

Abstract
Thalidomide is emerging as a potentially important therapeutic option in the treatment of metastatic prostate cancer. Although the mechanism of action of this agent remains elusive in malignancies of the prostate, recent data has indicated that thalidomide may play a role in inflammation, immunomodulation, and anti-angiogenesis. Lenalidomide (CC-5013), a thalidomide analogue with improved activity and safety profile in certain disease contexts, is in the early stages of development in prostate cancer. This review will provide the current status of the history, mechanism, metabolism, and clinical use of thalidomide in metastatic prostate cancer. It will also describe the mechanism and clinical use of lenalidomide as it pertains to malignancies of the prostate.
Keywords: Prostate cancer, thalidomide, lenalidomide, CC-5013, angiogenesis inhibition
INTRODUCTION
Despite numerous advances in drug treatment, prostate cancer has the second-highest mortality rate of all cancers in men living within the USA, with 28,660 deaths estimated in 2008 [1] This malignancy is very difficult to treat, and there are few meaningful therapeutic strategies once the disease develops into androgen independent prostate cancer (AIPC, also known as hormone-refractory prostate cancer, HRPC). Docetaxel-based therapy, which is the current standard-of-care for AIPC, only provides an approximate 2-to 3-month survival benefit over palliative care with mitoxantrone plus prednisone [2, 3] Therefore, therapeutic strategies that can increase survival, alone or in combination with docetaxel are urgently needed. Thalidomide (Fig. 1) has recently reemerged as a potential therapy for men with prostate cancer, and many thalidomide analogues, such as lenalidomide, may also have some activity in malignancies of the prostate. While these agents are not necessarily cytotoxic, thalidomide and its analogues possess antiangiogenic, anti-inflammatory, and immunomodulatory activity useful in the treatment in many diseases, including prostate cancer. This review will summarize the current status of thalidomide and its analogue, CC-5013 (lenalidomide) (Fig. 1) with respect to metabolism, mechanism of action, and clinical treatment with these agents in men with prostate cancer.
Fig. (1).

Thalidomide, CC-5013, CC4047.
HISTORY OF THALIDOMIDE
Thalidomide was originally prescribed in the late 1950’s as a sedative and antiemetic treatment for morning sickness. Unfortunately, thalidomide was widely used in pregnant women prior to the discovery of its teratogenic properties, resulting in severe congenital defects in over 10,000 infants exposed to the drug during gestation,[4, 5] and leading to the agent’s withdrawal from the market in 1961. While thalidomide has a tragic history in this context, the drug was rediscovered in 1965 when it was found to relieve the painful skin condition, erythema nodosum leprosum (ENL), associated with leprosy [6], It was later discovered that the inhibition of lipopolysaccharide-induced monocyte secretion of tumor necrosis alpha (TNF-α), a cytokine involved in the pathogenesis of several autoimmune and infectious diseases, was the mechanism behind the success of thalidomide treatment in ENL [7], and was approved by the FDA in 1998 for this indication. Indeed, the inhibition of TNF-α is also the primary mechanism of action of thalidomide in the treatment in multiple myeloma, where thalidomide in combination with dexamethasone has been responsible for major advances in treatment. This led to FDA approval for thalidomide in the treatment of multiple myeloma in 2006.
Some have hypothesized that the inhibition of limb bud formation by thalidomide was due to drug-induced decreases in angiogenesis at these sites, and this realization was responsible for the exploration of thalidomide as an anti-angiogenesis agent. Another, non-mutually exclusive, hypothesis states that thalidomide induces oxidative stress in limb buds that subsequently enhances signaling through bone-morphogenic-proteins (BMPs), thereby altering limb patterning and early skeletal formation through the DKKl/Wnt/ beta-catenin pathway [8]. Others have hypothesized that thalido-mide-induced oxidative stress alters NF-ĸB targets involved in proliferation and differentiation [9]. More recently, it has been suggested that loss of immature blood vessels is the primary cause for thalidomide-induced teratogenesis [10]; therefore, thalidomide’s teratogenic mechanism remains controversial. Nonetheless, in 1994, D’Amato et al. determined that thalidomide is an active antiangiogenesis agent, [11] and this realization led to the exploration of thalidomide treatment in solid tumors. Since then thalidomide has been shown to have utility in treating prostate cancer, a disease that requires the formation of new blood vessels for continued growth and progression [12], However, thalidomide may be quite pleiotropic, influencing not only angiogenesis, but also immuno-genecity, inflammation, and possibly several other pathways that remain unexplored. To date, thalidomide has also been used with some success in the treatment of multiple myeloma, graft-versus-host disease, Waldenstrom’s macroglobulinemia, AL amyloidosis, myelofibrosis with myeloid metaplasia, myelodysplastic disorders, acute myeloid leukemia, brain tumors, renal cell carcinoma, melanoma, Kaposi’s sarcoma, and other tumors as reviewed in [13]. The present review will focus on the use of thalidomide in prostate cancer.
IMMUNOMODULATORY AND ANTI-INFLAMMATORY ACTIVITY
Overview
Prostate tumors are heavily dependent upon growth factors and cytokines that regulate proliferation, anti-apopotosis, chemoresistance, and metastasis. Many of these same cytokines play a role in angiogenesis, alter the tumor microenvironment, facilitate stromalepithelial interactions, promote attachment of metastatic cells, and promote growth of tumor cells at distant sites. Perhaps some of the best characterized cytokines explored in prostate cancer to date have been, IL1, IL6, IL8, and TNF-α, that signal through NF-ĸB, MAPK (NF-ĸB, Elk-1, ATF-2, Erkl/2), JAK/STAT, AP-1, AR, HIF1A, and other pathways that are known to promote tumor progression in the prostate [14–18], Such signaling pathways are ultimately responsible for the cellular synthesis of proteins that mediate the complex process of tumor progression, microenvironmental changes, and drug resistance. COX2 regulation of arachadonic acid seems to also be important in inflammatory dysregulation of prostate tumors [19]. Finally, growth factor signaling through PDGF, and bFGF, also mediate prostate tumor progression, angiogenesis, and metastatic state [20, 21], Targeting these pathways may halt, or attenuate disease progression and allow for chemosensitization of tumor cells. It is interesting that thalidomide has been shown to play a role in altering many of these signaling pathways, although the implications of thalidomide’s ability to change these pathways in prostate cancer, and other solid tumors, is rather poorly understood. Here we will highlight the current status of thalidomide’s participation in altering several of the aforementioned pathways.
TNF-α
TNF-α is classically thought to induce proinflammatory and death signaling through IKK, and MKK7/JNK, and caspase 8 resulting in signaling through NF-ĸB, AP-1 and Bid/caspase 3. However, other evidence suggests that TNF-α expression may promote growth, proliferation, differentiation and interactions between stromal cells and the tumor. Interestingly, TNF-α signaling is also often associated with signaling through IL-1, IL-6, and M-CSF as is the case in many prostate tumors [22], suggesting that the TNF-α cytokine is involved in global dysregulation of prostate tumors.
The presence of high serum concentrations of TNF-α are associated with the extent of the disease in patients with prostate cancer [23], although it seems that serum levels of TNF-α are not always altered between benign prostatic hyperplasia, hormone sensitive prostate cancer, or AIPC [24], However, others have observed high levels of TNF-α in prostatic fluid and serum-free peripheral blood mononuclear cells (PBMCs) in men with prostate cancer [25, 26], Therefore, it appears that serum concentrations are not sufficient to determine differences in TNF-α levels within the prostate, but those having high serum TNF-α are more likely to have a poor prognosis. TNF-α receptors (TNFRI, and TNRII) are also upregulated within prostate tumors as compared to normal tissues suggesting that tissue-specific concentrations of TNF-α may be more important to consider than serum concentrations. Some have observed that TNF-α serum concentrations are elevated prior to PSA progression [23], while others have demonstrated no relationship between PSA and TNF-α levels [27]. Others have observed that TNF-α levels are associated with loss of androgen responsiveness [22], A study comparing men treated with thalidomide versus an untreated control cohort found that TNF-α is actually increased within the tumor microenvironment in biopsy samples, and in serum samples following thalidomide therapy [28], Serum TNF-α levels also appeared to increase in patients with advanced prostate cancer receiving thalidomide as monotherapy [29], Therefore, while thalidomide treatment seems to alter TNF-α signaling in prostate tumors, it remains unclear what effect thalidomide has on TNF-α production in men with prostate cancer, and if this cytokine is related to the success of thalidomide treatment in men with prostate cancer as is the case for other diseases such as multiple myeloma and ENL.
Interleukins (IL6, IL8)
Thalidomide has been shown to increase or decrease the serum concentrations of several interleukins, including IL1β, IL2, IL4, IL5, IL6, IL8, IL10, and IL12 [28, 30]. Such interactions may alter the prostate tumor cell, stromal epithelial interactions, metastasis (especially to cytokine-rich bone), angiogenesis, and immune response. Therefore, the interaction between thalidomide and interleukins may be central to the multiplicity of effects that thalidomide has in the treatment of cancer. Here, we will discuss IL6, and IL8.
