelets is CD40.
Figure 3.
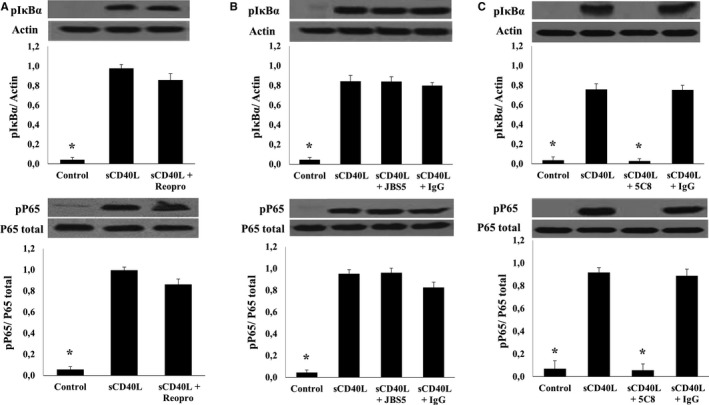
Platelet NF‐κB activation by sCD40L is CD40‐dependent. Washed human platelets (1000×106/mL) were treated with (A) 10 μg/mL of ReoPro, (B) 10 μg/mL of JBS5, or (C) 10 μg/mL of 5C8 and corresponding immunoglobulin Gs for 5 minutes at 37°C. Pretreated platelets were then stimulated with 1000 ng/mL of sCD40L for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for pIκBα and pP65. Actin blot is from stripped membranes of pIκBα blot; and P65 blot is from stripped membranes of pP65 blot. Blots are representative of 4 independent experiments. Histograms represent the mean of data, expressed in optical density (n=4, mean±SEM). IgG indicates immunoglobulin G. *P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs the sCD40L group).
Figure 4.
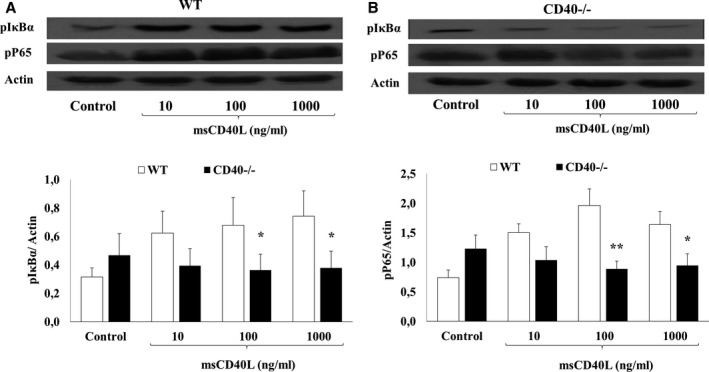
NF‐κB activation in response to msCD40L is absent in CD40−/− mouse platelets. Washed platelets (1000×106/mL) from wild‐type or CD40−/− mice were stimulated with different concentrations of msCD40L for 5 minutes at 37°C. Platelet lysates were resolved in 10% SDS–PAGE and assessed for (A) pIκBα and (B) pP65. Actin blot is from stripped membranes of either pIκBα blot or pP65 blot. Blots are representative of 6 independent experiments. WT indicates wild type. Histograms represent the mean of data, expressed in optical density (n=6, mean±SEM). *P<0.05; † P<0.01 (Paired t test; CD40−/− vs wild‐type).
sCD40L Primes Platelets and Potentiates Platelet Aggregation Via CD40
To obtain insights into the role of the sCD40L/CD40 axis in platelet function, we examined its effect on platelet aggregation. As shown previously,12 we first sought to confirm that sCD40L did not affect platelet aggregation by itself. Treatment of platelets with sCD40L alone did not affect aggregation independently of its concentration (Figure 5A). However, pretreatment of platelets with sCD40L for 5, 15, or 30 minutes lead to a significant and dose‐dependent increase of aggregation in response to a suboptimal dose of thrombin (Figure 5B). For example, pretreatment of platelets with sCD40L for 30 minutes significantly potentiates the response to a suboptimal dose of thrombin from ≈15% in the absence of sCD40L to 40%, 75%, and 85% with 10, 100, and 1000 ng/mL of sCD40L, respectively. This indicates that sCD40L primes platelets and predisposes them to increased aggregation in response to a suboptimal concentration of platelet agonists. Moreover, sCD40L potentiated platelet aggregation in the presence of a suboptimal dose of thrombin receptor activator peptide 1 (TRAP‐1) replicating in a similar fashion the potentiation observed with a suboptimal dose of thrombin, indicating that the action of thrombin is related to its platelet‐activating property, independently from its proteolytic activity (Figure S3).
Figure 5.
