

A broad spectrum of primary sarcoma subtypes are now recognized to occur intracranially, presumably arising from mesenchymal progenitor cells within the meningeal covering of the brain and along perivascular Virchow-Robin spaces. Here we report the clinical, radiologic, histologic, and molecular features of three patients with intracranial sarcomas that are unified by the presence of primary intracranial location with meningeal involvement, pleomorphic morphology, high proliferation index, prominent eosinophilic cytoplasmic globules, focal immunophenotypic evidence of myogenic differentiation, and the combination of DICER1 and TP53 mutations, along with ATRX inactivation and genetic alterations causing activation of the MAP kinase signaling pathway. One patient has neurofibromatosis type 1 (NF1) with multiple cutaneous neurofibromas, cafe-au-lait macules, an optic pathway glioma, and other brain parenchymal lesions characteristic of NF1. This patient was found to have a heterozygous germline nonsense mutation in the NF1 tumor suppressor gene, along with a second somatic mutation of NF1 in the primary intracranial sarcoma, indicating that this tumor arose due to biallelic NF1 gene inactivation. Genome-wide methylation profiling performed on two of the cases revealed that they clustered with the recently described group of tumors termed “primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features, DICER1 mutant” [7]. However, while the three tumors in our cohort demonstrate myogenic differentiation as evidenced by focal desmin immunopositivity, none contain identifiable rhabdomyoblasts, they uniformly lack myogenin expression, and are all morphologically best characterized as pleomorphic rather than predominantly spindled. Thus, this patient cohort expands the histologic spectrum and association with familial tumor predisposition syndromes of the primary intracranial sarcomas that cluster with this new methylation subgroup. As such, we suggest a broader designation for this new proposed entity: “Primary intracranial sarcoma, DICER1-mutant”.
The one female and two male patients ranged in age at time of initial surgery from 11–20 years (Figure 1a). Patients #1 and #3 had been previously healthy and had no family history of cancer or cutaneous, visceral, or brain lesions other than the primary intracranial sarcomas suggestive of a tumor predisposition syndrome. Patient #2 had a clinical diagnosis of NF1 with multiple cutaneous neurofibromas, café-au-lait macules, an optic pathway glioma, and multifocal T2 signal abnormality within the thalamus, brainstem, and cerebellum (Supplemental Figure 1 [Online Resource 1]). The patients had initially presented with either seizure or headache. Brain imaging demonstrated complex, solid and cystic, heterogeneously enhancing masses centered within the frontal or parietal lobes with meningeal involvement in all three patients (Figure 1b, Supplemental Figure 1 [Online Resource 1], and Supplemental Table 1 [Online Resource 2]). All tumors demonstrated evidence of prior hemorrhage and were associated with peritumoral edema. Surgical resection was performed for each patient.
Figure 1.