IL-6 is a proinflammatory cytokine that acts as both an autocrine and paracrine growth factor in AIPC tumors [31]. IL6 is stimulated by the activation of NF-ĸB and is involved in cell proliferation, cell survival, crosstalk between the tumor and inflammatory cells, and angiogenesis [32], Elevated IL-6 is found in the serum of patients with metastatic prostate cancer and is associated with poor prognosis in two different studies [23, 33]. Prostate cancer cell lines also secrete IL-6, although some controversy remains as to whether or not the androgen-dependent LNCaP line produces IL-6 [14]. IL-6 levels are decreased by thalidomide in the prostate microenvironment [28], and thalidomide also decreases the levels of IL-6 secreted by mitogen-stimulated PBMCs [34].
IL-8 is also an inflammatory chemokine that normally regulates the immune response. However, IL-8 can also promote tumor progression in prostate cancer largely through the induction of matrix metalloproteases (MMPs) that remodel the tumor microenvironment thereby promoting angiogenesis and metastasis [14]. Increases in IL-8 levels have been associated with MMP induction, high Gleason score, and metastatic prostate cancer [35–37], although a direct causal relationship between IL-8 and MMPs remains to be established [14]. Thalidomide treatment has been related to increases in IL-8 within the tumor microenvironment, and this has been attributed to a stress response that may compensate for microvessel density decreases following thalidomide treatment [28].
NF-ĸB Inactivation
NF-ĸB is a DNA-binding factor that participates in the transcription of a number of proinflammatory genes. In unstimulated cells, NF-ĸB is localized within the cytoplasm and bound to inhibitory proteins, such as IkB. Stimulation of NF-ĸB occurs through several extracellular signals, including IL 113 and TNF-α [38], NF-ĸB transcribes IL-8, IL-12, TNF-α, regulates genes that promote cell growth, suppresses apoptosis, and promotes inflammation and metastasis [39]. However, TNF-α can also promote apoptosis under certain circumstances [22]. Thalidomide appears to inhibit NF-ĸB activity by suppressing IkB kinase activity [39], although thalidomide is only able to block NF-ĸB activation induced by TNF-α and peroxide, while it is unable to prevent NF-ĸB activation through lipopolysaccharides and phorbol esters. This suggests that NF-ĸB activation can occur through multiple mechanisms, some of which are not prevented by thalidomide treatment [20], and these compensatory pathways may be behind thalidomide-resistance in certain patients.
Prostate cells are sensitized to the pro-apoptotic effects of TNF-α secretion by macrophages once NF-ĸB transcription is blocked [40]. Thus, thalidomide may promote apoptosis of these cells while suppressing pro inflammatory pathways that mediate cell survival and migration. The ability of thalidomide to inhibit NF-ĸB activation through TNF-α may also be behind the paradoxical increase of TNF-α levels in the prostate cancer microenvironment [28], and in human serum [29] following thalidomide treatment. In these cases, it could be hypothesized that TNF-α may be involved in a concentration-dependent immunosupression of tumor growth once thalidomide blocks the proinflammatory potential of the cytokine. It is also possible that increases in intratumoral TNF-α are related to a compensatory response to the antiangiogenic effects of thalidomide since TNF-α is proangiogenic [41]. Such inhibition of inflammation and angiogenesis may not be sufficient to cause regression of tumors alone, but may be responsible for the effectiveness of combinations of thalidomide and cytotoxic drugs [42, 43], Nonetheless, the NF-ĸB pathway seems to be a major downstream player in the multiplicity of effects that thalidomide has in the treatment of several diseases, including prostate cancer.
COX2 Inhibition
Cyclooxygenase-2 (COX2) is involved in the synthesis of prostaglandins, and is highly expressed in prostate cancer [19]. COX2 mediates proinflammatory processes, apoptosis, and promotes angiogenesis as is apparent from experiments conducted in prostate cells treated in vitro and in vivo with COX2 inhibitors [44], Thalidomide inhibits the lipopolysaccharide-mediated induction of COX2 by decreasing COX2 mRNA stability, and results in a dose-dependent downregulation of prostaglandin E2 (PGE2) [45], Interestingly, it was recently shown that the mechanism behind the downregulation of COX2 by thalidomide is related to posttranscriptional alteration of COX2 mRNA by inhibiting the MAPK pathway thereby blocking the nuclear translocation of HuR, an mRNA-stabilizing protein [46], However, it must be noted that the former experiment was conducted in a human macrophage cell line, while the latter was conducted in Caco-2 cells, a colon carcinoma line, and it is unclear how well these results can be translated to prostate cancer. Others have observed that the thalidomide analogue, CPS-49, actually increased signaling through activation of MAPK that correlated with a direct cytotoxic effect in prostate cancer cells [47]. Therefore, clarification of the role of thalidomide on COX2 metabolism of PGEs is warranted in the context of prostate cancer.
NK-Cell Activation and Costimulation of T-Cells
Natural Killer (NK) cells are well known to be involved in the immune response towards tumors by induction of perforin- and granzyme-mediated apopotosis and necrosis of target cells, and NK cells are critical to antitumor immunity [48], Elevated numbers of CD56+ NK cells has been related with a better prognosis in men with prostate cancer undergoing androgen deprivation therapy [49]. Thalidomide treatment has been shown to increase lysis of multiple myeloma cell lines, and increases PBMC-mediated apoptosis in K562 cell lines [50], Increases in NK cell-induced apoptosis have been related to the production of IL-2 by T-cells that are costimulated by immunomodulatory agents related to thalidomide [50], However, any potential link between NK cell infiltration and clinical outcome in patients with prostate cancer has not yet been established.
Initial steps evaluating thalidomide’s effect on T-cell migration into prostate tumors have been made. Following androgen deprivation therapy, large numbers of T-cells infiltrate the prostate, and increases in peripheral lymphoid T-cell populations are observed [14]. These cells have a low activation threshold, and it appears that androgens may negatively regulate inflammation, or that T-cell activation and migration occurs in response to inflammatory pathways (i.e. NF-ĸB activation), and increases in the number of apop- totic cells (i.e. in hypoxic tumors, or following androgen withdrawal) [14]. Thalidomide costimulates human CD4+ and CD8+ T- cell production, increases IL-2 serum concentrations, and interferon-gamma (IFN-γ) production [13]. Moreover, the immune response may also be modulated by the ability of thalidomide to downregulate COX2 [51]. Although the consequences of T-cell infiltration into prostate tissue is unclear, it has been hypothesized that these immune cells may be auto-reactive and induce further inflammation by damaging the epithelium resulting in a loss of tolerance towards normal prostate antigens [14]. Therefore, while the functional aspects of T-cell infiltration into the prostate remain to be clarified, thalidomide may influence both the migration and activation of leukocytes through its ability to modulate cytokines and COX2.
ANTIANGIOGENIC ACTIVITY
Vascular and Endothelial Growth Factor Inhibition
Prostate cancer is heavily dependent upon neovascularization for growth and progression to metastatic disease, and angiogenesis also plays an important role following the development of metastatic disease. Weidner et al. demonstrated that patients who progressed to metastatic disease, and had poorly differentiated prostate tumors, had an approximate 2-fold increase in microvessel density (MVD) within histological specimens of prostate carcinoma stained for endothelial cells [52], The MVD was also much higher within invasive tumors as compared to surrounding normal tissue within the same patient [53], Moreover, the effectiveness of antiangiogenesis therapy in hormone-dependent prostate cancer and AIPC has been observed in several drugs that target VEGF, the VEGF receptor, and inhibit signaling events that regulate the migration, adhesion, and survival of endothelial cells, as reviewed in [54].
Thalidomide has been shown to mediate anti-angiogenic activity by inhibiting pathways related to extracellular matrix degradation, endothelial cell proliferation, and endothelial cell migration by decreasing the macrophage-associated secretion of several proangiogenic factors (e.g. acidic fibroblast growth factor (aFGF), basic fibroblast growth factor (bFGF), VEGF, granulocyte-macrophage colony stimulating factor (GM-CSF), TNF-α, IL-6, and IL-8 [55]). Thalidomide has also been shown to inhibit b-FGF and VEGF in human bone marrow endothelial cells, and in the mouse corneal model [56, 57]. Moreover, thalidomide has demonstrated anti- angiogenesis activity in the rabbit cornea micropocket assay [11, 56]. Interestingly, only those biological models utilizing intact mice or human liver microsomes have demonstrated thalidomide-related antiangiogenic activity [58]; therefore, it appears that thalidomide must undergo enzymatic metabolism by certain drug metabolizing enzymes in order to mediate an anti-angiogenic effect. Thalidomide also appears to induce defects in tumor vasculature characterized by altered morphology, and the loss of receptors such as, the VEGF receptor, neuropilin-1, and Flk-1 that may further degrade the angiogenic signal [59]. A recent study showed that in addition to VEGF downregulation, thalidomide treatment also altered the levels of several markers of angiogenesis, including IL-6, IL-8, and bFGF, in men with prostate adenocarcinoma receiving thalidomide, versus a similar cohort of men not receiving thalidomide [28]. Two animal models have also recently been published demonstrating that thalidomide can enhance the docetaxel-mediated killing of circulating endothelial cells, showing that thalidomide may also have a direct effect on the precursors of the tumor vasculature when combined with cytotoxic agents [43, 60]. Therefore, although the exact role of thalidomide activity in prostate cancer remains to be elucidated, the anti-angiogenic properties of the agent appear to be very important for the treatment of prostate cancer.