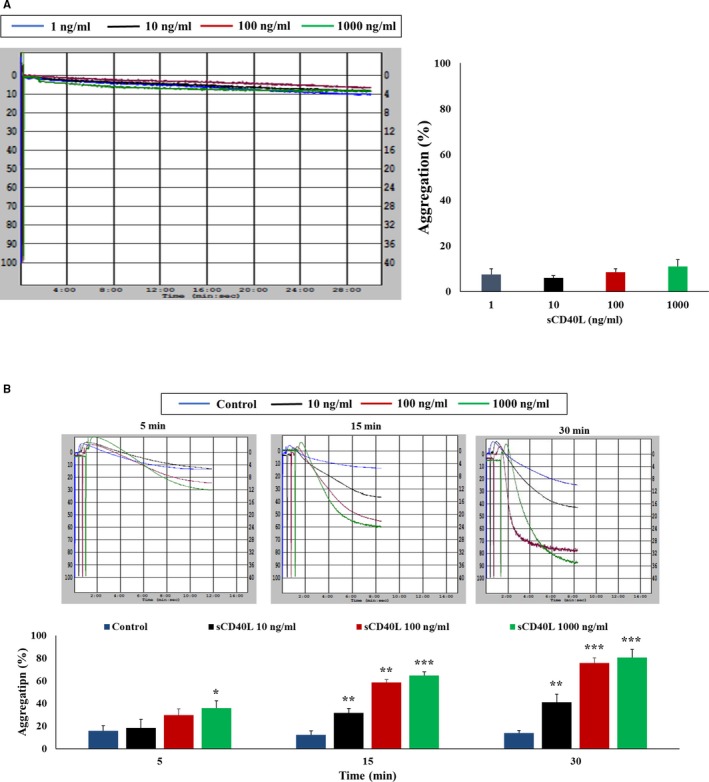
sCD40L potentiates platelet aggregation in response to a suboptimal dose of thrombin. A, Washed human platelets (250×106/mL) were treated with sCD40L (10, 100, and 1000 ng/mL) for 30 minutes. Histograms represent the mean of aggregations (n=4, mean±SEM). B, Platelets were treated with sCD40L (10, 100, and 1000 ng/mL) for 5, 15, and 30 minutes at 37°C. Aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL). Histograms represent the mean of aggregations (n=4, mean±SEM). *P<0.05; † P<0.01; ‡ P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs control).
Next, we sought to determine if such response involves CD40. Effectively, as shown in Figure 6A, blockade of the CD40L/CD40 axis before treatment with sCD40L abolished the potentiation in platelet aggregation in response to a suboptimal dose of thrombin; in contrast, α5β1 blockade did not (Figure 6B). Blockade of αIIbβ3 with ReoPro cannot be tested to observe the effect of sCD40L on platelet potentiation, because it is a platelet aggregation inhibitor that hinders the binding of fibrinogen to αIIbβ3 receptors, which is a necessary step for platelet aggregation. Finally, we confirmed the implication of NF‐κB in platelet priming through the use of BAY 11‐7082, a specific inhibitor of NF‐κB, which hinders the potentiating role of sCD40L in platelet aggregation (Figure 6C).
Figure 6.
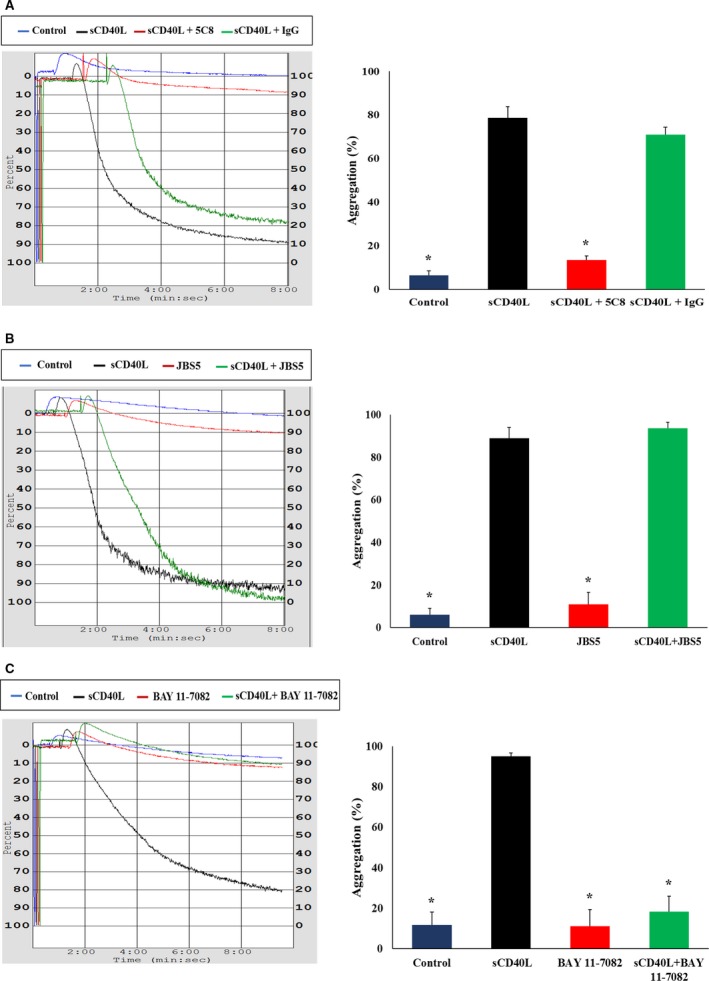
sCD40L primes platelets via CD40 and NF‐κB. Washed human platelets (250×106/mL) were pretreated with (A) 10 μg/mL of Fab fragment of 5C8 and its immunoglobulin G, (B) 10 μg/mL JBS5, or (C) 10 μmol/L BAY 11‐7082 for 5 minutes then stimulated with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL) in the presence or absence of sCD40L. Histograms represent means of aggregation (n=4, mean±SEM). IgG indicates immunoglobulin G. *P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs the sCD40L group).
