

Abstract
Annexin proteins function as Ca2+-dependent regulators of membrane trafficking and repair that may also modulate membrane curvature. Here, using high-resolution confocal imaging, we report that the intestine-specific annexin A13 (ANX A13) localizes to the tips of intestinal microvilli and determined the crystal structure of the ANX A13a isoform to 2.6 Å resolution. The structure revealed that the N terminus exhibits an alternative fold that converts the first two helices and the associated helix–loop–helix motif into a continuous α-helix, as stabilized by a domain-swapped dimer. We also found that the dimer is present in solution and partially occludes the membrane-binding surfaces of annexin, suggesting that dimerization may function as a means for regulating membrane binding. Accordingly, as revealed by in vitro binding and cellular localization assays, ANX A13a variants that favor a monomeric state exhibited increased membrane association relative to variants that favor the dimeric form. Together, our findings support a mechanism for how the association of the ANX A13a isoform with the membrane is regulated.
Keywords: annexin, protein structure, protein folding, membrane fusion, oligomerization, calcium regulation, intestinal microvilli, membrane curvature, structure-function
Introduction
Annexins (ANXs)5 comprise a large superfamily of proteins that promote Ca2+-dependent modulation of membrane curvature (1). This fundamental process underlies biological activities such as membrane organization, membrane repair, the separation of membranes during cell division, and formation of the multivesicular body (2, 3). A mechanism for how annexin proteins influence membrane curvature was suggested by the crystal structure of ANX A5 (4), which revealed a core fold of four separate four-helix bundles that form a distinctively curved, convex surface. Subsequent annexin structures identified that each of the four helical bundles is associated with up to three Ca2+ ions on the convex surface (5, 6). These Ca2+ ions bind to conserved clusters of backbone carbonyls and acidic side chains (7, 8). Cocrystallization of ANX A5 with Ca2+ and phosphoserine further identified a cleft within the C-terminal helical bundle that could accommodate lipid head groups, with Ca2+ bridging the interaction between phosphoserine and the annexin protein (5). The interaction between the membrane and this convex surface was proposed to physically adjust the membrane curvature to conform to the annexin surface (9), a function proposed to be conserved across annexin proteins.
ANX A136 is transcribed as two isoforms (A13a and A13b) that were originally identified in a screen for myristoylated intestine-specific genes (10). Cellular localization studies of ANX A13 homologs from rabbit (11) and fluke (12) confirmed the narrow intestine-specific expression pattern. More recently, RT-PCR of two human colorectal carcinoma cell lines showed that both the ANX A13a and ANX A13b isoforms are expressed in cells (13). The enrichment of ANX A13 in vesicles shedding from the lumenal tips of intestinal microvilli (14) suggests a possible biological role in the creation of these exosome-like vesicles.
In exploring the function of ANX A13, one unusual biochemical property particularly stands out. Although ANX A13 is proposed as the common ancestor of mammalian annexins (15), it is the only annexin to be N-terminally myristoylated. As assessed in the ANX A13b isoform, this post-translational lipidation allows constitutive, Ca2+-independent association with membranes (16). This result initially seemed paradoxical because it implied that ANX A13 isoforms localize to the membrane without using the canonical membrane-remodeling surface. An alternative suggestion is that the membrane-remodeling surface of ANX A13 does not function in high-affinity membrane binding. However, it is also possible that both the myristoylation and the canonical membrane-binding surface facilitate membrane interactions cooperatively and/or under different biological situations. The interplay between these two possible mechanisms of membrane interaction has not been evaluated. Here, we combine X-ray crystallography and biochemical measurements with localization studies in tissues and cells to explore structure-function relationships in ANX A13a. These findings identify new mechanisms controlling annexin membrane association and support a model where the canonical membrane-binding surface is functionally relevant in ANX A13 isoforms.
Results
In vivo localization of ANX A13 in mouse intestine shows preference toward brush border
Prior studies used immunolabeling and fluorescence microscopy to reveal that ANX A13 exhibits prominent enrichment at the apical surface of intestinal epithelial cells (10–12). We used high-resolution confocal imaging of tissue sections to more closely examine the distribution of ANX A13 in this tissue; the antibody used for these labeling studies does not differentiate between ANX A13a and ANX A13b. Confocal imaging showed a strong signal corresponding to the apical brush border (Fig. 1), with striking punctate enrichment that mapped to the highly curved membranes at the tips of microvilli. Microvillar tip labeling makes biological sense given that a previous proteomic analysis of brush border–derived lumenal vesicles (14) identified ANX A13 as a major cargo. Because these lumenal vesicles are predicted to have intestinal alkaline phosphatase (IAP) as a major cargo, we asserted that ANX A13 and IAP colocalize, which we confirmed with costaining (Fig. 1).
Figure 1.

Localization of ANX A13 in mouse intestinal tissue. Laser scanning confocal image of mouse small intestine tissue section labeled with anti-ANX A13 (green); anti-IAP, a protein that is enriched in luminal vesicles (magenta); and phalloidin to highlight F-actin (blue). Panels at right show higher magnification views from region marked Zoom. Arrowheads in merge panel mark puncta with high levels of ANX A13 and IAP colocalization.
Structure of an ANX A13a dimer reveals an elongated α-helix
Crystals of the nonmyristoylated ANX A13a were grown in the absence of Ca2+ and diffracted to 2.6 Å resolution (Fig. 2 and Table 1). Reasonable electron density was apparent for the residues corresponding to the annexin core fold (residues 13–316). In contrast, the N-terminal regulatory region, including residues 2–12 and the canonical site of myristate attachment at Gly-2, was disordered (Fig. 2, A and B).
Figure 2.