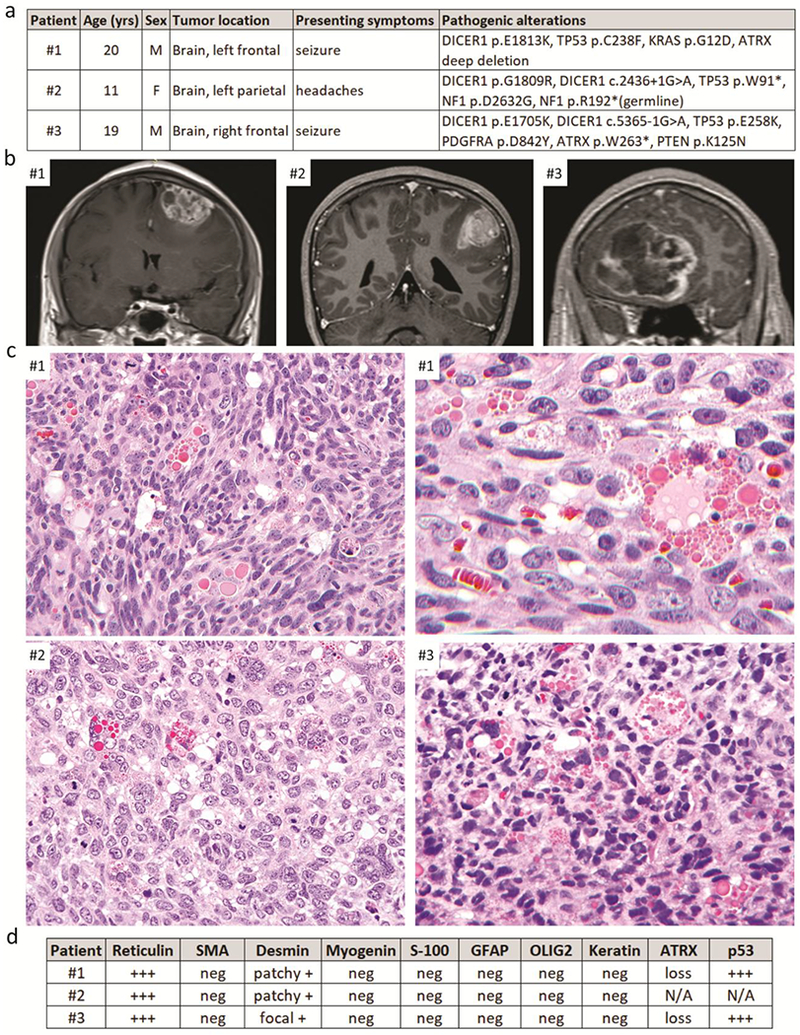
Clinical, radiologic, histologic, and molecular features of three patients with primary intracranial pleomorphic sarcomas with myogenic differentiation and DICER1 mutation. a, Clinical features and pathogenic molecular alterations identified. b, Pre-operative T1-weighted post-contrast MR images. c, Representative images from H&E stained sections showing pleomorphic sarcomas with brisk mitotic activity and prominent eosinophilic cytoplasmic globules. d, Summary of reticulin staining and immunohistochemical results.
Histologic analysis of the three tumors revealed sarcomatous neoplasms that were highly cellular with brisk mitotic activity, foci of necrosis, and intratumoral hemorrhage. Morphology was variable but predominantly consisted of sheets of pleomorphic tumor cells without a discernible growth pattern. A notable histologic feature shared by all three cases was the presence of prominent variablysized brightly eosinophilic cytoplasmic globules (Figure 1c and Supplemental Figures 2-4 [Online Resource 1]). Similar eosinophilic cytoplasmic globules were present at least focally in the majority of the 22 tumors reported by Koelsche et al [7], Case #3 contained a small focus of cartilaginous differentiation. Rhabdomyoblasts with cytoplasmic striations (“strap cells”) were not identifiable in any of the cases, nor was production of osteoid matrix or bone. No associated diffuse glioma was seen in any case to warrant classification as gliosarcoma, although the possibility of a sarcoma-predominant gliosarcoma was considered a diagnostic possibility prior to molecular profiling. Reticulin staining demonstrated abundant intercellular basement membrane deposition in each tumor. Immunohistochemistry demonstrated focal or patchy staining for desmin in all cases, but no myogenin or smooth muscle actin expression was observed (Figure 1d and Supplemental Figures 2-4). Tumor cells were uniformly negative for GFAP, OLIG2, S-100, SOX10, and cytokeratin immunostaining. The Ki67 labeling index was greater than 50% in all three cases. Tumors #1 and #3 demonstrated strong nuclear p53 positivity in the majority of tumor cells, as well as loss of ATRX expression.