Potential for a TNF-α Related Antiangiogenic Effect
It has been suggested that TNF-α secretion by macrophages is a major proangiogenic stimulus in prostate cancer [55]. Conflicting data by Fujita et al. and Kenyon et al. have been presented as to whether thalidomide confers the anti-angiogenic effect as a direct result of TNF-α downregulation [61], or if the effect is independent of TNF-α [56], However, the study by Fujita et al. was conducted ex vivo, and never established a causative relationship between angiogenesis and TNF-α, only that angiogenesis and TNF-α suppression were highly correlated. On the other hand, the study by Kenyon et al. was conducted in intact mice and demonstrated that TNF-α inhibition did not alter neovascularization. Therefore, more direct evidence suggests that angiogenesis is not likely to be related to TNF-α suppression, although it remains unclear if this relationship exists in the context of certain diseases. For example, it appears that TNF-α is an important inducer of angiogenic factors such as IL-8 and VEGF in human prostate cancer [62–64], although a direct link between the downregulation of TNF-α by thalidomide resulting in inhibition of VEGF and IL-8 remains to be established in humans. However, TNF-α also has proinflammatory activity in prostate cancer as will be discussed in the next section; therefore downregulation of this cytokine may affect both angiogenesis and tumor growth simultaneously.
THALIDOMIDE METABOLISM
Thalidomide Hydrolysis and Metabolism
The four amide bonds in thalidomide are rapidly hydrolyzed in aqueous solution, and under physiological conditions to form several hydrolysis products. Hydrolysis occurs more slowly at lower pH, and for this reason studies evaluating thalidomide pharmacokinetics have stabilized the drug in patient samples by acidification [65]. Studies on thalidomide pharmacokinetics and metabolite formation in mice, rabbits, and patients with multiple myeloma identified three hydrolysis metabolites in plasma and urine using liquid chromatography-mass spectrometry: 2-phthalimidoglutaramic acid, α-(o-carboxybenzamido)glutarimide and 4-phthalimidoglutaramic acid. Other hydrolysis metabolites that are produced in aqueous solution, have yet to been identified in humans (Fig. 2; [66]).
Fig. (2).
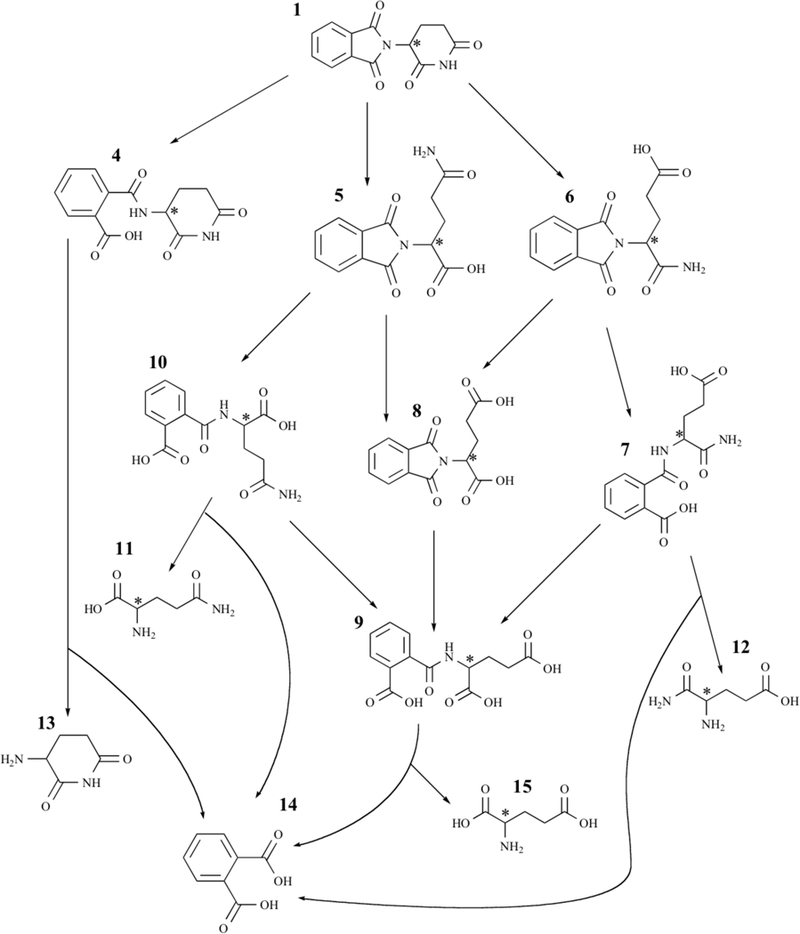
Thalidomide hydrolysis products. Adapted from [55]. The names of all compounds are listed in Table 1 alongside their respective numbers.
A series of studies have been performed in vitro and in vivo to elucidate the structures of the enzyme-mediated metabolites, and to understand the interspecies differences in metabolism, which may be responsible for the striking inter-species differences in biological effects (Fig. 3; [58]). The monohydroxylated metabolites 5-hydroxythalidomide and 5’-hydroxythalidomide have been of particular interest, and these have been identified in both plasma and urine obtained from human subjects [67]. The 5-hydroxy- thalidomide metabolite is formed by hydroxylation of the phthalimide ring, and 5’-hydroxythalidomide is formed by hydroxylation of the glutarimide ring [58]. Ando et al., found that thalidomide was converted into 5-hydroxythalidomide in 32% of patients with advanced prostate cancer, while 4 cis-5’- hydroxythalidomide in the plasma of 48% AIPC patients [67]. Others have detected only some of the above hydroxylated metabolites after thalidomide therapy in different groups of human subjects treated with thalidomide, while others have detected no metabolites [58].
Fig.(3).
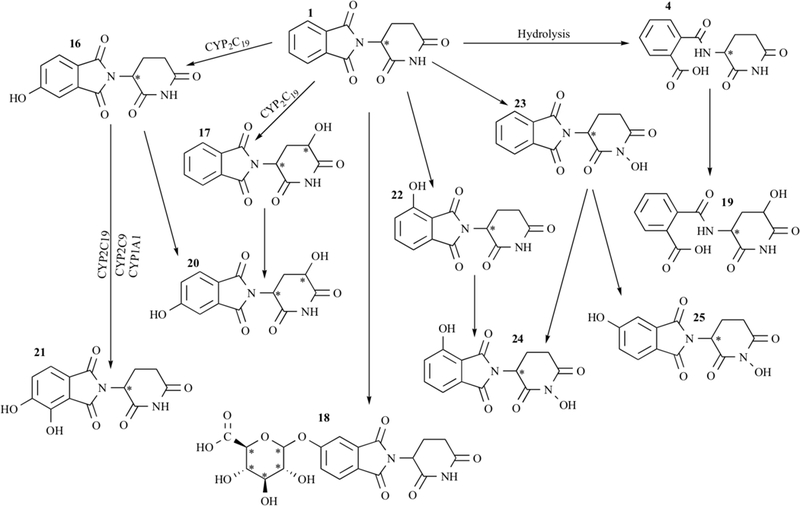
Thalidomide metabolites. Adapted from[55]. The names of all compounds are listed in Table 1alongside their respective numbers.
The polymorphic enzyme CYP2C19 is primarily responsible for 5- and 5’-hydroxylated metabolites of thalidomide in humans [68]. However, polymorphisms in CYP2C19 have not been related to inter-individual differences in metabolism or clinical outcome. CYP2B6 and CYP2C9 are also involved in the formation of cis-5 hydroxythalidomide but to a lesser extent. The 5-hydroxythali- domide was subsequently hydroxylated to 5,6-dihydroxy- thalidomide by CYP2C19, CYP2C9 and CYP1A1 in humans. Other metabolites have been proposed, including dihydroxylated metabolites, and a glucuronide conjugate of thalidomide that was identified in mouse urine and rabbit plasma [69]. The mechanism behind thalidomide activity is still poorly understood. It is clear however, that thalidomide must undergo enzymatic metabolism in order to exhibit certain activities, as listed in Table 1.