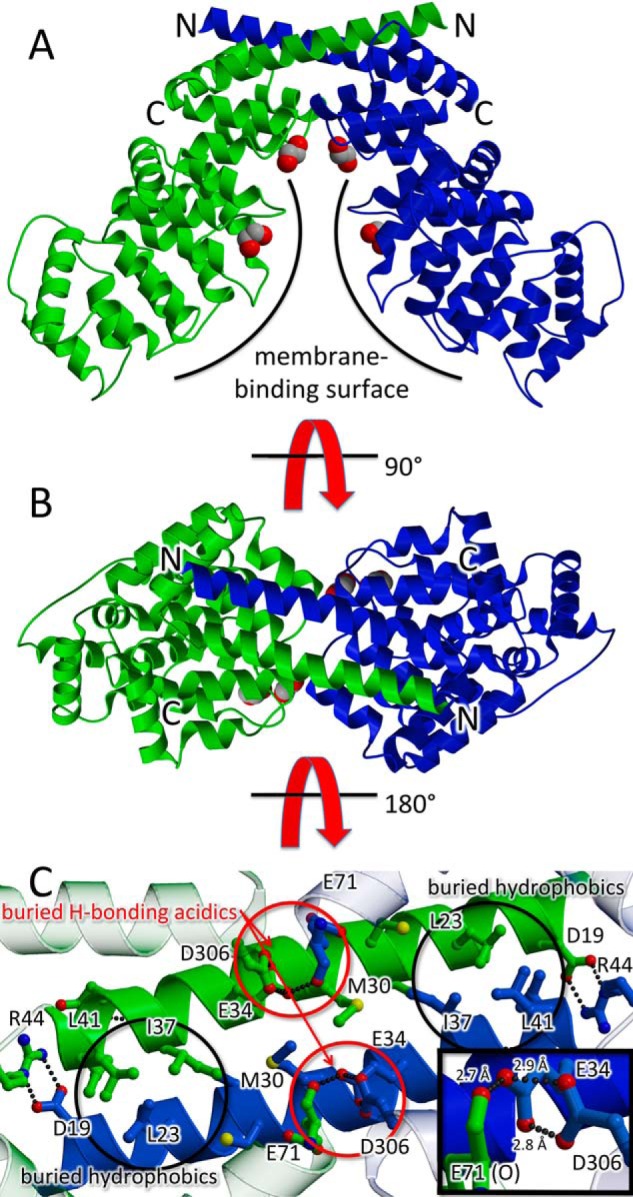
Domain swapped dimer of ANX A13a. In the ribbon diagram, one protomer is colored blue, one protomer is colored green, and the N and C termini are labeled. Putative ethylene glycol molecules from the cryoprotectant that are bound to the predicted lipid sites are shown as space-filling models. The two views are separated by 90°. A, view of the domain-swapped dimer perpendicular to the molecular 2-fold axis. The membrane-binding surface is marked with a curved line. Access to the predicted membrane-binding surface is occluded by dimer formation. B, rotation of the view by 90° shows the domain-swapped dimer down the molecular 2-fold axis. C, interactions of the extended helices. The view is from the protein interior such to highlight buried side chains. This identifies hydrophobic interactions (black circles) and six unusual hydrogen bonds between acidic residues (three in each of the red circles). The acidic hydrogen bonding would suggest that Glu-34 and Asp-306 share at least two protons. The inset highlights these hydrogen-bonds; the view of the inset is rotated 30° around the y axis as compared with the rest of the panel.
Table 1.
Crystallographic data collection and refinement statistics
The values in parentheses are for the highest resolution shell. No reflections were excluded from the refinement.
| Data collection | |
| PDB entry | 6B3I |
| SBGrid entrya | 353 |
| Beamline | 21-ID-G |
| Resolution | 50.0–2.6 Å (2.64–2.60 Å) |
| Completeness | 98.5% (98.5%) |
| I/σ | 15.6 (1.9) |
| Rsym | 0.053 (0.448) |
| Rpim | 0.039 (0.334) |
| CC½ | 0.758 |
| Number of reflections | 126,525 |
| Unique reflections | 49,419 |
| Rfree reflections | 2,485 |
| Refinement | |
| Resolution | 50.0–2.6 Å (2.68–2.60 Å) |
| Rcryst | 0.187 (0.283) |
| Rfree | 0.225 (0.316) |
| Number of atoms | 7,144 |
| RMS bonds | 0.002 Å |
| RMS angles | 0.49° |
| Average B-factor (protein) | 79.5 Å2 |
| Average B- factor (solvent) | 66.1 Å2 |
| Ramachandran | |
| Most favored | 97.1% |
| Additionally allowed | 2.3% |
| Outliers | 0.6% |
a Raw data can be accessed at doi: 10.15785/SBGRID/353.
Intriguingly, the first α-helix (residues 13–44) of all three copies of ANX A13a in the asymmetric unit deviates significantly from the structure observed in all other annexin crystal structures reported to date (Fig. 3A). Here, ANX A13a lacks the helix–turn–helix motif that normally allows these 30 residues to participate in the core fold. It instead adopts an extended conformation, which results in the first α-helix doubling its length from ∼20 to ∼40 Å (Figs. 2, A and B, and 3A). In this extended conformation, the N terminus of the first helix now completes the first four-helical bundle of a second ANX A13a molecule, such that the molecule forms a domain swapped dimer (Fig. 2). The dimer buries 1,850 Å2 of surface area per protomer, a value consistent with the formation of a stable biological assembly. We further investigated the potential relevance of this interaction by calculating shape complementarity of the dimer interface. A mean value of 0.74 is consistent with physiological protein oligomer interfaces, which range from 0.7 to 0.76 (17). Finally, the dimer does not appear to be influenced by crystal packing interactions, which could conceivably support either the N-terminal helix in either the canonical conformation or the extended conformation (Fig. S1, A and B).
Figure 3.
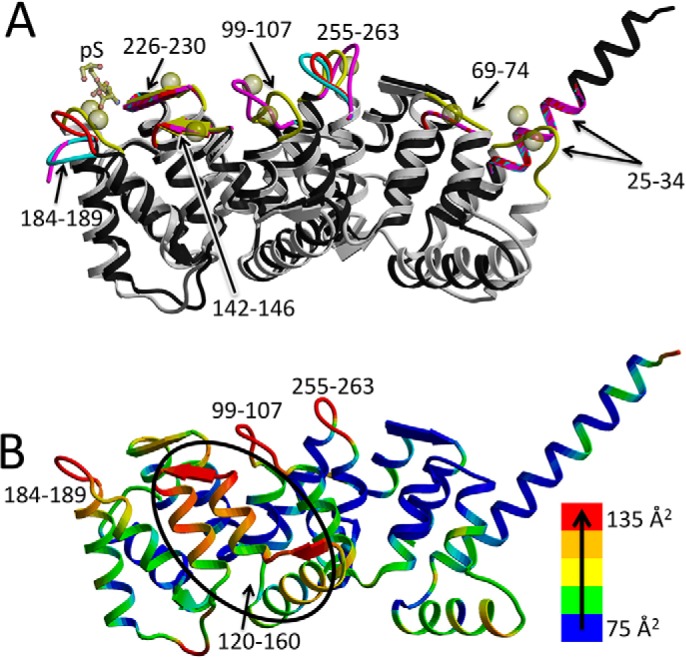
Ca2+-binding sites of ANX A13a. A, overlay of the structure of the three molecules of ANX A13a in each asymmetric unit (black with red, cyan, and magenta Ca2+-binding loops) with ANX A5 (Protein Data Bank entry 1A8A (5); gray backbone with yellow loops) bound of Ca2+ (yellow spheres) and phosphoserine (yellow sticks). The positions of the Ca2+-binding loops are marked and are among the regions of greater structural dissimilarity. B, relative temperature factors of ANX A13a. The structure of ANX A13a (A chain) is colored according to temperature factor, with the lowest temperature factor in blue and ramping via green, yellow, and orange to the highest temperature factor in red. Sequence regions containing the highest temperature factors are labeled and correlate with the Ca2+-binding loops.