TAK1 is Required in CD40L/CD40/NF‐κB Signaling and Platelet Priming
After demonstrating the involvement of CD40L/CD40/NF‐κB signaling in platelet priming, we sought to determine if TAK1 is the effector protein involved upstream of NF‐κB. We showed that TAK1 is present in platelets and is phosphorylated upon sCD40L activation (Figure 7). Moreover, we demonstrated that TAK1 is upstream of NF‐κB because its inhibitors, ZOL and Takinib, in a dose‐dependent manner, abolish IκB phosphorylation when stimulated with sCD40L. Indeed, at 100 nmol/L, ZOL inhibits entirely TAK1 and IκB phosphorylation (Figure 8A). Similarly, at 10 μmol/L, Takinib suppresses TAK1 and IκB phosphorylation (Figure 8B). Such results highlight the requirement of TAK1 for sCD40L‐induced NF‐κB activation in platelets. Furthermore, blockade of TAK1 with ZOL (Figure 9A) or Takinib (Figure 9B) dose dependently impairs the potentiating action of sCD40L on platelet aggregation, highlighting for the first time the key involvement of TAK1 in CD40L activation of NF‐κB and priming of platelets. It should be noted that ZOL or Takinib alone have no effects on platelets aggregation (Figure S4).
Figure 7.
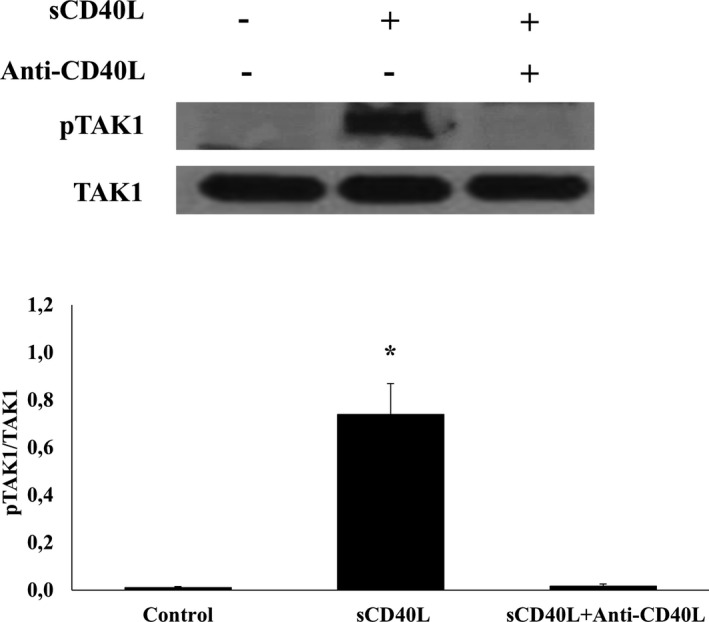
TAK1 is present in platelets and phosphorylated by sCD40L. Washed human platelets (1000×106/mL) were treated with 10 μg/mL of blocking polyclonal anti‐CD40L 37°C, then stimulated with 1000 ng/mL of sCD40L for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for pTAK1. TAK1 blot is from stripped membranes of pTAK1 blot. Blots are representative of 4 independent experiments. Histograms represent the mean of data, expressed in optical density (n=4, mean±SEM). pTAK1 indicates phospho transforming growth factor‐β‐activated kinase 1. *P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs control).
Figure 8.
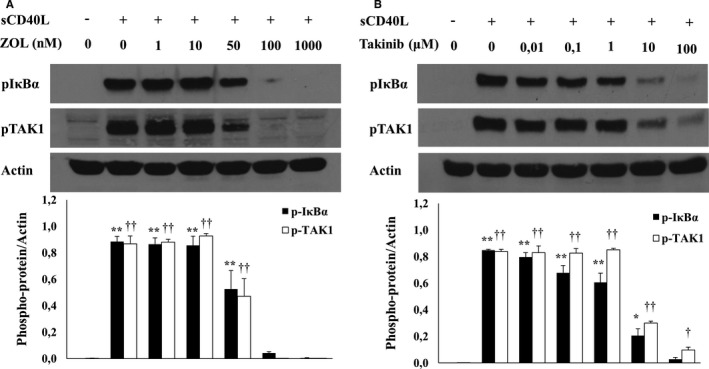
sCD40L activates TAK1/NF‐κB in platelets. Washed human platelets (1000×106/mL) were treated with several concentrations of (A) ZOL or (B) Takinib for 5 minutes at 37°C, and then stimulated with 1000 ng/mL of sCD40L for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for pIκBα and pTAK1. Actin blot is from stripped membranes of pIκBα and pTAK1. Blots are representative of 4 independent experiments. Histograms represent the mean of data, expressed in optical density (n=4, mean±SEM). For pIκBα: **P<0.001, *P<0.01, vs unstimulated platelets. For pTAK1: †† P<0.001, † P<0.01, vs unstimulated platelets (one‐way ANOVA followed by Dunnett's multiple comparisons test vs the unstimulated groups). Zol indicates 5Z‐7‐Oxozeaenol.
Figure 9.
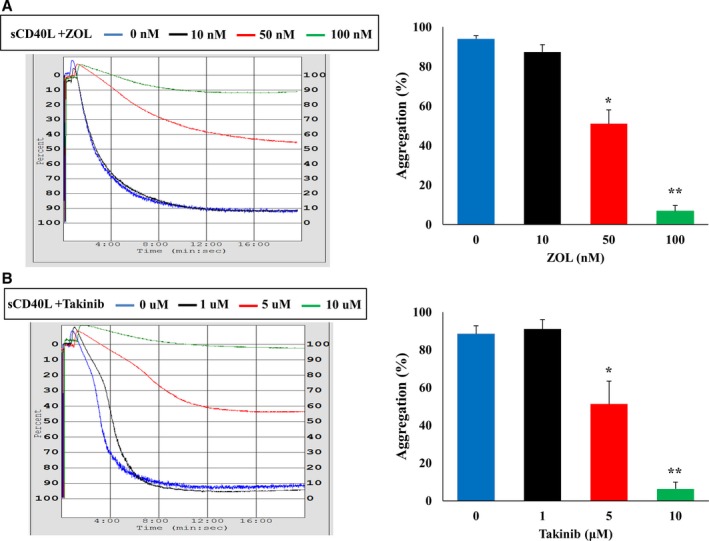
TAK1 inhibition reduces the potentiation action of sCD40L on platelet aggregation. Washed human platelets (250×106/mL) were pretreated with 3 doses of (A) ZOL (10, 50, and 100 nmol/L) or (B) Takinib (1, 5, and 10 μmol/L), then stimulated with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL). Histograms represent means of aggregation (n=4, mean±SEM). *P<0.01, † P<0001 (one‐way ANOVA followed by Dunnett's multiple comparisons test vs the control groups without ZOL (5Z‐7‐Oxozeaenol) or Takinib).