In order to assist with diagnostic classification, comprehensive genetic profiling was performed on genomic DNA extracted from formalin-fixed, paraffin-embedded tumor tissue from the three patients, as well as a peripheral blood sample from patient #2, using the UCSF500 Cancer Panel, which assesses approximately 500 cancer-associated genes for mutations, copy number alterations, and structural variants, as well as evaluation of chromosomal copy number changes, microsatellite stability, and somatic mutation burden (Supplementary Table 2 [Online Resource 2] and refs. 3, 4, 5, 8, 9, 10, 11, 12). All three cases demonstrated hotspot missense mutations in exons 24 or 25 of the DICER1 gene (p.E1705K, p.G1809R, and p.E1813K) located within the Ribonuclease III domain near the C-terminus of the encoded microRNA processing enzyme (Supplemental Table 3 [Online Resource 2]), which have been recurrently seen as confirmed somatic mutations in numerous cystic nephromas, Sertoli-Leydig cell tumors, and other neoplasms known to be driven by DICER1 mutation (Catalog Of Somatic Mutations In Cancer [COSMIC] database, version 87 release). The DICER1 mutation in the tumor from patient #1 was homozygous due to copy-neutral loss of heterozygosity of chromosome 14q that eliminated the remaining wildtype allele. In the tumors from patients #2 and #3, there was a second splice site mutation in the DICER1 gene (c.2436+1G>A and c.5365-1G>A) likely to be present in trans and causing inactivation of the remaining wildtype allele. Such inactivating mutations affecting one allele together with a hotspot missense mutation affecting the other allele are commonly seen in tumor types known to be driven by DICER1 mutations [2]. Additionally, all three cases demonstrated inactivating mutations affecting the TP53 tumor suppressor gene (p.W91*, p.C238F, and p.E258K) accompanied by loss of the remaining wildtype alleles. Two cases demonstrated inactivation of the ATRX tumor suppressor gene, one with deep deletion and one with a nonsense mutation (p.W263*). Lastly, each case harbored pathogenic mutations predicted to cause activation of the MAP kinase signaling pathway. The tumor from patient #1 harbored an activating hotspot mutation in the KRAS oncogene (p.G12D), and the tumor from patient #3 harbored a hotspot missense mutation in the PDGFRA oncogene (p.D842Y) located within the intracellular tyrosine kinase domain that has been recurrently seen as a confirmed somatic mutation in numerous gastrointestinal stromal tumors (COSMIC database, version 87 release). Patient #2 was found to harbor a heterozygous truncating nonsense mutation in NF1 tumor suppressor gene (p.R192*) in the germline, genetically confirming the clinical diagnosis of NF1. A second somatic missense mutation in the NF1 gene (p.D2632G) was seen in the tumor, indicating that the sarcoma likely arose, at least in part, due to biallelic NF1 inactivation. As tumor tissue only without a paired normal sample was sequenced for patients #1 and #3, the somatic versus germline status of the identified DICER1 and TP53 mutations could not be determined, but the DICER1 and TP53 mutations in patient #2 were both confirmed to be somatic (tumor-specific). All three tumors demonstrated markedly aneuploid genomes with partial gains and losses involving portions of most chromosomes (Supplementary Figure 5 [Online Resource 1]). Genome-wide DNA methylation profiling was performed on cases #1 and #2 as previously described [1, 6] using the Illumina MethylationEPIC BeadChip (850k array) to further characterize these primary intracranial sarcomas with myogenic differentiation and DICER1 mutation. Unsupervised clustering of DNA methylation patterns demonstrated that both cases clustered with the recently described group of tumors termed “primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features, DICER1 mutant” – calibrated score for case #1 was 0.84 and for case #2 was 0.99 (Supplementary Figure 6 [Online Resource 1]).
Following gross total resection, patient #1 was treated with craniospinal radiation and adjuvant chemotherapy with vincristine. He was alive without evidence of disease recurrence at last clinical follow-up approximately six weeks after initial resection. Following gross total resection, patient #2 was treated with cranial radiation and adjuvant chemotherapy with temozolomide. Disease recurrence adjacent to the resection cavity in the left parietal lobe was seen on follow-up imaging at four years after initial resection, and a second resection was recently performed confirming recurrent pleomorphic sarcoma with myogenic differentiation and DICER1 mutation. Patient #3 was only recently diagnosed and is currently beginning adjuvant therapy.