Table 1.
Activities of Metabolites and Hydrolysis Products
| Numbering* | Compound | Identification | Ref. |
TNF-Altering Activity |
Ref. |
Anti-Angiogenesis Activity |
Assay Type | Ref. |
Anti- Proliferative Activity |
Assay Type |
Ref. |
Tubulin Polymerization- Inhibitory Activity |
Ref. |
| 1 | Thalidomide | N/A | |||||||||||
| S(−) Thalidomide | chiral synthesis | inhibits | [88] | inhibits bFGF and VEGF | rabbit cornea neovascularization | [56] | |||||||
| blocks vascular sprout formation | tube formation assay | [78] | |||||||||||
| R(+) Thalidomide | chiral synthesis | no | tube formation assay | [78] | |||||||||
| Hydrolysis Products | |||||||||||||
| 4 | α-(o- carboxyben- zamido) glu- tarimide | Human urine, mouse urine, mouse plasma | [69, 89] | inactive | rat aortic ring assay | [90] | |||||||
| rabbit plasma and urine | [89] | ||||||||||||
| in vitro | [91] | ||||||||||||
| 5 | 2- phthalimidogluta- ramic acid (phthaloylglu- tamine) |
Human urine, mouse urine, mouse plasma | [69, 89] | inactive | rat aortic ring assay | [90] | |||||||
| rabbit plasma | [89] | ||||||||||||
| in vitro | [91] | ||||||||||||
| 6 | 4- phthalimidogluta- ramic acid (phthaloylisoglu- tamine) |
Human urine, mouse urine, mouse plasma | [69, 89] | inactive | rat aortic ring assay | [90] | |||||||
| rabbit plasma and urine | [89] | ||||||||||||
| in vitro | [91] | ||||||||||||
| 7 | 4-(o- carboxyben- zamido) gluta- ramic acid | mouse urine | [69, 89] | ||||||||||
| rabbit plasma and urine | [89] | ||||||||||||
| in vitro | [91] | ||||||||||||
| 8 | 2- phthalimidoglu- taric acid (phtha- loylglutamic acid) |
aqueous solution | [58] | inactive | rat aortic ring assay | [90] | YES (100% mean inhibition at 333uM) | HUVEC | [92] | ||||
| effects on blood vessel density | CAM assay | [92] | |||||||||||
| activity | rabbit cornea neovascularization | [56] | |||||||||||
| 9 | 2-(o- carboxyben- zamido) glutaric acid | aqueous solution | [58] | inactive | rat aortic ring assay | [90] | |||||||
| 10 | 2-(o- carboxyben- zamido) gluta- ramic acid | aqueous solution | [58] | ||||||||||
| 11 | 2- aminoglutaramic acid (glutamine) |
aqueous solution | [58] | ||||||||||
| 12 | 4- aminoglutaramic acid (isoglutamine) |
aqueous solution | [58] | ||||||||||
| 13 | α- aminoglutarimide |
aqueous solution | [58] | ||||||||||
| 14 | phthalic acid | aqueous solution | [58] | ||||||||||
| 15 | 2-aminoglutaric acid (glutamic acid) | aqueous solution | [58] | ||||||||||
| Metabolites | |||||||||||||
| 16 | 5- hydroxythalido- mide S-5- hydroxythalido- mide R-5- hydroxythalido- mide |
human plasma | [67] | no inhibition | rat aortic ring assay | [93] | moderate activity | [94] | |||||
| mouse urine | [69, 89] | no inhibition | human saphenous vein model | [93] | |||||||||
| rabbit plasma | [89] | no inhibition | endothelial cell tube formation assay | [93] | |||||||||
| in vitro | [68,91] | effects on blood vessel density | CAM assay | [92] | |||||||||
| activity | HUVEC tube formation assay | [95] | |||||||||||
| weak inhibition on vascular sprout formation, R enantiomer slightly more potent than S | preliminary? tube formation assay | [77] | |||||||||||
| 17 | cis,trans −5 hydroxythalido- mide | cis- enantiomer human plasma | [67] | inhibition at high concentration | rat aortic ring assay | [93] | |||||||
| mouse plasma and urine. | [69, 89] | no inhibition | human saphenous vein model | [93] | |||||||||
| rabbit plasma | [89] | no inhibition | endothelial cell tube formation assay | [93] | |||||||||
| in vitro | [68,91] | cis-5’- hydroxythalidomide: no inhibition | preliminary? tube formation assay | [78] | |||||||||
| 18 | thalidomide-5-O- glucuronide |
mouse urine | [69, 89] | ||||||||||
| rabbit plasma | [89] | ||||||||||||
| 19 | cis,trans-5 ‘- hydroxy-N-(o- carboxyben- zoyl)glutamic acid | mouse urine (proposed) | [69, 89] | moderate activity | [94] | ||||||||
| rabbit plasma | [89] | ||||||||||||
| in vitro (proposed) | [91] | ||||||||||||
| 20 | cis,trans-5,5’- dihydroxy thalidomide | in vitro (proposed) | [91] | ||||||||||
| 21 | 5,6- dihydroxy thalidomide | in vitro | [68] | ||||||||||
| 22 | 4- hydroxythalido- mide |
no inhibition | rat aortic ring assay | [93] | YES (100% mean inhibition at 333uM) | HUVEC | [92] | ||||||
| no inhibition | human saphenous vein model | [93] | |||||||||||
| no inhibition | endothelial cell tube formation assay | [93] | |||||||||||
| weak effects on blood vessel density | CAM assay | [92] | |||||||||||
| 23 | N- hydroxythalido- mide |
effects on blood vessel density | CAM assay | [92] | YES (87% mean inhibition at 333uM) | HUVEC | [92] | activity | [94] | ||||
| 24 | 5,N- dihydroxy thalidomide | effects on blood vessel density | CAM assay | [92] | YES (88% mean inhibition at 333uM) | HUVEC | [92] | ||||||
| 25 | 4,N- dihydroxy thalidomide | weak effects on blood vessel density | CAM assay | [92] | |||||||||
Influence of Chirality on Activity
Thalidomide has one chiral center, and the agent consists of a racemic mixture of S(−) and R(+) enantiomers, that inter-convert rapidly under physiological conditions. The enantiomers show differences in biological properties [70, 71], and are metabolized to a number of chiral and achiral metabolites that may have different activities, although only some of these metabolites have been studied in vitro [72]. The differences in efficacy of the enantiomers of thalidomide and thalidomide metabolites could reflect requirement for a specific possession at the binding site of target proteins or metabolizing enzymes [20], R(+)-thalidomide appears to be responsible for sedative effects [73, 74] while S(−)-thalidomide may be responsible for teratogenic effects [75, 76], The antiangiogenic properties of thalidomide in bFGF- and VEGF- induced corneal neovascularization model showed the S(−)-enantiomer to be more effective than the R(+)-enantiomer [56], That is consistent with data obtained from asymmetric synthesis of both enantiomers of the major thalidomide metabolites cis-5’-hydroxythalidomide and 5- hydroxythalidomide. The cis-5’-hydroxythalidomide metabolite also shows resistance to racemization and epimerization at physiological pH, while 5-hydroxythalidomide was also found to be con- figurationally more stable than thalidomide under the same conditions [77, 78].
Both enantiomers of thalidomide, racemic cis-5’-hydroxythalidomide, and both (3’R,5’S)cis-5’-hydroxythalidomide and (3’S, 5 ‘R)cis-5’-hydroxythalidomide have been tested for their ability to inhibit endothelial cell tube formation, providing some insight into the antiangiogenic capability of these compounds [77, 78], Racemic thalidomide and (S)-thalidomide blocked vascular sprout formation in high concentration, while the others failed to show antiangiogenesis activity [78]. However, the concentration needed to block vascular sprout formation in this experiment was well above the concentration range achieved in the plasma of patients treated with thalidomide [65]. Later experiments determined that racemic thalidomide and (S)-thalidomide blocked vascular sprout formation in high concentration, and (R)-thalidomide and both enantiomers of 5- hydroxythalidomide showed weak inhibitory activity, with the R enantiomer slightly more potent than S [77]. The in vitro antitumor activities of enantiomers of thalidomide and 5-hydroxythalidomide against KB cells from oral cancer, MCF-7 from breast cancer, and multiple myeloma cell lines, have also been evaluated [77]. The results revealed no significant differences between enantiomers, but 5-hydroxythalidomide showed anti-proli-ferative effects in KB and MCF-7 cells and inhibited multiple myeloma cell line growth markedly whereas thalidomide showed only weak antitumor activities. Thus, it appears that metabolism of thalidomide into 5- hydroxythalidomide is necessary for the antitumor activity of this agent, and that stereoisomers of thalidomide may only contribute to minor differences in activity. However, these experiments are limited by the elusiveness of the true mechanism of action of thalidomide, and these experiments may not have evaluated an, as yet, unknown activity. It is hoped that in vivo models will be useful in determining the activity of many of these compounds.