In evaluating the side chains that mediate dimerization, many are buried hydrophobic residues (Fig. 2C). However, near the center of the extended helix is an interaction of note that involves Glu-34, Glu-71, and Asp-306. Glu-34 is positioned within hydrogen-bonding distance of the Glu-71 carbonyl and is poised to make two strong hydrogen-bonding interactions (2.7 and 2.9 Å) to the Asp-306 and the Glu-71 carbonyl oxygen (2.8 Å; Fig. 2C). The side chain of Glu-71 is similarly making a bidentate interaction with the carbonyl of Cys-27 (2.8 and 2.9 Å; not shown). The observed interactions are unusual because glutamate and aspartate side chains are normally negatively charged at physiological pH, and the observed distances would require that the Glu-34, Glu-71, and Asp-306 side chains are protonated. This may have been facilitated by the mildly acidic crystallization conditions, may be a result of altered pKa values of these side chains, or may be a combination of these two possibilities.
The sequence motifs that bind to Ca2+ and phospholipids are biochemically well-defined in homologous annexins, and the electrostatic surface potential is consistent with the presence of Ca2+-binding sites (Fig. S1, C and D). This allows us to propose putative binding sites for these molecules on the convex surface of ANX A13a. For example, in ANX A5, side chains analogous to Glu-34 and Glu-71 are among those that contribute to the coordination sphere of three Ca2+ ions (5, 18). The conformational change of ANX A13a that extends the N-terminal helix relocates Glu-34 and Glu-71 such that these putative Ca2+ sites are now eliminated in the ANX A13a dimer (Fig. 3A and Table 2). Comparisons between the structure of ANX A13a and ANX A5 bound to phosphoserine as bridged by Ca2+ (5, 18) identify that four of the seven remaining Ca2+-binding loops differ in conformation despite conservation of the Ca2+-coordinating residues. Thus, this ANX A13a structure would require conformational adjustment to support Ca2+ binding. Analysis of the three copies of ANX A13a in the asymmetric unit suggests that flexibility of these Ca2+-binding regions could allow them to sample a conformation capable of interacting with Ca2+. Specifically, whereas the RMS deviation in pair-wise comparisons of each of the chains in the asymmetric unit is ∼0.3 Å, there are significant conformational differences in the loops comprising residues 184–189 and 255–263, which are putative Ca2+-binding regions. These loops have a maximal displacement of 10.0 and 10.6 Å, respectively (Fig. 3A). Elevated temperature factors of these loops also suggest that they are flexible in the absence of Ca2+ (Fig. 3B).
Table 2.
Summary of putative Ca2+-binding sites in the ANX A13a dimer
| Residues | Ca2+ bound to ANX A5 | Comments |
|---|---|---|
| 25–34 | 2 | Eliminated in A13a dimer by the elongation of helix 1 |
| 69–74 | 1 | Eliminated in A13a dimer by contacts to the adjacent protomer |
| 97–106 | 2 | Conformation in A13a differs from that of Ca2+-bound ANX A5 |
| 142–146 | 1 | Conformation in A13a appears competent to bind Ca2+ |
| 180–190 | 2 + p-Ser | Ca2+-binding loop is flexible |
| 226–230 | 1 | Conformation in A13a appears competent to bind Ca2+ |
| 257–261 | 1 | Ca2+-binding loop is flexible |
Although the putative Ca2+ and phospholipid-binding sequence motifs could conceivably bind Ca2+ with conformational adjustments, neither soaking preformed crystals nor cocrystallizing the protein prebound to 1 mm Ca2+ resulted in the appearance of electron density consistent with Ca2+ at these locations. We posit that the mildly acidic conditions (pH 5.05) used for crystallization could preclude Ca2+ binding by protonating the acidic side chains of the binding sites. Nevertheless, two of the putative Ca2+-associated phospholipid-binding clefts were associated with electron density for a nonprotein ligand. This electron density was tentatively assigned as ethylene glycol, which was used as a cryoprotectant (10% v/v) during data collection.
ANX A13a exists as a pH-dependent monomer–dimer in solution
Because the dimer observed in the crystal structure requires an alternative fold that is unprecedented in crystal structures of ANX family proteins, we tested whether ANX A13a dimerizes in solution and whether this occurs at physiological pH. To do this, we monitored the average Stokes radius of purified ANX A13a using size-exclusion chromatography (Fig. 4A). At pH 8, ANX A13a eluted as a doublet, with calculated molecular masses of 37.9 ± 3.7 and 74.5 ± 6.4 kDa. These values are consistent with the theoretical molecular masses of an ANX 13a monomer (35.4 kDa) and dimer (70.8 kDa). The eluted protein from each of the two peaks was evaluated as >95% pure using SDS-PAGE (Fig. 5) and validated as ANX A13a by MS.
Figure 4.
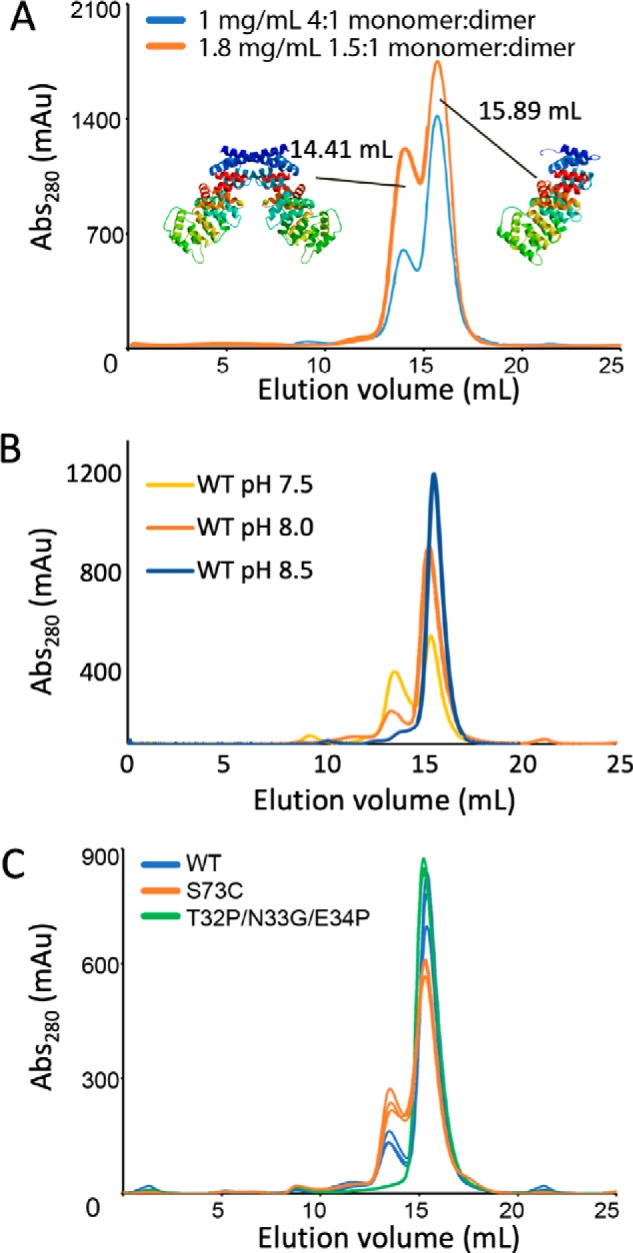
Monomer–dimer equilibrium of WT and variant ANX A13a in solution. A, evaluation of the elution profile of WT ANX A13a at 1 and 1.8 mg/ml. WT ANX A13a eluted as a doublet, with calculated molecular masses of 37.9 ± 3.7 and 74.5 ± 6.4 kDa. These values are consistent with the theoretical molecular masses of an ANX 13a monomer (35.4 kDa) and dimer (70.8 kDa). At a concentration of 1 mg/ml (blue trace), the monomer:dimer ratio is 4:1, whereas increasing the concentration to 1.8 mg/ml (orange trace) increases the percentage of the dimer such that the monomer:dimer ratio is now 1.5:1. Analysis was done in triplicate with a representative replicate shown. B, evaluation of the elution profile of 1 mg/ml wild-type ANX A13a at pH 7.0, 7.5, and 8.5. The peak corresponding to the ANX A13a dimer is reduced as pH increases. Analysis was performed in triplicate with a representative chromatogram shown. C, comparison of dimerization propensity of purified WT and variant ANX A13a at a concentration of 0.5 mg/ml. WT ANX A13a (blue trace) exhibits a distinct monomer:dimer distribution. The ANX A13aT32P/N33G/E34P (green trace) designed monomer exhibits no detectable dimer. The ANX A13aS73C (orange trace) designed dimer exhibits an increase in dimerization over WT but is not fully converted to the dimeric form under the conditions tested. Analysis was performed in triplicate with each replicate shown.