Discussion
Platelets are vital players in immune and inflammatory reactions.39, 40 In particular, platelet CD40L, expressed on the surface of activated platelets and cleaved to generate most of the sCD40L within the blood circulation, induces inflammatory and thrombotic responses in the vascular system.4, 5, 20, 41 However, the mechanisms involved in its modulation of platelet function remain unclear. Our previous studies have shown that sCD40L alone, independently of the presence of agonists such as thrombin or collagen, primes platelets via different signaling mechanisms, which includes a CD40‐dependent TRAF‐2/Rac1/p38 MAPK signaling pathway12. In fact, the effect of sCD40L is CD40 dependent, since a mutated form of sCD40L that does not bind CD40 (sCD40LR/Y), and CD40−/− mouse platelets failed to elicit such responses. More recently, our group also showed that sCD40L alone triggers a strong NF‐κB activation in platelets13 Inhibition of IκBα reverses sCD40L‐induced IκBα phosphorylation without affecting p38 MAPK phosphorylation. On the other hand, inhibition of p38 MAPK has no effect on IκBα phosphorylation, indicating a divergence in the signaling pathway originating from CD40L. However, it remains unclear if sCD40L triggers NF‐κB activation via CD40 alone or with its other receptors. In addition, the downstream signaling and their impact on platelet aggregation remain unexplored.
In nucleated cells, CD40L/CD40 axis induces activation of the NF‐κB signaling pathway to trigger the transcription of key genes involved in inflammation that may lead to increased cardiovascular events.42 Although the presence of NF‐κB in nucleated cells has been well documented since its discovery in immune cells >30 years ago43 it was not until 2002 that Liu et al demonstrated the expression NF‐κB in platelets and how thrombin‐induced platelet activation triggers the degradation of IκBα15 Treatment with pharmacological inhibitors of NF‐κB impairs platelet function, thus highlighting the non‐genomic roles of NF‐κB in platelet biology16, 44 Our group also showed that sCD40L could activate NF‐κB and thus predisposes platelets to enhanced aggregation in response to a thrombotic stimulus.13 However, CD40L has multiple receptors on platelets that might be involved in platelet function23, 24
We first confirmed that the priming action of sCD40L alone, via either p38MAPK or NF‐κB, is insufficient to trigger platelet aggregation but potentiates platelet aggregation in response to suboptimal concentrations of platelets agonists, like thrombin or collagen, which are unable themselves to activate p38MAPK or NF‐κB. Furthermore, we showed the ability of sCD40L, independently of the aggregation process, to act as a potent activator of platelet NF‐κB through the phosphorylation of IκBα and p65. Such activation could be involved in platelet secretion, as we demonstrated that the main activator of NF‐κB involved in platelet secretion, IKKβ, is phosphorylated upon sCD40L stimulation and its inhibition reduces the translocation of P‐selectin from α granules to the platelet surface.13 IKKβ implication on platelet secretion is related to the phosphorylation of synaptosomal‐associated protein‐23 (SNAP‐23), which acts as a regulator of granule secretion.45 Wei et al46 who show that platelets with IKKβ deficiency exhibit decreased secretion and activation, confirmed this observation.
Since its discovery, CD40L was thought to possess only 1 receptor, CD4047 However, several studies have indicated the existence of additional receptors, such as αIIbβ3, α5β1 and αMβ2 or Mac‐117, 18, 23, 48 Of the 4 known CD40L receptors, CD40, α5β1 and αMβ2 are expressed on a wide variety of cell types, whereas αIIbβ3 is exclusively expressed on platelets and megakaryocytes. We first confirmed the presence, expression and release of sCD40L by activated platelets and demonstrated the presence and expression of its receptors CD40, αIIbβ3 and α5β1; however, we did not find the presence of αMβ2, which is expressed on leukocytes.49 To elucidate whether αIIbβ3 or α5β1 triggers platelet NF‐κB activation in response to CD40L, we used two relevant antagonists: ReoPro for αIIbβ317 and JBS5 for α5β124 We observed no changes in the phosphorylation of IκBα and p65 with or without the antagonists following sCD40L stimulation. To test the involvement of CD40, we used 5C8, which is an anti CD40L/CD40 antagonist.34 Interestingly, NF‐κB activation was abolished entirely by 5C8 following CD40L stimulation, which demonstrates that CD40L induces platelet NF‐κB activation exclusively through CD40. To strengthen our observation, we confirmed those findings in a knockout mouse model, and similar to the CD40/CD40L blockade, NF‐κB activation by msCD40L was absent in the CD40−/− mouse platelets. We had previously demonstrated that injection of msCD40L exacerbates thrombosis in wild‐type mice but not in CD40−/− mice, indicating that enhanced levels of CD40L prime platelets in a CD40‐dependent manner and thus predispose them to enhanced thrombus formation12 In the present study, we show that sCD40L triggers its intrinsic platelet secretion via CD40. In fact, minimal concentrations of sCD40L can induce significant intrinsic secretion of sCD40L from platelets compared with un‐stimulated platelets; this effect is inhibited by the blockade of CD40L/CD40 binding. Moreover, blockade of CD40L/CD40 and NF‐κB before treatment with sCD40L abolished the potentiation of platelet aggregation in response to a suboptimal dose of thrombin. Kuijpers et al50 who claim that CD40 is not involved in CD40L‐induced platelet priming, dispute this finding. However, several studies, including our own,11, 12, 51, 52 yield opposite results in this regard.