CLINICAL USE OF THALIDOMIDE IN PROSTATE CANCER
Clinical Trials
Thalidomide has been tested in men with stage DO prostate (defined as rising PSA following tumor resection and x-ray therapy (XRT), but with no evidence of metastasis [79]. Androgen deprivation therapy (ADT) with leuprolide was given for 6-months followed by randomization to either thalidomide or placebo until PSA progression. Following PSA progression, the study subjects were given ADT for another 6 months and men that had received thalidomide crossed over to the placebo arm and vice versa. Although the trial was stopped before reaching the accrual target, a clear trend was noted; PSA progression was delayed in men receiving thalidomide (15 months versus 9.6 months; P = 0.21), and individuals that crossed over to the thalidomide arm had a longer time to PSA progression than the placebo group (17.1 months versus 6.6 months; P = 0.0002). Although the overall survival benefit of thalidomide administered in early metastatic prostate cancer could not be evaluated, thalidomide appears to delay PSA progression in this disease.
More extensive research has been conducted evaluating thalidomide treatment in men with more advanced, androgen independent prostate cancer (AIPC). Thalidomide was initially administered to a cohort of 63 men with AIPC in an open-label Phase II clinical trial comparing low-dose (200 mg/d; n = 50) and high dose (up to 1200 mg/d; n = 13) thalidomide. In this trial, thalidomide therapy had minimal activity based on PSA progression measures (28% of patients had a >40% PSA decrease), and was not well tolerated in the high dose arm due to the development of toxicities, such as sedation and fatigue [29]. A similar study evaluating lOOmg of thalidomide at bedtime found that 37.5% of patients had a 48% PSA decrease [80]. However, when thalidomide was administered in combination with docetaxel for the treatment of metastatic AIPC (200 mg thalidomide at bedtime, plus 30mg/m2 docetaxel), the combination was well tolerated, and was found to significantly improve docetaxel therapy with an 16% greater PSA response over docetaxel alone [42], Moreover, when overall survival was evaluated after 18 months, the overall survival in the docetaxel group was 42.9%, while 68.2% of the patients receiving the combination were still alive [42], Based on these encouraging results, a clinical trial evaluating the combination of docetaxel, thalidomide, and estramustine was conducted [43], This combination was rather toxic, with 24 grade 3 or 4 toxicities associated with the regimen, however, 18 of 20 patients had a PSA decline >50%, and 10 of 20 patients with soft-tissue lesions achieved a partial response. Therefore, although a definitive Phase III trial has not yet been published, it appears that thalidomide significantly improves standard-of-care docetaxel therapy in AIPC.
Thalidomide Toxicity
Thalidomide treatment is associated with several different toxicities, many of which are dose limiting. The major dose limiting toxicity stemming from thalidomide treatment has historically been the development of sensory peripheral neuropathy, observed in approximately 30% of patients [13]. While peripheral neuropathy was not a major issue in men with prostate cancer being treated with thalidomide alone, constipation, dizziness, lightheadedness, edema, and fatigue, and mood changes were more common or severe [29], However, given that thalidomide treatment is more successful when combined with docetaxel, it is important to note that the combination of thalidomide and docetaxel is associated with more peripheral neuropathies than in docetaxel alone. Thalidomide also decreases the time taken to achieve a peripheral neuropathy in patients treated with both drugs [42, 81]. The combination also increased the incidence of thromboembolic events (0/25 patients on docetaxel alone versus 12/43 patients on the combination) that were cause of some concern, although no further events were seen after prophylactically administering low-molecular weight heparin [42].
Approximately 20% of men treated with combination bevaci- zumab, thalidomide, docetaxel, and prednisone developed osteonecrosis of the jaw (ONJ). However, the severity of ONJ was moderate in most patients, and the toxicity was associated with cotherapy with bisphosphonates that can lead to faulty bone remodeling. It appears that ONJ develops because the jaw is subject to significant microtrauma and is highly vascularized resulting in the concentration of bisphosphonates. When the vasculature in the bone becomes involuted, osteoclasts are not resorbed leading to acellularity and necrosis [82],
THALIDOMIDE ANALOGUES
Initially, thalidomide analogues were developed in order to increase the immunomodulatory activity and improve the safety profile of thalidomide. This effort led to the identification of several amino-phthaloyl-substituted thalidomide analogues, where an amino group was added to carbon 4 of the aromatic phthaloyl ring of thalidomide. This class of drugs was called the “IMiDs,” and lenalidomide (CC-5013; (RS)-3-(4-amino-l-oxo-3H-isoindol-2- yl)piperidine-2,6-dione), approved for use in multiple myeloma and certain myelodysplastic syndromes, has also been under investigation for the treatment of prostate cancer [83], CC-4047 (4-amino-2- (2,6-dioxopiperidin-3-yl)isoindole-l,3-dione; Fig. 1), has also undergone clinical testing for the treatment of metastatic prostate cancer although the final results of this study have yet to be published [84], Another class of thalidomide analogues, termed the selective cytokine inhibitory drugs (SelCIDs), have also been developed. These drugs only inhibit a few cytokines (e.g. TNFot), have little immunomodulatory activity, and appear to have the greatest utility as anti-inflammatory agents for the treatment of inflammatory conditions such as Crohn’s disease [12]. For these reasons, this review will only focus on the IMiDs.
Mechanism of Action
Although the true mechanism of action behind the thalidomide analogues is unknown, the iMIDs are very similar to thalidomide. The iMIDs are also strong immunomodulators, inhibiting IL 1(3, IL6, and IL12 production while increasing the production of IL10 [12]. The iMIDs were also designed to inhibit TNFa more effectively than thalidomide, and have strong anti-angiogenesis, and T- cell costimulatory activity, while unlike thalidomide, are also cytotoxic to certain tumor cells at biologically relevant concentrations [12, 85], However, the anti-angiogenic properties of the iMIDs are somewhat controversial as lenalidomide is not very active in the rat aortic ring model, although lenalidomide has an additive effect in combination with sunitinib and sorafenib [86, 87]. However, it is important to note that most of the efforts to elucidate the effects of the iMIDs has been worked out in hematological malignancies, while little research has been conducted the mechanism of action of these agents in solid tumors, such as prostate cancer.
The only published preclinical study to evaluate the effects of lenalidomide and CC-4047 in a PC3/PBMC coculture found that these agents increase natural killer (NK) and natural killer T (NKT) cell populations. The iMIDs were neither cytotoxic or cytostatic in PC-3 cells. However, when the iMIDS were evaluated in a PC3/PBMC coculture model, both lenalidomide and CC-4047 induced PC-3 apoptosis while CC-4047 was approximately 1.3 to 2 times more effective across the concentration ranges tested [85]. When docetaxel was added to the PC3/PBMC coculture in combination with the iMIDS, it was determined that docetaxel had an additive or synergistic effect on PC-3 apoptosis [85]. Therefore, while there are minimal published data evaluating the iMIDs in preclinical models of prostate cancer, the iMIDs may be quite useful-especially as cotherapy with docetaxel.
Clinical Use of iMIDs in Prostate Cancer
Although lenalidomide is approved for hematological diseases, dosing and toxicity information may not be relevant in individuals with solid tumors and the agent may also have a different toxicity profile. Thus, the study of lenalidomide as a potential therapeutic strategy for prostate cancer and other solid tumors is in its infancy. Indeed, only a single Phase I study has been published that has used lenalidomide in the context of metastatic prostate cancer. This study determined that lenalidomide has potential activity in prostate cancer, with 9 of 35 individuals with prostate cancer responding with stable disease following dose escalation of lenalidomide, and found that lenalidomide was generally well tolerated. PSA response has also been observed in men with AIPC treated with CC4047 [83]. Therefore, while the iMIDs have shown some promise in the treatment of prostate cancer, there is little preclinical or clinical data to determine whether or not the iMIDs will be more or less effective than thalidomide.
CONCLUSION
Thalidomide has been typically thought of as an antiangiogenic agent in the context of prostate cancer and other solid tumors. However, it is becoming increasingly clear that thalidomide actually changes the tumor microenvironment in many ways, and although the exact mechanism is unknown, thalidomide appears to also prevent inflammation within the prostate. The iMIDs were designed to increase the anti-tumor effects, and mitigate the side effects of thalidomide. While these drugs have had much success in the treatment of hematological diseases, such as multiple myeloma, their utility in the treatment of solid tumors is still unknown. Therefore, future efforts should focus on increasing the clinical utility of thalidomide and establishing a true mechanism of action behind this drug in order to better exploit its clear usefulness in prostate cancer. Preclinical, and early clinical investigations establishing a role for the iMIDs in the treatment of prostate cancer are also needed in order to determine whether or not the iMIDs will be useful in the treatment of solid tumors, such as prostate cancer.