Figure 5.
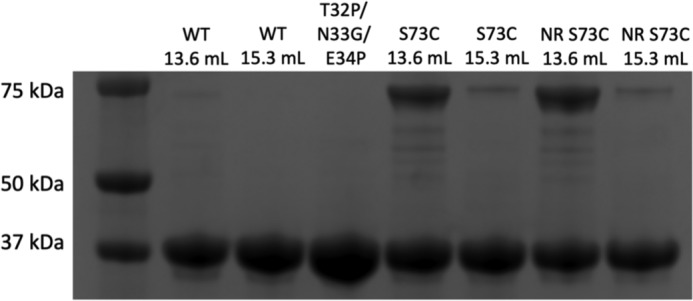
SDS-PAGE analysis for WT and variant ANX A13a. The numbers refer to the elution volume from the size-exclusion column shown in Fig. 4. Both the monomer and dimer peak migrate at the same molecular weight. For the S73C variant, fractions were evaluated under both reducing (unlabeled) and non-reducing (NR) conditions, showing that disulfide is resistant to reduction.
We further wanted to identify whether there was a dynamic equilibrium between monomers and dimers. Again, using size-exclusion chromatography, we measured the monomer–dimer equilibrium at multiple concentrations of ANX A13a protein. The relative quantities of the monomer and dimer appear to be concentration-dependent (Fig. 4A), with an increase in dimer observed upon an increase in protein concentration. Reinjection of either the isolated monomer peak or an isolated dimer peak resulted in a similar monomer–dimer distribution (Fig. S2). Taken together, this suggests that the protein is in a dynamic monomer–dimer equilibrium at slightly basic pH and in solution.
The dimer interface of the crystal structure involved unusual protonation of acidic side chains. Protonation of these residues is anticipated to increase as pH values are decreased; thus, we anticipate increased dimer formation at lower pH values. To test the influence of pH on the monomer–dimer equilibrium of ANX A13a protein, we again used size-exclusion chromatography (Fig. 4B). Increasing the pH to 8.5 resulted in a near complete conversion of ANX A13a to a monomer, whereas decreasing the pH to 7.5 increased the dimeric population. Decreasing the pH further resulted in protein insolubility (not shown) that prevented additional analysis. These observations indicate that ANX A13a oligomerization is pH-dependent.
Because the dimer interface also involved restructuring of the putative Ca2+-binding sites, we assessed whether the addition of Ca2+ affected dimerization. At the ANX A13a concentrations necessary to evaluate this effect, the addition of Ca2+ resulted in protein precipitation under the conditions tested. Solubility could not be restored through the addition of detergent (not shown).
Oligomerization of AnxA13a influences liposome binding
The dimer observed in the crystal structure brings two copies of ANX A13a together in a manner that occludes a significant portion of the membrane-binding surface. There are two reasonably plausible competing hypotheses for how this dimer might contribute to the regulation of membrane remodeling (Fig. 6). One possibility is that the dimer inhibits association between the membrane remodeling surface of ANX A13a and the membrane; in this scenario, the ANX A13a dimer could retain membrane association via the N-terminal myristoylation. Disassociation of the dimer into monomers would allow the membrane-remodeling surface to be available for interaction with the membrane. An alternative possibility is that the dimer would bind to the membrane across the two monomers; in this latter scenario, the membrane would be pinched between the two protomers of the dimer, potentially inducing an opposite membrane curvature than the annexin monomer.
Figure 6.
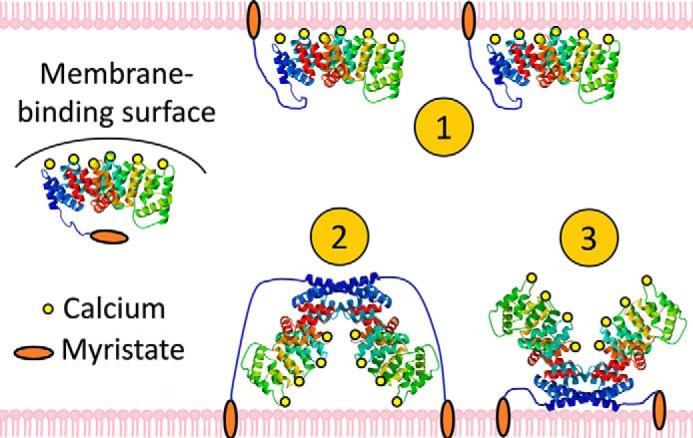
Hypotheses for membrane binding by ANX A13a. The canonical membrane-binding surface of ANX A13a is shielded by the domain-swapped dimer, but constitutive membrane association is promoted by the N-terminal myristoylation. Multiple membrane-interacting modes could be envisioned: 1) ANX A13a disassociates into monomers and interacts with the membrane via both the myristoylation and the membrane-remodeling surface; 2) the dimer interacts with the membrane weakly via the canonical membrane-binding surface and strongly via the myristoylation; and 3) the dimer remains membrane-associated via myristoylation, but the canonical membrane-remodeling surface is unable to interact with membranes in the oligomer.
To distinguish between these possibilities, we constructed variants of ANX A13a that we predicted would bias the equilibrium toward monomers or dimers and tested liposome binding in vitro. We used several strategies to develop a designed monomer, including targeted mutagenesis and chimeragenesis. Of these variants, ANX A13a with Thr-32, Asn-33, and Glu-34 replaced by Pro–Gly–Pro expressed, folded, and exclusively formed monomers when analyzed by size-exclusion chromatography (Figs. 4C and 5). The rationale behind this ANX A13aT32P/N33G/E34P variant is that the introduced prolines should prevent the first helix–loop–helix motif from adopting the extended helix.