Finally, we demonstrated the critical role of TAK1 in platelet priming downstream of CD40L/CD40 and upstream of NF‐κB. In fact, we showed the presence of TAK1 in platelets; CD40 blockade inhibits its phosphorylation upon sCD40L ligation. Moreover, 2 TAK1 inhibitors, ZOL and Takinib, in a dose‐dependent manner, abolish NF‐κB activation following CD40L activation and block the priming action of CD40L on platelet aggregation. Previous studies have shown that TAK1 plays a central role in regulating proinflammatory signaling cascades in several cell types26, 28 The evidence for the involvement of TAK1 in CD40‐induced NF‐κB activation was first reported in carcinoma cells. Stimulated CD40, through its association with TRAFs, triggers the engagement of the TAK1/IKKβ/IκBα cascade, which leads to the mobilization of p65 to the interferon regulator factor 1 promoter and the stimulation of its transcriptional activation.53 Furthermore, TAK1 mediates the production of ROS as well as the production of inflammatory mediators in a manner that implies the CD40L/CD40/NF‐κB pathway in vascular smooth muscle cells.54
We recognize that this study has some minor limitations that should be mentioned. sCD40L is released in humans in vivo in thrombotic conditions, although it attains circulating concentrations lower than those required in vitro to prime platelets. Indeed, at least 100 ng/mL of sCD40L was necessary to induce platelet NF‐κB activation; whereas the levels of sCD40L in the circulating blood approximate 1 to 5 ng/mL and increased 10‐folds up to 50 ng/mL in diabetic or in patients with thrombotic events.55, 56 However, the level of sCD40L is predicted to be higher at the site of vascular injury. Indeed, the environment that forms in the gaps between aggregated platelets and the injured vessel wall might allow attaining active higher localized concentrations,57 than those found in the circulating blood. This study may be also limited by the fact that we did not assess the involvement of CD40L in TAK1 induced NF‐κB activation and platelet priming in in vivo experimental thrombosis model, which is planned in further investigations.
Conclusions
In summary, the present study adds more insights on the role of CD40L in platelet function showing that 1) activation of platelet NF‐κB by sCD40L is CD40‐dependent; 2) sCD40L/CD40 axis activates TAK1 upstream of NF‐κB; and 3) CD40L/CD40/TAK1/NF‐κB signaling primes platelets and potentiates aggregation in response to thrombotic stimuli. Elevated levels of sCD40L are now considered reliable predictors of cardiovascular diseases,7 particularly in patients with diabetes mellitus,55, 58 hypercholesterolemia,59 atherosclerosis,60 and acute coronary syndrome.10, 56, 61 Thus, targeting CD40L/CD40/TAK1/NF‐κB limits platelet priming and activation and may ultimately represent a therapeutic target in the treatment of thromboinflammatory diseases.
Sources of Funding
This study was supported by a grant from the Research Foundation of the Montreal Heart Institute.
Disclosures
None.
Supporting information
Figure S1. Dose‐response curve of platelet aggregation in response to thrombin. Washed human platelets (250×106/mL) were stimulated with 4 doses of thrombin: 0.0125 U/mL, 0.025 U/mL, 0.05 U/mL, and 0.1 U/mL. (n=25, mean±SEM).
Figure S2. Comparison of the action of sCD40L versus suboptimal concentrations of thrombin or collagen on platelet NF‐κB and p38 MAPK activation. Washed human platelets (1000×106/mL) were stimulated with thrombin (0.025 U/mL), collagen (0.25 μg/mL), or sCD40L (1000 ng/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for (A) pIκBα. and (B) pP38 blot. Actin blot is from stripped membranes of pIκBα and pP38 blots. Blots are representative of 4 independent experiments, expressed in optical density (n=4, mean±SEM). *P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test versus control).
Figure S3. Comparison of the effects of sCD40L on platelet aggregation in response to suboptimal dose of thrombin or TRAP‐1. Washed human platelets (250×106/mL) were treated or not with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL) or TRAP‐1 (1±0.5 μmol/L). Histograms represent means of aggregation (n=3, mean±SEM). *P<0.001 vs Thr, † P<0.001 versus TRAP‐1 (one‐way ANOVA followed by Dunnett's multiple comparisons test versus the control groups).
Figure S4. ZOL and Takinib have no effect on platelet aggregation in the absence of sCD40L. Washed human platelets (250×106/mL) were pretreated with (A) 100 nmol/L ZOL or (B) 10 μmol/L Takinib for 5 minutes and then stimulated with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL) in the presence or absence of sCD40L. Histograms represent means of aggregation (n=4, mean±SEM). *P<0.001, (one‐way ANOVA followed by Dunnett's multiple comparisons test versus the control group).
Acknowledgments
The authors thank all blood donors and the nursing department of the Montreal Heart Institute bio‐bank for their valuable technical support in blood sampling.
(J Am Heart Assoc. 2018;7:e009636 DOI: 10.1161/JAHA.118.009636.)