ACKNOWLEDGEMENTS
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E*. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. *E.R.G.
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
REFERENCES
- [1].Jemal A; Siegel R; Ward E; Hao Y; Xu J; Murray T; Thun MJ Cancer statistics:2008. CA Cancer J. Clin., 2008, 58(2), 71–96. [DOI] [PubMed] [Google Scholar]
- [2].Petrylak DP; Tangen CM; Hussain MH; Lara PN Jr.; Jones JA; Taplin ME; Burch PA; Berry D; Moinpour C; Kohli M; Benson MC; Small EJ; Raghavan D; Crawford ED Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med, 2004,351(15), 1513–20. [DOI] [PubMed] [Google Scholar]
- [3].Tannock IF; de Wit R; Berry WR; Horti J; Pluzanska A; Chi KN; Oudard S; Theodore C; James ND; Turesson I; Rosenthal MA; Eisenberger MA Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351(15), 1502–12. [DOI] [PubMed] [Google Scholar]
- [4].Lenz W; Pfeiffer R; Kosenow W; Hayman D Thalidomide and [26] congenital abnormalities. Lancet, 1962, 27945–46. [Google Scholar]
- [5].McBride W Thalidomide and congenital abnormalities. Lancet, 1961, [Google Scholar]
- [6].Sheskin J Thalidomide in the treatment of lepra reactions. Clin. Pharmacol. Ther., 1965, 6303–6. [DOI] [PubMed] [Google Scholar]
- [7].Sampaio EP; Samo EN; Galilly R; Cohn ZA; Kaplan G Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med, 1991, 173(3), 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Knobloch J; Shaughnessy JD Jr.; Ruther U Thalidomide in- [29] duces limb deformities by perturbing the Bmp/Dkkl/Wnt signaling pathway. FASEBJ., 2007, 21(7), 1410–21. [DOI] [PubMed] [Google Scholar]
- [9].Hansen JM; Harris C A novel hypothesis for thalidomide- induced limb teratogenesis: redox misregulation of the NF-kappaB pathway. Antioxid. Redox Signal, 2004, 6(1), 1–14. [DOI] [PubMed] [Google Scholar]
- [10].Therapontos C; Erskine L; Gardner ER; Figg WD; Varges-son N Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl. Acad. Sci. USA 2009, 106(21), 8573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].D’Amato RJ; Loughnan MS; Flynn E; Folkman J Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA, 1994, 91(9), 4082–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aragon-Ching JB; Li H; Gardner ER; Figg WD Thalidomide analogues as anticancer drugs. Recent Pat. Anticancer Drug Discov., 2007, 2(2), 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bamias A; Dimopoulos MA Thalidomide and immunomodulatory drugs in the treatment of cancer. Expert Opin. Investig. Drugs,2005, 14(1), 45–55. [DOI] [PubMed] [Google Scholar]
- [14].Haverkamp J; Charbonneau B; Ratliff TL Prostate inflammation and its potential impact on prostate cancer: a current review. J. CellBiochem, 2008,103(5), 1344–53. [DOI] [PubMed] [Google Scholar]
- [15].Heinrich PC; Behrmann I; Haan S; Hermanns HM; Muller Newen G; Schaper F Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J, 2003, 374(Pt 1), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ko S; Shi L; Kim S; Song CS; Chatterjee B Interplay of nuclear factor-kappaB and B-myb in the negative regulation of androgen receptor expression by tumor necrosis factor alpha. Mol. Endocrinol., 2008, 22(2), 273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maxwell PJ; Gallagher R; Seaton A; Wilson C; Scullin P;Pettigrew J; Stratford IJ; Williams KJ; Johnston PG; Waugh DJ HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene, 2007, 26(52), 7333–45. [DOI] [PubMed] [Google Scholar]
- [18].Royuela M; Rodriguez-Berriguete G; Fraile B; Paniagua R TNF-alpha/IL-1/NF-kappaB transduction pathway in human cancer prostate. Histol. Histopathol, 2008, 23(10), 1279–90. [DOI] [PubMed] [Google Scholar]
- [19].Daniel TO; Liu H; Morrow JD; Crews BC; Mamett LJ Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res., 1999, 59(18),4574–7. [PubMed] [Google Scholar]
- [20].Macpherson GR; Franks M; Tomoaia-Cotisel A; Ando Y;Price DK; Figg WD Current status of thalidomide and its role in the treatment of metastatic prostate cancer. Crit. Rev. Oncol. Hematol., 2003, 46 (Suppl), S49–57. [DOI] [PubMed] [Google Scholar]
- [21].Ng SS; MacPherson GR; Gutschow M; Eger K; Figg WD Antitumor effects of thalidomide analogs in human prostate cancer xenografts implanted in immunodeficient mice. Clin. Cancer Res, 2004, 10(12 Pt 1), 4192–7. [DOI] [PubMed] [Google Scholar]
- [22].Balkwill F Tumor necrosis factor or tumor promoting factor? Cytokine. Growth Factor Rev., 2002,13(12), 135–41. [DOI] [PubMed] [Google Scholar]
- [23].Michalaki V; Syrigos K; Charles P; Waxman J Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer, 2004, 90(12), 2312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wise GJ; Marella VK; Talluri G; Shirazian D Cytokine variations in patients with hormone treated prostate cancer. J. Urol., 2000,164(3 Pt 1), 722–5. [DOI] [PubMed] [Google Scholar]
- [25].Li SP; Meng SY; Li R Clinical evaluation of four cytokines in serum and pro static fluid in chronic abacterial prostatitis. Zhonghua Nan Ke Xue, 2006, 12(1), 25–7. [PubMed] [Google Scholar]
- [26].Perambakam SM; Srivastava R; Peace DJ Distinct cytokine patterns exist in peripheral blood mononuclear cell cultures of patients with prostate cancer. Clin. Immunol., 2005, 117(1), 94–9. [DOI] [PubMed] [Google Scholar]
- [27].Pfitzenmaier J; Vessella R; Higano CS; Noteboom JL; Wallace D Jr.; Corey E Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer, 2003, 97(5), 1211–6. [DOI] [PubMed] [Google Scholar]
- [28].Efstathiou E; Troncoso P; Wen S; Do KA; Pettaway CA; Pisters LL; McDonnell TJ; Logothetis CJ Initial modulation of the tumor microenvironment accounts for thalidomide activity in prostate cancer. Clin. Cancer Res., 2007, 13(4), 1224–31. [DOI] [PubMed] [Google Scholar]
- [29].Figg WD; Dahut W; Duray P; Hamilton M; Tompkins A; Steinberg SM; Jones E; Premkumar A; Linehan WM; Floeter MK; Chen CC; Dixon S; Kohler DR; Kruger EA; Gubish E; Pluda JM; Reed E A randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen- independent prostate cancer. Clin. Cancer Res., 2001, 7(7), 1888–93. [PubMed] [Google Scholar]
- [30].Leonard GD; Dahut WL; Gulley JL; Arlen PM; Figg WD Docetaxel and thalidomide as a treatment option for androgen- independent, nonmetastatic prostate cancer. Rev. Urol., 2003, 5 (Suppl), 3S65–70. [PMC free article] [PubMed] [Google Scholar]
- [31].Kurokouchi K; Kambe F; Yasukawa K; Izumi R; Ishiguro N; Iwata H; Seo H TNF-alpha increases expression of IL-6 and ICAM-1 genes through activation of NF-kappaB in osteoblast-like ROS 17/2.8 cells. J. Bone Miner. Res., 1998,13(8), 1290–9. [DOI] [PubMed] [Google Scholar]
- [32].Culig Z; Steiner H; Bartsch G; Hobisch A Interleukin-6 regulation of prostate cancer cell growth. J. Cell Biochem., 2005, 95(3), 497–505. [DOI] [PubMed] [Google Scholar]
- [33].Stark JR; Li H; Kraft P; Kurth T; Giovannucci EL; Stampfer MJ; Ma J; Mucci LA Circulating prediagnostic inter- leukin-6 and C-reactive protein and prostate cancer incidence and mortality. Int. J. Cancer, 2009, 124(11), 2683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rowland TL; McHugh SM; Deighton J; Dearman RJ; Ewan PW; Kimber I Differential regulation by thalidomide and dex- amethasone of cytokine expression in human peripheral blood mononuclear cells, lmmunopharmacology, 1998, 40( 1), 11–20. [DOI] [PubMed] [Google Scholar]