We also designed several variants intended to stabilize the dimer. Although we were not able to develop a variant that fully converted ANX A13a to the dimeric form, ANX A13aS73C expressed, folded, and modestly increased the population of the dimer as compared with the WT protein under the conditions tested (Figs. 4C and 5). The ANX A13aS73C variant is proposed to introduce a disulfide between protomers of the dimer, which would lock the dimeric state. We considered ANX A13aT32P/N33G/E34P (designed monomer) and ANX A13aS73C (enhanced dimer) suitable for additional analyses.
To identify whether dimerization impacts the ability of the membrane-remodeling surface of ANX A13a to interact with membranes, we measured Ca2+-dependent binding of nonmyristoylated WT, ANX A13aT32P/N33G/E34P, and ANX A13aS73C to liposomes in vitro. Because myristoylation promotes constitutive membrane binding in ANX A13b (16), we used nonmyristoylated protein. This now allows membrane association to be a reporter for the interaction of the membrane-remodeling surface with liposomes. We used three types of liposomes. The first is pure 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), which is not anticipated to bind annexin proteins, even in the presence of Ca2+. We also used two different lipid mixtures that each had 70% DOPC but also contained either 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS) or 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA). Prior studies show that these acidic phospholipids support Ca2+-dependent binding of ANX proteins via the membrane-remodeling surface (16).
Nonmyristoylated ANX A13a (dynamic monomer–dimer equilibrium) exhibited Ca2+-dependent binding to liposomes containing acidic phospholipids (Fig. 7 and Fig. S3). Under the conditions that we tested, membrane recruitment was incomplete, consistent with previously published studies (13, 16). The ANX A13aS73C (enhanced dimer) had subtly decreased membrane association relative to WT, which may be anticipated because this mutation only modestly increased the percentage of dimer (Fig. 4C). In contrast, ANX A13aT32P/N33G/E34P (designed monomer) appeared to be fully recruited to liposomes containing acidic phospholipids in the presence of Ca2+. Similar treatment of purified WT and variant AnxA13a proteins in the absence of lipid resulted in all protein remaining in solution, indicating that the amount of pelleted protein was not influenced by a population of protein that was insoluble. Taken together, these studies indicate that the ANX A13a monomer has an improved capacity to interact with acidic phospholipids in the presence of Ca2+.
Figure 7.
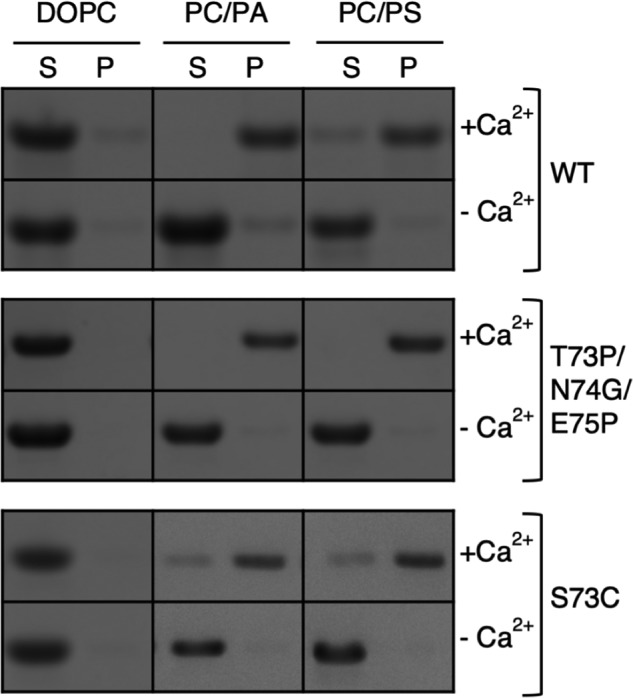
Ca2+-dependent binding of ANX A13a to phospholipids. Phospholipids with varied compositions (100% DOPC, 70% PC/30% PA, 70% PC/30% PS) were incubated with purified WT ANX A13a, the designed monomer (ANX A13aT32P/N33G/E34P), and the enhanced dimer (ANX A13aS73C) at a ratio of 500:1 and in the presence or absence of 0.5 mm CaCl2 (± Ca2+). Samples were centrifuged at 100,000 × g, and the resultant supernatant (S) and pellet (P) were separated by SDS-PAGE. The assay was performed in triplicate, with a representative gel shown.
Designed variants of ANX A13 isoforms dictate membrane association in epithelial cells
To understand how ANX A13a oligomerization affected membrane association in cells, we heterologously expressed WT and variant EGFP-tagged ANX A13a in LS174T-W4 epithelial cells. These LS174T-W4 cells exhibit a low surface area to volume ratio, making it easy to assess the extent of plasma membrane targeting for expressed proteins. ANX A13a-EGFP was strongly enriched on the plasma membrane with a peripheral:cytoplasmic intensity ratio of 3.5:1 (Fig. 8 and Table 3). Conversely, nonmyristoylated ANX A13aG2A-EGFP appeared to be purely cytoplasmic, with little observable enrichment at the plasma membrane (Fig. 8 and Table 3). The effect of myristoylation can be inferred by the direct comparison between ANX A13a-EGFP and ANX A13aG2A-EGFP, where the loss of myristoylation reduces ANX A13a at the phospholipid membrane of the cell.
Figure 8.

Localization of ANX A13a variants expressed in LS174T-W4 cells. A, single plane confocal images of individual LS174T-W4 cells expressing EGPF-tagged ANX A13a, A13aG2A (nonmyristoylated), ANX A13aT32P/N33G/E34P (designed monomer), and A13aS73C (enhanced dimer). Higher intensities are encoded with warmer colors. WT ANX A13a is robustly localized at the cell surface, whereas nonmyristoylated ANX A13a (ANX A13aG2A) is distinctly localized in the cytoplasm. Expression of the designed monomeric form of ANX A13a (ANX A13T32P/N33G/E34P) results in a subtle decrease in surface levels compared with WT ANX A13a. The enhanced dimeric form of ANX A13a exhibits increased cytoplasm localization relative to WT ANX A13a. All boxes are 25 × 25 μm. B, peripheral:cytoplasmic intensity ratios derived from 11–13 cells representing two biological replicates. Pair-wise p values are listed in Table 3.