References
- 1. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 2. Hansson GK, Libby P. The immune response in atherosclerosis: a double‐edged sword. Nat Rev Immunol. 2006;6:508–519. [DOI] [PubMed] [Google Scholar]
- 3. Hassan GS, Merhi Y, Mourad WM. CD154 and its receptors in inflammatory vascular pathologies. Trends Immunol. 2009;30:165–172. [DOI] [PubMed] [Google Scholar]
- 4. Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, Wijnands E, Goossens P, van Kruchten R, Thevissen L, Boon L, Flavell RA, Noelle RJ, Gerdes N, Biessen EA, Daemen MJ, Heemskerk JW, Weber C, Lutgens E. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood. 2010;116:4317–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Michel NA, Zirlik A, Wolf D. CD40L and its receptors in atherothrombosis‐an update. Front Cardiovasc Med. 2017;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mazzei GJ, Edgerton MD, Losberger C, Lecoanet‐Henchoz S, Graber P, Durandy A, Gauchat JF, Bernard A, Allet B, Bonnefoy JY. Recombinant soluble trimeric CD40 ligand is biologically active. J Biol Chem. 1995;270:7025–7028. [DOI] [PubMed] [Google Scholar]
- 7. Andre P, Nannizzi‐Alaimo L, Prasad SK, Phillips DR. Platelet‐derived CD40L: the switch‐hitting player of cardiovascular disease. Circulation. 2002;106:896–899. [DOI] [PubMed] [Google Scholar]
- 8. Danese S, Katz JA, Saibeni S, Papa A, Gasbarrini A, Vecchi M, Fiocchi C. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut. 2003;52:1435–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. [DOI] [PubMed] [Google Scholar]
- 10. Heeschen C, Dimmeler S, Hamm CW, van den Brand MJ, Boersma E, Zeiher AM, Simoons ML, Investigators CS. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. 2003;348:1104–1111. [DOI] [PubMed] [Google Scholar]
- 11. Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92:1041–1048. [DOI] [PubMed] [Google Scholar]
- 12. Yacoub D, Hachem A, Theoret JF, Gillis MA, Mourad W, Merhi Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40‐dependent tumor necrosis factor receptor‐associated factor‐2/Rac1/p38 mitogen‐activated protein kinase signaling pathway. Arterioscler Thromb Vasc Biol. 2010;30:2424–2433. [DOI] [PubMed] [Google Scholar]
- 13. Hachem A, Yacoub D, Zaid Y, Mourad W, Merhi Y. Involvement of nuclear factor kappaB in platelet CD40 signaling. Biochem Biophys Res Comm. 2012;425:58–63. [DOI] [PubMed] [Google Scholar]
- 14. Gilmore TD. Introduction to NF‐kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. [DOI] [PubMed] [Google Scholar]
- 15. Liu F, Morris S, Epps J, Carroll R. Demonstration of an activation regulated NF‐kappaB/I‐kappaBalpha complex in human platelets. Thromb Res. 2002;106:199–203. [DOI] [PubMed] [Google Scholar]
- 16. Malaver E, Romaniuk MA, D'Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M. NF‐kappaB inhibitors impair platelet activation responses. J Thromb Haemost. 2009;7:1333–1343. [DOI] [PubMed] [Google Scholar]
- 17. Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8:247–252. [DOI] [PubMed] [Google Scholar]
- 18. Leveille C, Bouillon M, Guo W, Bolduc J, Sharif‐Askari E, El‐Fakhry Y, Reyes‐Moreno C, Lapointe R, Merhi Y, Wilkins JA, Mourad W. CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J Biol Chem. 2007;282:5143–5151. [DOI] [PubMed] [Google Scholar]
- 19. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, Ahrens I, Ernst S, Bassler N, Missiou A, Patko Z, Aikawa M, Schonbeck U, Bode C, Libby P, Peter K. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac‐1. Circulation. 2007;115:1571–1580. [DOI] [PubMed] [Google Scholar]
- 20. Henn V, Steinbach S, Buchner K, Presek P, Kroczek RA. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood. 2001;98:1047–1054. [DOI] [PubMed] [Google Scholar]
- 21. Garlichs CD, Eskafi S, Raaz D, Schmidt A, Ludwig J, Herrmann M, Klinghammer L, Daniel WG, Schmeisser A. Patients with acute coronary syndromes express enhanced CD40 ligand/CD154 on platelets. Heart. 2001;86:649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abu el‐Makrem MA, Mahmoud YZ, Sayed D, Nassef NM, Abd el‐Kader SS, Zakhary M, Ghazaly T, Matta R. The role of platelets CD40 ligand (CD154) in acute coronary syndromes. Thromb Res. 2009;124:683–688. [DOI] [PubMed] [Google Scholar]
- 23. Prasad KS, Andre P, He M, Bao M, Manganello J, Phillips DR. Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside‐in signaling. Proc Natl Acad Sci USA. 2003;100:12367–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simic D, Bogdan N, Teng F, Otieno M. Blocking alpha5beta1 Integrin Attenuates sCD40L‐Mediated Platelet Activation. Clin Appl Thromb Hemost. 2017;23:607–614. [DOI] [PubMed] [Google Scholar]
- 25. Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF‐beta signal transduction. Science. 1995;270:2008–2011. [DOI] [PubMed] [Google Scholar]
- 26. Ninomiya‐Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK‐I kappaB as well as the MAP kinase cascade in the IL‐1 signalling pathway. Nature. 1999;398:252–256. [DOI] [PubMed] [Google Scholar]
- 27. Hayden MS, Ghosh S. Signaling to NF‐kappaB. Genes Dev. 2004;18:2195–2224. [DOI] [PubMed] [Google Scholar]