- [35].Inoue K; Slaton JW; Eve BY; Kim SJ; Perrotte P; Balbay MD; Yano S; Bar-Eli M; Radinsky R; Pettaway CA; Dinney CP Interleukin 8 expression regulates tumorigenicity and me- tastases in androgen-independent prostate cancer. Clin. Cancer Res., 2000, 6(5), 2104–19. [PubMed] [Google Scholar]
- [36].Uehara H; Troncoso P; Johnston D; Bucana CD; Dinney C; Dong Z; Fidler IJ; Pettaway CA Expression of interleukin-8 gene in radical prostatectomy specimens is associated with advanced pathologic stage. Prostate, 2005, 64(1), 40–9. [DOI] [PubMed] [Google Scholar]
- [37].Wang JM; Taraboletti G; Matsushima K; Van Damme J; Mantovani A Induction of haptotactic migration of melanoma cells by neutrophil activating protein/interleukin-8. Biochem. Bio- phys. Res. Commun., 1990, 169(1), 165–70. [DOI] [PubMed] [Google Scholar]
- [38].Mercurio F; Manning AM Multiple signals converging on NF- kappaB. Curr. Opin. Cell Biol., 1999, 11(2), 226–32. [DOI] [PubMed] [Google Scholar]
- [39].Keifer JA; Guttridge DC; Ashbumer BP; Baldwin AS Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J. Biol. Chem., 2001, 276(25), 22382–7. [DOI] [PubMed] [Google Scholar]
- [40].Muenchen HJ; Lin DL; Walsh MA; Keller ET; Pienta KJ Tumor necrosis factor-alpha-induced apoptosis in prostate cancer cells through inhibition of nuclear factor-kappaB by an IkappaBal- pha “super-repressor”. Clin. Cancer Res., 2000, 6(5), 1969–77. [PubMed] [Google Scholar]
- [41].Sunderkotter C; Steinbrink K; Goebeler M; Bhardwaj R; Sorg C Macrophages and angiogenesis. J. Leukoc. Biol., 1994, 55(3), 410–22. [DOI] [PubMed] [Google Scholar]
- [42].Dahut WL; Gulley JL; Arien PM; Liu Y; Fedenko KM; Steinberg SM; Wright JJ; Pames H; Chen CC; Jones E; Parker CE; Linehan WM; Figg WD Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer. J. Clin. Oncol., 2004, 22(13), 2532–9. [DOI] [PubMed] [Google Scholar]
- [43].Figg WD; Li H; Sissung T; Retter A; Wu S; Gulley JL; Arlen P; Wright JJ; Pames H; Fedenko K; Latham L; Steinberg SM; Jones E; Chen C; Dahut W Pre-clinical and clinical evaluation of estramustine, docetaxel and thalidomide combination in androgen-independent prostate cancer. BJU Int., 2007, 99(5), 1047–55. [DOI] [PubMed] [Google Scholar]
- [44].Kirschenbaum A; Liu X; Yao S; Levine AC The role of cyclooxygenase-2 in prostate cancer. Urology, 2001, 58(2 Suppl 1),127–31. [DOI] [PubMed] [Google Scholar]
- [45].Fujita J; Mestre JR; Zeldis JB; Subbaramaiah K; Dannenberg AJ Thalidomide and its analogues inhibit lipopolysaccha- ride-mediated Iinduction of cyclooxygenase-2. Clin. Cancer Res.,2001, 7(11), 3349–55. [PubMed] [Google Scholar]
- [46].Jin SH; Kim TI; Yang KM; Kim WH Thalidomide destabilizes cyclooxygenase-2 mRNA by inhibiting p38 mitogen-activated protein kinase and cytoplasmic shuttling of HuR. Eur. J. Pharmacol., 2007, 558(1–3), 14–20. [DOI] [PubMed] [Google Scholar]
- [47].Warfel NA; Lepper ER; Zhang C; Figg WD; Dennis PA Importance of the stress kinase p38alpha in mediating the direct cytotoxic effects of the thalidomide analogue, CPS49, in cancer cells and endothelial cells. Clin. Cancer Res., 2006, 12(11 Pt 1), 3502–9. [DOI] [PubMed] [Google Scholar]
- [48].Smyth MJ; Hayakawa Y; Takeda K; Yagita H New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer, 2002, 2(11), 850–61. [DOI] [PubMed] [Google Scholar]
- [49].Gannon PO; Poisson AO; Delvoye N; Lapointe R; Mes- Masson AM; Saad F Characterization of the intra-pro static immune cell infiltration in androgen-deprived prostate cancer patients.J. Immunol. Methods., 2009, 348( 1–2), 9–17. [DOI] [PubMed] [Google Scholar]
- [50].Galustian C; Dalgleish A Lenalidomide: a novel anticancer drug with multiple modalities. Expert Opin. Pharmacother., 2009,10( 1), 125–33. [DOI] [PubMed] [Google Scholar]
- [51].Wang W; Bergh A; Damber JE Chronic inflammation in benign prostate hyperplasia is associated with focal upregulation of cyclooxygenase-2, Bcl-2, and cell proliferation in the glandular epithelium. Prostate, 2004, 61(1), 60–72. [DOI] [PubMed] [Google Scholar]
- [52].Weidner N; Carroll PR; Flax J; Blumenfeld W; Folkman J Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol, 1993,143(2), 401–9. [PMC free article] [PubMed] [Google Scholar]
- [53].Siegal JA; Yu E; Brawer MK Topography of ne o vascularity in human prostate carcinoma. Cancer, 1995, 75(10), 2545–51. [DOI] [PubMed] [Google Scholar]
- [54].Aragon-Ching JB; Dahut WL The role of angiogenesis inhibitors in prostate cancer. Cancer J., 2008, 14(1), 20–5. [DOI] [PubMed] [Google Scholar]
- [55].Joseph IB; Isaacs JT Macrophage role in the anti-prostate cancer response to one class of antiangiogenic agents. J. Natl. Cancer Inst., 1998, 90(21), 1648–53. [DOI] [PubMed] [Google Scholar]
- [56].Kenyon BM; Browne F; D’Amato RJ Effects of thalidomide and related metabolites in a mouse comeal model of neovascularization. Exp. Eye Res, 1997, 64(6), 971–8. [DOI] [PubMed] [Google Scholar]
- [57].Vacca A; Scavelli C; Montefusco V; Di Pietro G; Neri A; Mattioli M; Bicciato S; Nico B; Ribatti D; Dammacco F;Corradini P Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma.J. Clin. Oncol., 2005, 23(23), 5334–46. [DOI] [PubMed] [Google Scholar]
- [58].Lepper ER; Smith NF; Cox MC; Scripture CD; Figg WD Thalidomide metabolism and hydrolysis: mechanisms and implications. Curr. Drug Metab., 2006, 7(6), 677–85. [DOI] [PubMed] [Google Scholar]
- [59].Yabu T; Tomimoto H; Taguchi Y; Yamaoka S; Igarashi Y;Okazaki T Thalidomide-induced antiangiogenic action is mediated by ceramide through depletion of VEGF receptors, and is antagonized by sphingosine-1-phosphate. Blood, 2005, 106(1), 125–34. [DOI] [PubMed] [Google Scholar]
- [60].Li H; Raia V; Bertolini F; Price DK; Figg WD Circulating endothelial cells as a therapeutic marker for thalidomide in combined therapy with chemotherapy drugs in a human prostate cancer model. BJU. Int., 2008,101(7), 884–8. [DOI] [PubMed] [Google Scholar]
- [61].Fujita K; Asami Y; Murata E; Akita M; Kaneko K Effects of thalidomide, cytochrome P-450 and TNF-alpha on angiogenesis in a three-dimensional collagen gel-culture. Okajimas. Folia. Anat. Jpn.,2002, 79(4), 101–6. [DOI] [PubMed] [Google Scholar]
- [62].De AK; Kodys K; Puyana JC; Fudem G; Savoie P; Miller- Graziano CL Elevated IL-8 production by trauma patients’ monocytes is associated with elevated secretion of TNF alpha. Shock,1995, 4(3), 171–7. [DOI] [PubMed] [Google Scholar]
- [63].Ferrer FA; Miller LJ; Andrawis RI; Kurtzman SH; Albertsen PC; Laudone VP; Kreutzer DL Angiogenesis and prostate cancer: in vivo and in vitro expression of angiogenesis factors by prostate cancer cells. Urology, 1998, 51(1), 161–7. [DOI] [PubMed] [Google Scholar]
- [64].Wilmer JL; Luster MI Chemical induction of interleukin-8, a pro inflammatory chemokine, in human epidermal keratinocyte cultures and its relation to cytogenetic toxicity. Cell Biol. Toxicol.,1995. 11(1), 37–50. [DOI] [PubMed] [Google Scholar]
- [65].Figg WD; Raje S; Bauer KS; Tompkins A; Venzon D; Berg an R; Chen A; Hamilton M; Pluda J; Reed E Pharmacokinetics of thalidomide in an elderly prostate cancer population. J. Pharm. Sci., 1999, 88(1), 121–5. [DOI] [PubMed] [Google Scholar]