Table 3.
p values and significance for pair-wise comparisons of peripheral:cytoplasmic intensity ratios
A Student's t test was used to compare the peripheral:cytoplasmic intensity ratios in Fig. 6. Numeric p values are given in the lower left, and the levels of significance are listed in the upper right. NS, not significant; *, p < 0.05; ***, p < 0.001.
| WT | G2A | T32P/N33G/E34P | S73C | |
|---|---|---|---|---|
| WT | — | *** | * | *** |
| G2A | <0.001 | — | *** | NS |
| T32P/N33G/E34P | 0.02 | <0.001 | — | *** |
| S73C | <0.001 | 0.06 | <0.001 | — |
To test how oligomerization affects annexin localization, we next expressed the constitutive monomer A13aT32P/N33G/E34P-EGFP and enhanced dimer ANX A13aS73C-EGFP. A13aT32P/N33G/E34P-EGFP (designed monomer) distinctly favors localization at the cell membrane, with a peripheral:cytoplasmic intensity ratio similar to WT A13a-EGFP. The slight shift in localization of this monomeric variant to the cytoplasm as compared with WT might reflect that the designed monomer mutagenizes residues of a Ca2+-binding site, which may reduce phospholipid-binding activity. In contrast, the A13aS73C-EGFP (enhanced dimer) exhibited a reduced peripheral:cytoplasmic intensity ratio. These results strengthen the premise that ANX A13a dimerization inhibits interaction between the membrane-remodeling surface and the phospholipid bilayer.
Discussion
Annexin proteins have long been recognized for their roles in membrane remodeling. However, each of the 12 mammalian isoforms of annexin (ANX A1–13, with ANX A12 unassigned) performs distinct biological roles associated with specific biophysical properties (3, 19). The location of ANX A13a at the apical brush border of intestines (Fig. 1) is consistent with a biological role in forming lumenal vesicles (14).
The structures of ANX A13 isoforms have been proposed to center around a highly conserved core of helical bundles common to all characterized annexins (13, 16). Surprisingly, the crystal structure of ANX A13a reveals only three of the four expected helical bundles because the N-terminal helical bundle exhibits an alternative fold. Here, a unique extended α-helix at the N terminus mediates dimerization (Fig. 2).
An annexin oligomer is not uncommon. Indeed, one biophysical property that differs in the various annexin isoforms is its oligomerization. For example, both ANX A5 and ANX B12 form trimers (20, 21). The trimers of ANX A5 pack into two-dimensional arrays on membranes (22, 23), with this close association proposed to assist in local membrane organization. However, a domain-swapped dimer has not previously been reported. Many annexins can also form heteromeric assemblies facilitated by a partner protein (24). This strikingly different type of assembly is exemplified by ANX A2 where the N-terminal sequence forms a complex with an oligomeric partner protein, S100 (25–29). Because S100 proteins can self-associate with multiple geometries, heteromeric annexin oligomerization can allow one annexin protein to approach multiple membrane surfaces at different angles, which is anticipated to induce different membrane curvatures.
In stark contrast to these oligomers that assist in membrane remodeling, the ANX A13a dimer (Figs. 2 and 4) appears to negatively regulate remodeling (Figs. 7 and 8). Notably, the monomer is sufficient to interact with liposomes in vitro (Fig. 7), but membrane interaction also requires myristoylation in cells (Fig. 8). In contrast, the dimer reduces lipid association both in vitro and in cells (Fig. 7 and 8). The apparently contradictory finding that nonmyristoylated protein can still associate with the membrane in vitro (Fig. 7) but has significantly reduced membrane interaction in cells (Fig. 8) suggests that the monomer–dimer ratio (and/or membrane association) may be additionally regulated in cells. There are several possibilities for how this could occur. Our finding that the monomer–dimer ratio is concentration-dependent (Fig. 4A) suggests that one possibility is via local annexin concentrations. Indeed, a high local concentration of myristoylated ANX A13a could shift the equilibrium toward dimerization as occurs in vitro (Fig. 4A), and one interpretation of our results is that the local concentration of ANX A13a on the cell membrane is higher than was used in our in vitro assays. An alternative possibility for regulating membrane association in cells could be via an interacting protein. Prior proteomics analysis of intestinal exosomes identified that in addition to ANX A13a, these vesicles contain a range of proteins that could serve this function (14). Finally, the ANX A13a structure shows that the dimer interface involves unusual hydrogen-bonding interactions of glutamates and aspartates (Fig. 2C), an observation that indicates that protonation likely accompanies the formation of the dimer. Because these same residues are predicted to form a Ca2+-binding site in the canonical annexin fold, one may posit that a local decrease in either pH or Ca2+ concentrations could convert monomers to dimers. Conversely, a local increase in pH and/or Ca2+ concentration could convert dimers to monomers by deprotonating the acidic residues and exposing the membrane-remodeling Ca2+-binding sites. Combinations of these mechanisms are also possible.
Although data support the dimeric form of ANX A13a as a negative regulator of membrane remodeling, they do not exclude other physiological functions for the dimer or other alternatively folded ANX proteins. One speculative possibility arises from the consideration of early studies of annexins, which showed that purified annexins can insert into lipid bilayers and conduct Ca2+ current under mildly acidic conditions (30–33). In the context of the canonical annexin architecture, a plausible mechanism for Ca2+ channel activity is difficult to envision. However, site-directed spin labeling combined with EPR spectroscopy in Hydra ANX B12 (34) identified that acidic conditions induced a large conformational change for α-helix 5 and its associated helix–loop–helix motif that converted these into a continuous α-helix. Extrapolation of this finding to all helices allowed a proposal where the annexin sequence supports two distinct alternative folds: a membrane-remodeling fold represented by the available crystal structures, and a Ca2+ channel fold that involves the straightening and extension of alternative helix–loop–helix motifs into seven continuous membrane-spanning α-helices (34).
Although the ANX A13a dimer is clearly not a channel, the alternative fold of the N terminus (Fig. 2) provides direct evidence that the first helical bundle can undergo the elongation proposed to be necessary for conversion to a channel. Because the structural conversion to a channel likely requires protonation of acidic residues (Fig. 2C), it should be enhanced by lowered pH, as previously shown (30–33); however, the observation of a monomer–dimer equilibrium at pH 8 in solution (Fig. 4A) suggests that this structural rearrangement is partially achievable at neutral pH in ANX A13a. The intimate contacts observed in the dimer allow us to further posit that a channel form of ANX proteins could involve a symmetric, intercalated dimer with each monomer containing seven transmembrane helices (14 transmembrane helices for the dimer). An alternative fold associated with a distinct function is observed in other moonlighting proteins. For example, an alternative fold of the RfaH protein converts it from a transcription factor to a translation factor (35). The structural results on ANX A13a further support the general possibility that some moonlighting proteins have more than one fold.
These studies reveal that the ANX A13 domain-swapped dimer in the crystal structure converts a helix–turn–helix motif into an uninterrupted, extended helix, providing direct evidence that the annexin sequence can support two architectures in this region. Our results determine that ANX A13 localizes to the intestinal brush border and suggest that both myristoylation and an annexin monomer (canonical fold) are required for efficient membrane binding. Conversely dimerization (alternative N-terminal fold) prevents the membrane-remodeling surface of the annexin protein from interacting with acidic phospholipids in a Ca2+-regulated fashion.