- 28. Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33:522–530. [DOI] [PubMed] [Google Scholar]
- 29. Sakurai H, Miyoshi H, Toriumi W, Sugita T. Functional interactions of transforming growth factor beta‐activated kinase 1 with IkappaB kinases to stimulate NF‐kappaB activation. J Biol Chem. 1999;274:10641–10648. [DOI] [PubMed] [Google Scholar]
- 30. Karim ZA, Vemana HP, Khasawneh FT. MALT1‐ubiquitination triggers non‐genomic NF‐kappaB/IKK signaling upon platelet activation. PLoS One. 2015;10:e0119363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Stoppelaar SF, Claushuis TA, Schaap MC, Hou B, van der Poll T, Nieuwland R, van't Veer C. Toll‐like receptor signalling is not involved in platelet response to streptococcus pneumoniae in vitro or in vivo. PLoS One. 2016;11:e0156977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ledbetter JA, Grosmaire LS, Hollenbaugh D, Aruffo A, Nadler SG. Agonistic and antagonistic properties of CD40 mAb G28‐5 are dependent on binding valency. Circ Shock. 1994;44:67–72. [PubMed] [Google Scholar]
- 33. D'Souza BN, Edelstein LC, Pegman PM, Smith SM, Loughran ST, Clarke A, Mehl A, Rowe M, Gelinas C, Walls D. Nuclear factor kappa B‐dependent activation of the antiapoptotic bfl‐1 gene by the Epstein‐Barr virus latent membrane protein 1 and activated CD40 receptor. J Virol. 2004;78:1800–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karpusas M, Lucci J, Ferrant J, Benjamin C, Taylor FR, Strauch K, Garber E, Hsu YM. Structure of CD40 ligand in complex with the Fab fragment of a neutralizing humanized antibody. Structure. 2001;9:321–329. [DOI] [PubMed] [Google Scholar]
- 35. Nakajima M, Kawaguchi M, Ota K, Fujita J, Matsukura S, Huang SK, Morishima Y, Ishii Y, Satoh H, Sakamoto T, Hizawa N. IL‐17F induces IL‐6 via TAK1‐NFkappaB pathway in airway smooth muscle cells. Immun Inflamm Dis. 2017;5:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ninomiya‐Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. A resorcylic acid lactone, 5Z‐7‐oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–18490. [DOI] [PubMed] [Google Scholar]
- 37. Totzke J, Gurbani D, Raphemot R, Hughes PF, Bodoor K, Carlson DA, Loiselle DR, Bera AK, Eibschutz LS, Perkins MM, Eubanks AL, Campbell PL, Fox DA, Westover KD, Haystead TAJ, Derbyshire ER. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF‐alpha inhibition for cancer and autoimmune disease. Cell Chem Biol. 2017;24:1029–1039.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu TY, Uemura Y, Suzuki M, Narita Y, Hirata S, Ohyama H, Ishihara O, Matsushita S. Distinct subsets of human invariant NKT cells differentially regulate T helper responses via dendritic cells. Eur J Immunol. 2008;38:1012–1023. [DOI] [PubMed] [Google Scholar]
- 39. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jenne CN. Platelets: crossroads of immunity and hemostasis. Blood. 2014;124:671–672. [DOI] [PubMed] [Google Scholar]
- 41. Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi‐functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost. 2009;102:206–214. [DOI] [PubMed] [Google Scholar]
- 42. Antoniades C, Bakogiannis C, Tousoulis D, Antonopoulos AS, Stefanadis C. The CD40/CD40 ligand system: linking inflammation with atherothrombosis. J Am Coll Cardiol. 2009;54:669–677. [DOI] [PubMed] [Google Scholar]
- 43. Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF‐kappa B transcription factor. Science. 1988;242:540–546. [DOI] [PubMed] [Google Scholar]
- 44. Fuentes E, Rojas A, Palomo I. NF‐kappaB signaling pathway as target for antiplatelet activity. Blood Rev. 2016;30:309–315. [DOI] [PubMed] [Google Scholar]
- 45. Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, Roche PA, Whiteheart SW. IkappaB kinase phosphorylation of SNAP‐23 controls platelet secretion. Blood. 2013;121:4567–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wei S, Wang H, Zhang G, Lu Y, An X, Ren S, Wang Y, Chen Y, White JG, Zhang C, Simon DI, Wu C, Li Z, Huo Y. Platelet IkappaB kinase‐beta deficiency increases mouse arterial neointima formation via delayed glycoprotein Ibalpha shedding. Arterioscler Thromb Vasc Biol. 2013;33:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hollenbaugh D, Grosmaire LS, Kullas CD, Chalupny NJ, Braesch‐Andersen S, Noelle RJ, Stamenkovic I, Ledbetter JA, Aruffo A. The human T cell antigen gp39, a member of the TNF gene family, is a ligand for the CD40 receptor: expression of a soluble form of gp39 with B cell co‐stimulatory activity. EMBO J. 1992;11:4313–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li G, Sanders JM, Bevard MH, Sun Z, Chumley JW, Galkina EV, Ley K, Sarembock IJ. CD40 ligand promotes Mac‐1 expression, leukocyte recruitment, and neointima formation after vascular injury. Am J Pathol. 2008;172:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weber C, Springer TA. Neutrophil accumulation on activated, surface‐adherent platelets in flow is mediated by interaction of Mac‐1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet‐activating factor. J Clin Invest. 1997;100:2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuijpers MJ, Mattheij NJ, Cipolla L, van Geffen JP, Lawrence T, Donners MM, Boon L, Lievens D, Torti M, Noels H, Gerdes N, Cosemans JM, Lutgens E, Heemskerk JW. Platelet CD40L modulates thrombus growth via phosphatidylinositol 3‐kinase beta, and not via CD40 and IkappaB kinase alpha. Arterioscler Thromb Vasc Biol. 2015;35:1374–1381. [DOI] [PubMed] [Google Scholar]