- [66].Jonsson NA Chemical structure and teratogenic properties. 3. A review of available data on structure-activity relationships and mechanism of action of thalidomide analogues. Acta. Pharm. Suec., 1972, 9(6), 521–42. [PubMed] [Google Scholar]
- [67].Ando Y; Price DK; Dahut WL; Cox MC; Reed E; Figg WD Pharmacogenetic associations of CYP2C19 genotype with in vivo metabolisms and pharmacological effects of thalidomide. Cancer Biol. Ther., 2002, 1(6), 669–73. [DOI] [PubMed] [Google Scholar]
- [68].Ando Y; Fuse E; Figg WD Thalidomide metabolism by the CYP2C subfamily. Clin. Cancer Res., 2002, 8(6), 1964–73. [PubMed] [Google Scholar]
- [69].Lu J; Palmer BD; Kestell P; Browett P; Baguley BC; Muller G; Ching LM Thalidomide metabolites in mice and patients with multiple myeloma. Clin. Cancer Res., 2003, 9(5), 1680–8. [PubMed] [Google Scholar]
- [70].Diggle GE Thalidomide: 40 years on. Int. J. Clin. Pract., 2001, 55(9), 627–31. [PubMed] [Google Scholar]
- [71].Eriksson T; Bjorkman S; Hoglund P Clinical pharmacology of thalidomide. Eur. J. Clin. Pharmacol., 2001, 57(5), 365–76. [DOI] [PubMed] [Google Scholar]
- [72].Bosch ME; Sanchez AJ; Rojas FS; Ojeda CB Recent advances in analytical determination of thalidomide and its metabolites. J. Pharm. Biomed. Anal., 2008, 46(1), 9–17. [DOI] [PubMed] [Google Scholar]
- [73].Eriksson T; Bjorkman S; Roth B; Hoglund P Intravenous formulations of the enantiomers of thalidomide: pharmacokinetic and initial pharmacodynamic characterization in man. J. Pharm. Pharmacol., 2000, 52(7), 807–17. [DOI] [PubMed] [Google Scholar]
- [74].Hoglund P; Eriksson T; Bjorkman S A double-blind study of the sedative effects of the thalidomide enantiomers in humans. J. Pharmacokinet. Biopharm., 1998, 26(4), 363–83. [DOI] [PubMed] [Google Scholar]
- [75].Blaschke G; Kraft HP; Fickentscher K; Kohler F [Chromatographic separation of racemic thalidomide and teratogenic activity of its enantiomers (author’s transl)]. Arzneimittelforschung, 1979,29(10), 1640–2. [PubMed] [Google Scholar]
- [76].Heger W; Schmahl HJ; Klug S; Felies A; Nau H; Merker HJ; Neubert D Embryotoxic effects of thalidomide derivatives in the non-human primate callithrix jacchus. IV. Teratogenicity of micrograms/kg doses of the EM 12 enantiomers. Teratog. Carcinog. Mutagen, 1994,14(3), 115–22. [DOI] [PubMed] [Google Scholar]
- [77].Yamamoto T; Shibata N; Sukeguchi D; Takashima M; Nakamura S; Torn T; Matsunaga N; Hara H; Tanaka M; Obata T; Sasaki T Synthesis, configurational stability and stereochemical biological evaluations of (S)- and (R)-5-hydroxythalidomides. Bioorg. Med. Chem. Lett., 2009, 19(14), 3973–6. [DOI] [PubMed] [Google Scholar]
- [78].Yamamoto T; Shibata N; Takashima M; Nakamura S; Torn T; Matsunaga N; Hara H Enzymatic resolution and evaluation of enantiomers of cis-5’-hydroxythalidomide. Org. Biomol. Chem,2008. 6(9), 1540–3. [DOI] [PubMed] [Google Scholar]
- [79].Figg WD; Hussain MH; Gulley JL; Arlen PM; Aragon- Ching JB; Petrylak DP; Higano CS; Steinberg SM; Chatta GS;Pames H; Wright JJ; Sartor O; Dahut WL A doubleblind randomized crossover study of oral thalidomide versus placebo for androgen dependent prostate cancer treated with intermittent androgen ablation. J. Urol, 2009, 181(3), 1104–13; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Drake MJ; Robson W; Mehta P; Schofield I; Neal DE; Leung HY An open-label phase II study of low-dose thalidomide in androgen-independent prostate cancer. Br. J. Cancer, 2003, 88(6), 822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sissung TM; Baum CE; Deeken J; Price DK; Aragon-Ching J; Steinberg SM; Dahut W; Sparreboom A; Figg WD ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin. Cancer Res., 2008, 14(14), 4543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Aragon-Ching JB; Ning YM; Chen CC; Latham L; Guadagnini JP; Gulley JL; Arlen PM; Wright JJ; Parnes H; Figg WD; Dahut WL Higher incidence of Osteonecrosis of the Jaw (ONJ) in patients with metastatic castration resistant prostate cancer treated with anti-angiogenic agents. Cancer Invest.,2008. 27(2), 221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dahut WL; Aragon-Ching JB; Woo S; Tohnya TM; Gulley JL; Arlen PM; Wright JJ; Ventiz J; Figg WD Phase I study of oral lenalidomide in patients with refractory metastatic cancer. J. Clin. Pharmacol., 2009, 49(6), 650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Floissac M; Knight R; Levin LM; Glode ED; Crawford ED Phase II Study of CC-4047 in Patients (pts) with Metastatic Hormone-Refractory Prostate Cancer (HRPC). J. Clin. Oncol., 2005, 23(16S, Part I of II), [Google Scholar]
- [85].Zhu D; Corral LG; Fleming YW; Stein B Immunomodulatory drugs Revlimid (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol. Immunother., 2008, 57(12), 1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Blansfield JA; Caragacianu D; Alexander HR 3rd; Tangrea MA; Morita SY; Lorang D; Schafer P; Muller G; Stirling D; Royal RE; Libutti SK Combining agents that target the tumor microenvironment improves the efficacy of anticancer therapy. Clin. Cancer Res., 2008,14(1), 270–80. [DOI] [PubMed] [Google Scholar]
- [87].Mangiameli DP; Blansfield JA; Kachala S; Lorang D;Schafer PH; Muller GW; Stirling DI; Libutti SK Combination therapy targeting the tumor microenvironment is effective in a model of human ocular melanoma. J. Transl. Med., 2007, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wnendt S; Finkam M; Winter W; Ossig J; Raabe G; Zwin- genberger K Enantioselective inhibition of TNF-alpha release by thalidomide and thalidomide-analogues. Chirality, 1996, 8(5), 390–6. [DOI] [PubMed] [Google Scholar]
- [89].Chung F; Lu J; Palmer BD; Kestell P; Browett P; Baguley BC; Tingle M; Ching LM Thalidomide pharmacokinetics and metabolite formation in mice, rabbits, and multiple myeloma patients. Clin. Cancer Res., 2004,10(17), 5949–56. [DOI] [PubMed] [Google Scholar]
- [90].Lepper ER; Ng SS; Gutschow M; Weiss M; Hauschildt S; Hecker TK; Luzzio FA; Eger K; Figg WD Comparative molecular field analysis and comparative molecular similarity indices analysis of thalidomide analogues as angiogenesis inhibitors. J. Med. Chem., 2004, 47(9), 2219–27. [DOI] [PubMed] [Google Scholar]
- [91].Lu J; Helsby N; Palmer BD; Tingle M; Baguley BC; Kestell P; Ching LM Metabolism of thalidomide in liver micro-somes of mice, rabbits, and humans. J. Pharmacol. Exp. Ther., 2004,310(2), 571–7. [DOI] [PubMed] [Google Scholar]
- [92].Marks MG; Shi J; Fry MO; Xiao Z; Trzyna M; Pokala V; Ihnat MA; Li PK Effects of putative hydroxylated thalidomide metabolites on blood vessel density in the chorioallantoic membrane (CAM) assay and on tumor and endothelial cell proliferation. Biol. Pharm. Bull., 2002, 25(5), 597–604. [DOI] [PubMed] [Google Scholar]
- [93].Price DK; Ando Y; Kruger EA; Weiss M; Figg WD 5’-OH- thalidomide, a metabolite of thalidomide, inhibits angiogenesis. Ther. DrugMonit, 2002, 24( 1), 104–10. [DOI] [PubMed] [Google Scholar]
- [94].Inatsuki S; Noguchi T; Miyachi H; Oda S; Iguchi T; Kizaki M; Hashimoto Y; Kobayashi H Tubulin-polymerization inhibitors derived from thalidomide. Bioorg. Med. Chem. Lett., 2005, 15(2), 321–5. [DOI] [PubMed] [Google Scholar]
- [95].Noguchi T; Fujimoto H; Sano H; Miyajima A; Miyachi H; Hashimoto Y Angiogenesis inhibitors derived from thalidomide. Bioorg. Med. Chem. Lett., 2005, 15(24), 5509–13. [DOI] [PubMed] [Google Scholar]