Experimental procedures
Protein expression and purification
The cDNA encoding human ANX A13a was obtained from the Mammalian Gene Collection (National Institutes of Health). Three single nucleotide polymorphisms (SNPs) associated with human ANX A13a are reported; sequencing identified that this clone contained two of the three SNPs that encode the minor variant (R86H and V272I). These SNPs were retained for this study and are referred to as WT ANX A13a in the text. ANX A13a was subcloned into the pPro ExHTa vector, encoding a His6 tag at the N terminus of the protein. Variant ANX A13a used in this study was created using site-directed mutagenesis (Mutagenex). WT or variant ANX A13a in this plasmid was transformed into Escherichia coli BL21(DE3) cells for overexpression and expressed and purified by the same protocol.
An overnight culture consisting of 50 ml of LB, 100 μg/ml ampicillin, and a single colony was grown at 30 °C with shaking. In the morning, 10 ml of overnight culture was used to inoculate 500 ml of LB supplemented with 100 μg/ml ampicillin. These cultures were then grown at 37 °C with shaking to an A600 of 0.6–0.9, when isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.5 mm. Cultures were allowed to continue shaking at 37 °C for an additional 3 h, and then cells were harvested by centrifugation at 6000 × g. The pellets were stored at −20 °C prior to purification.
Each cell pellet was resuspended in 60 ml of annexin buffer (100 mm KCl, 20 mm Tris, pH 8.5) supplemented with 4% glycerol, 10 mm β-mercaptoethanol, 10 mm imidazole, 100 μg/ml DNase, 5 μg/ml leupeptin, 1 μm pepstatin, and 1 mg/ml Pefabloc buffer. The cells were lysed by sonication, and the cellular debris was removed by centrifugation (1 h at 37,000 × g). The supernatant was then loaded onto a 1-ml HisTrap column. Following sample loading, the column was washed until the absorbance at 280 nm returned to baseline. The column was then washed with annexin buffer containing 40 mm imidazole, and the protein was eluted with 140 mm imidazole. Size-exclusion chromatography was performed on a Superdex S200 Increase 10/300 GL column (GE Healthcare) equilibrated with 25 mm Tris, pH 8.0, and 50 mm KCl.
Crystallization, data collection, and structure determination
ANX A13a was crystallized using the hanging-drop vapor-diffusion method by equilibrating 1.5 μl of ANX A13a (5 mg/ml in annexin buffer) and 1.5 μl of reservoir solution (0.8% w/v PEG 8000, 40 mm 1,2-butanediol, 0.1 m sodium acetate, pH 5.05) against 700 μl of reservoir solution and equilibrating the reactions at 20 °C. Crystals formed in space group C2, with unit cell dimensions of a = 190.6 Å, b = 93.8 Å, c = 94.9 Å, β = 106.0°, and three annexin protomers in each asymmetric unit. The crystals were cryoprotected in a solution containing all of the crystallization conditions plus 10% glycerol and 10% ethylene glycol and were cryocooled by plunging into liquid nitrogen. The data were collected at the Advanced Photon Source Beamline 21-ID-G at −180 °C using a Mar CCD detector. The data were processed and scaled using HKL2000 (36) and handled using programs within the CCP4 suite (37).
The structure of ANX A13a was determined by molecular replacement using the program MOLREP (38) and the structure of ANX A8 as the search model (Protein Data Bank entry 1W3W (39)). Model building was performed using COOT (40). The structure was refined using REFMAC (41), PHENIX (42), and CNS (43). Crystallographic data collection and statistics are listed in Table 1.
Assessment of oligomerization
Oligomerization of WT and variant ANX A13a was assessed in triplicate by injecting 0.5 ml of purified protein onto a Superdex S200 Increase column (GE Healthcare) equilibrated with 25 mm Tris, pH 8.0, and 50 mm KCl and a flow rate of 0.25 ml/min. For the pH dependence studies, ANX A13a at 1 mg/ml was injected into 25 mm Tris, 50 mm KCl at either pH 7.5, 8, or 8.5. Elution volumes were compared with a standard curve that was developed from proteins of known molecular mass.
Liposome binding assays
Large unilamellar vesicles composed of DOPC (100 mol%), DOPC/DOPS (70/30 mol%), or DOPC/DOPA (70/30 mol%) were prepared as follows. Lipids (Avanti Polar Lipids) in chloroform (CHCl3) were mixed, evaporated under a nitrogen stream, dried in a vacuum overnight, and then rehydrated in 10 mm HEPES, pH 7.2, and 100 mm KCl to a final concentration of 1 mg/ml. Lipid mixtures were vortexed, exposed to three cycles of freeze thaw using a dry ice/ethanol bath and 37 °C water bath, and then extruded 11 times through an 800-nm filter using an Avanti extruder. Large unilamellar vesicles were then frozen at −80 °C until further use.
Liposome binding assays (300 μl in annexin buffer) were performed with a constant phospholipid to protein ratio of 500:1; negative controls lacked liposomes. A final concentration of 0.5 mm CaCl2 was added to samples containing Ca2+. The samples were incubated at 22 °C for 30 min. The mixture was centrifuged at 100,000 × g for 45 min at 4 °C, and the supernatant was separated from the phospholipid–protein pellet. The pellet was reconstituted in 300 μl of annexin buffer. The supernatant and the pellet were then separated by SDS-PAGE.
Assessing localization of ANX A13a variants
EGFP-tagged variants of ANX A13a were created using standard molecular biological techniques to insert the ANX A13a coding sequence into pEGFP-N2 (Clontech). Maxiprep plasmid DNA was prepared with an endotoxin free method (Qiagen) and then used to transfect LS174T-W4 epithelial cells. LS174T-W4 cells were maintained and transfected according to our previously published protocols (44). For visualization of construct targeting, the cells were plated on no. 1.5 coverslips at single cell densities, thus reducing the prevalence of cell–cell contacts that ultimately confound plasma membrane enrichment analyses. LS174T-W4 cells were imaged using a Nikon A1r laser scanning confocal microscope equipped with a 100×/1.49 NA oil immersion objective. Peripheral:cytoplasmic ratios were derived from intensities measured using ImageJ (National Institutes of Health) as previously described (45)
Author contributions
K. M. M. and I. Y. formal analysis; K. M. M. and T. M. I. supervision; K. M. M., I. Y., D. A. S., R. E. M., N. J. F., P. K. S., and S. M. investigation; K. M. M. and T. M. I. writing-original draft; K. M. M., I. Y., D. A. S., R. E. M., N. J. F., P. K. S., S. M., M. J. T., and T. M. I. writing-review and editing; D. A. S., R. E. M., N. J. F., P. K. S., and S. M. methodology; M. J. T. and T. M. I. conceptualization; T. M. I. data curation; T. M. I. funding acquisition; T. M. I. project administration.
Supplementary Material
Acknowledgments
A portion of the experiments described here used the Vanderbilt PacVan robotic crystallization facility, which was supported by National Institutes of Health Grant S10 RR026915 and the Vanderbilt Center for Structural Biology. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy Office of Science by Argonne National Laboratory, was supported by the US Department of Energy under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor under Grant 085P1000817.