- 51. Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol. 2005;25:2428–2434. [DOI] [PubMed] [Google Scholar]
- 52. Danese S, de la Motte C, Reyes BM, Sans M, Levine AD, Fiocchi C. Cutting edge: T cells trigger CD40‐dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J Immunol. 2004;172:2011–2015. [DOI] [PubMed] [Google Scholar]
- 53. Moschonas A, Kouraki M, Knox PG, Thymiakou E, Kardassis D, Eliopoulos AG. CD40 induces antigen transporter and immunoproteasome gene expression in carcinomas via the coordinated action of NF‐kappaB and of NF‐kappaB‐mediated de novo synthesis of IRF‐1. Mol Cell Biol. 2008;28:6208–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song Z, Zhu X, Jin R, Wang C, Yan J, Zheng Q, Nanda A, Granger DN, Li G. Roles of the kinase TAK1 in CD40‐mediated effects on vascular oxidative stress and neointima formation after vascular injury. PLoS One. 2014;9:e101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Varo N, Vicent D, Libby P, Nuzzo R, Calle‐Pascual AL, Bernal MR, Fernandez‐Cruz A, Veves A, Jarolim P, Varo JJ, Goldfine A, Horton E, Schonbeck U. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: a novel target of thiazolidinediones. Circulation. 2003;107:2664–2669. [DOI] [PubMed] [Google Scholar]
- 56. Fong SW, Few LL, See Too WC, Khoo BY, Nik Ibrahim NN, Yahaya SA, Yusof Z, Mohd Ali R, Abdul Rahman AR, Yvonne‐Tee GB. Systemic and coronary levels of CRP, MPO, sCD40L and PlGF in patients with coronary artery disease. BMC Res Notes. 2015;8:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brass LF, Zhu L, Stalker TJ. Novel therapeutic targets at the platelet vascular interface. Arterioscler Thromb Vasc Biol. 2008;28:s43–s50. [DOI] [PubMed] [Google Scholar]
- 58. Varo N, Libby P, Nuzzo R, Italiano J, Doria A, Schonbeck U. Elevated release of sCD40L from platelets of diabetic patients by thrombin, glucose and advanced glycation end products. Diab Vasc Dis Res. 2005;2:81–87. [DOI] [PubMed] [Google Scholar]
- 59. Garlichs CD, John S, Schmeisser A, Eskafi S, Stumpf C, Karl M, Goppelt‐Struebe M, Schmieder R, Daniel WG. Upregulation of CD40 and CD40 ligand (CD154) in patients with moderate hypercholesterolemia. Circulation. 2001;104:2395–2400. [DOI] [PubMed] [Google Scholar]
- 60. de Lemos JA, Zirlik A, Schonbeck U, Varo N, Murphy SA, Khera A, McGuire DK, Stanek G, Lo HS, Nuzzo R, Morrow DA, Peshock R, Libby P. Associations between soluble CD40 ligand, atherosclerosis risk factors, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler Thromb Vasc Biol. 2005;25:2192–2196. [DOI] [PubMed] [Google Scholar]
- 61. Varo N, de Lemos JA, Libby P, Morrow DA, Murphy SA, Nuzzo R, Gibson CM, Cannon CP, Braunwald E, Schonbeck U. Soluble CD40L: risk prediction after acute coronary syndromes. Circulation. 2003;108:1049–1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dose‐response curve of platelet aggregation in response to thrombin. Washed human platelets (250×106/mL) were stimulated with 4 doses of thrombin: 0.0125 U/mL, 0.025 U/mL, 0.05 U/mL, and 0.1 U/mL. (n=25, mean±SEM).
Figure S2. Comparison of the action of sCD40L versus suboptimal concentrations of thrombin or collagen on platelet NF‐κB and p38 MAPK activation. Washed human platelets (1000×106/mL) were stimulated with thrombin (0.025 U/mL), collagen (0.25 μg/mL), or sCD40L (1000 ng/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for (A) pIκBα. and (B) pP38 blot. Actin blot is from stripped membranes of pIκBα and pP38 blots. Blots are representative of 4 independent experiments, expressed in optical density (n=4, mean±SEM). *P<0.001 (one‐way ANOVA followed by Dunnett's multiple comparisons test versus control).
Figure S3. Comparison of the effects of sCD40L on platelet aggregation in response to suboptimal dose of thrombin or TRAP‐1. Washed human platelets (250×106/mL) were treated or not with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL) or TRAP‐1 (1±0.5 μmol/L). Histograms represent means of aggregation (n=3, mean±SEM). *P<0.001 vs Thr, † P<0.001 versus TRAP‐1 (one‐way ANOVA followed by Dunnett's multiple comparisons test versus the control groups).
Figure S4. ZOL and Takinib have no effect on platelet aggregation in the absence of sCD40L. Washed human platelets (250×106/mL) were pretreated with (A) 100 nmol/L ZOL or (B) 10 μmol/L Takinib for 5 minutes and then stimulated with sCD40L (1000 ng/mL) for 30 minutes at 37°C. Platelet aggregation was induced by a suboptimal dose of thrombin (0.025±0.01 U/mL) in the presence or absence of sCD40L. Histograms represent means of aggregation (n=4, mean±SEM). *P<0.001, (one‐way ANOVA followed by Dunnett's multiple comparisons test versus the control group).