Overall, this series of three primary intracranial sarcomas has significant similarities with the group of “primary intracranial spindle cell sarcomas with rhabdomyosarcoma-like features, DICER1 mutant” that were recently reported including primary intracranial location, immunophenotypic evidence of focal myogenic differentiation, overlapping genome-wide methylation profile, co-occurring DICER1 and TP53 mutations, and alterations activating the MAP kinase signaling pathway. However, the tumors in our series have morphology best characterized as pleomorphic rather than spindled, uniformly lack rhabdomyoblasts and myogenin expression, and demonstrate frequent ATRX inactivation. As these tumors are all likely to be variants of the same entity we propose the broader nomenclature of “Primary intracranial sarcoma, DICER1-mutant”. Though unlikely to be specific, a histologic feature uniformly observed in our case series that should suggest consideration of this tumor entity in an intracranial sarcomatous neoplasm is the presence of prominent eosinophilic cytoplasmic globules. In addition to the previously reported association with the DICER1 pleuropulmonary blastoma tumor syndrome, we identify that these primary intracranial sarcomas with DICER1 mutation can also occur in the setting of NF1, thereby extending the tumor spectrum associated with this common familial tumor syndrome.
Supplementary Material
Acknowledgements
D.A.S. is supported by NIH Director’s Early Independence Award (DP5 OD021403) and the UCSF Physician-Scientist Scholar Program.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Compliance with ethical standards
This study was approved by the Committee on Human Research of the University of California, San Francisco, with a waiver of patient consent.
Conflict of interest
The authors declare that they have no competing interests related to this study.
Data availability
Scanned image files of the H&E stained slides from which representative images are presented are available for downloading and viewing at the following link: https://figshare.com/projects/Primary_intracranial_sarcoma_with_myogenic_differentiation_and_DICER1_mutation/57704. Sequencing and methylation data files are available from the authors upon request.
Terms of use and reuse: academic research for non-commercial purposes, see here for full terms. https://www.springer.com/aam-terms-v1
References
- 1.Capper D, Jones DTW, Sill M et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foulkes WD, Priest JR, Duchaine TF (2014) DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14:662–672 [DOI] [PubMed] [Google Scholar]
- 3.Goode B, Mondal G, Hyun M et al. (2018) A recurrent kinase domain mutation in PRKCA defines chordoid glioma of the third ventricle. Nat Commun 9:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iorgulescu JB, Van Ziffle J, Stevers M et al. (2018) Deep sequencing of WNT-activated medulloblastomas reveals secondary SHH pathway activation. Acta Neuropathol 135:635–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline CN, Joseph NM, Grenert JP et al. (2017) Targeted next-generation sequencing of pediatric neurooncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol 19:699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koelsche C, Hartmann W, Schrimpf D et al. (2018) Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol 31:1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koelsche C, Mynarek M, Schrimpf D et al. (2018) Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol 136:327–337 [DOI] [PubMed] [Google Scholar]
- 8.Lopez GY, Van Ziffle J, Onodera C et al. (2018) The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol [Epub ahead of print] September 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pekmezci M, Stevers M, Phillips JJ et al. (2018) Multinodular and vacuolating neuronal tumor of the cerebrum is a clonal neoplasm defined by genetic alterations that activate the MAP kinase signaling pathway. Acta Neuropathol 135:485–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pekmezci M, Villanueva-Meyer JE, Goode B et al. (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips JJ, Gong H, Chen K et al. (2018) The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol [Epub ahead of print] July 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon DA, Korshunov A, Sill M et al. (2018) Myxoid glioneuronal tumor of the septum pellucidum and lateral ventricle is defined by a recurrent PDGFRA p.K385 mutation and DNT-like methylation profile. Acta Neuropathol 136:339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.