This work was supported by pilot funds from Vanderbilt University Medical Center's Digestive Disease Research Center supported by National Institutes of Health Grant P30DK058404 (to T. M. I.). This work was also supported by National Institutes of Health Grants GM095633 (to T. M. I.) and DK075555, DK095811, and DK111949 (to M. J. T.), National Institutes of Health Training Grant in Molecular Biophysics T32GM008320 (to N. J. F.), and National Institutes of Health Training Grant in Mechanisms of Vascular Disease T32 HL007751 (to K. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
ANX A13 has historically been referred to as intestine-specific annexin or annexin XIII in the literature.
- ANX
- annexin
- RMS
- root mean square
- IAP
- intestinal alkaline phosphatase
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- DOPA
- 1,2-dioleoyl-sn-glycero-3-phosphate
- SNP
- single nucleotide polymorphism
- EGFP
- enhanced green fluorescent protein.
References
- 1. Creutz C. E., Pazoles C. J., and Pollard H. B. (1978) Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J. Biol. Chem. 253, 2858–2866 [PubMed] [Google Scholar]
- 2. Gerke V., Creutz C. E., and Moss S. E. (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461 10.1038/nrn1671, 10.1038/nrm1661 [DOI] [PubMed] [Google Scholar]
- 3. Lizarbe M. A., Barrasa J. I., Olmo N., Gavilanes F., and Turnay J. (2013) Annexin–phospholipid interactions: functional implications. Int. J. Mol. Sci. 14, 2652–2683 10.3390/ijms14022652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huber R., Berendes R., Burger A., Luecke H., and Karshikov A. (1992) Annexin V-crystal structure and its implications on function. Behring Inst. Mitt. 107–125 [PubMed] [Google Scholar]
- 5. Swairjo M. A., Concha N. O., Kaetzel M. A., Dedman J. R., and Seaton B. A. (1995) Ca2+-bridging mechanism and phospholipid head group recognition in the membrane-binding protein annexin V. Nat. Struct. Biol. 2, 968–974 10.1038/nsb1195-968 [DOI] [PubMed] [Google Scholar]
- 6. Favier-Perron B., Lewit-Bentley A., and Russo-Marie F. (1996) The high-resolution crystal structure of human annexin III shows subtle differences with annexin V. Biochemistry 35, 1740–1744 10.1021/bi952092o [DOI] [PubMed] [Google Scholar]
- 7. Capila I., Hernáiz M. J., Mo Y. D., Mealy T. R., Campos B., Dedman J. R., Linhardt R. J., and Seaton B. A. (2001) Annexin V-heparin oligosaccharide complex suggests heparan sulfate-mediated assembly on cell surfaces. Structure 9, 57–64 10.1016/S0969-2126(00)00549-9 [DOI] [PubMed] [Google Scholar]
- 8. Leow C. Y., Willis C., Osman A., Mason L., Simon A., Smith B. J., Gasser R. B., Jones M. K., and Hofmann A. (2014) Crystal structure and immunological properties of the first annexin from Schistosoma mansoni: Insights into the structural integrity of the schistosomal tegument. FEBS J. 281, 1209–1225 10.1111/febs.12700 [DOI] [PubMed] [Google Scholar]
- 9. Tait J. F., and Gibson D. (1992) Phospholipid binding of annexin V: effects of calcium and membrane phosphatidylserine content. Arch. Biochem. Biophys. 298, 187–191 10.1016/0003-9861(92)90111-9 [DOI] [PubMed] [Google Scholar]
- 10. Wice B. M., and Gordon J. I. (1992) A strategy for isolation of cDNAs encoding proteins affecting human intestinal epithelial cell growth and differentiation: characterization of a novel gut-specific N-myristoylated annexin. J. Cell Biol. 116, 405–422 10.1083/jcb.116.2.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Massey-Harroche D., Mayran N., and Maroux S. (1998) Polarized localizations of annexins I, II, VI and XIII in epithelial cells of intestinal, hepatic and pancreatic tissues. J. Cell Sci. 111, 3007–3015 [DOI] [PubMed] [Google Scholar]
- 12. Choi S. H., Kwon S. R., Lee E. H., and Kim K. H. (2009) Molecular cloning, functional characterization and localization of an annexin from a fish gill fluke Microcotyle sebastis (Platyhelminthes: Monogenea). Mol. Biochem. Parasitol. 163, 48–53 10.1016/j.molbiopara.2008.09.009 [DOI] [PubMed] [Google Scholar]
- 13. Fernández-Lizarbe S., Lecona E., Santiago-Gómez A., Olmo N., Lizarbe M. A., and Turnay J. (2017) Structural and lipid-binding characterization of human annexin A13a reveals strong differences with its long A13b isoform. Biol. Chem. 398, 359–371 10.1515/hsz-2016-0242 [DOI] [PubMed] [Google Scholar]
- 14. McConnell R. E., Higginbotham J. N., Shifrin D. A. Jr., Tabb D. L., Coffey R. J., and Tyska M. J. (2009) The enterocyte microvillus is a vesicle-generating organelle. J. Cell Biol. 185, 1285–1298 10.1083/jcb.200902147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iglesias J. M., Morgan R. O., Jenkins N. A., Copeland N. G., Gilbert D. J., and Fernandez M. P. (2002) Comparative genetics and evolution of annexin A13 as the founder gene of vertebrate annexins. Mol. Biol. Evol. 19, 608–618 10.1093/oxfordjournals.molbev.a004120 [DOI] [PubMed] [Google Scholar]
- 16. Turnay J., Lecona E., Fernández-Lizarbe S., Guzmán-Aránguez A., Fernández M. P., Olmo N., and Lizarbe M. A. (2005) Structure–function relationship in annexin A13, the founder member of the vertebrate family of annexins. Biochem. J. 389, 899–911 10.1042/BJ20041918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lawrence M. C, and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 10.1006/jmbi.1993.1648 [DOI] [PubMed] [Google Scholar]
- 18. Sopkova J., Renouard M., and Lewit-Bentley A. (1993) The crystal structure of a new high-calcium form of annexin V. J. Mol. Biol. 234, 816–825 10.1006/jmbi.1993.1627 [DOI] [PubMed] [Google Scholar]
- 19. Hatoum D., Yagoub D., Ahadi A., Nassif N. T., and McGowan E. M. (2017) Annexin/S100A protein family regulation through p14ARF-p53 activation: A role in cell survival and predicting treatment outcomes in breast cancer. PLoS One 10.1371/journal.pone.0169925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Concha N. O., Head J. F., Kaetzel M. A., Dedman J. R., and Seaton B. A. (1992) Annexin V forms calcium-dependent trimeric units on phospholipid vesicles. FEBS Lett. 314, 159–162 10.1016/0014-5793(92)80964-I [DOI] [PubMed] [Google Scholar]
- 21. Luecke H., Chang B. T., Mailliard W. S., Schlaepfer D. D., and Haigler H. T. (1995) Crystal structure of the annexin XII hexamer and implications for bilayer insertion. Nature 378, 512–515 10.1038/378512a0 [DOI] [PubMed] [Google Scholar]
- 22. Pigault C., Follenius-Wund A., Schmutz M., Freyssinet J. M., and Brisson A. (1994) Formation of two-dimensional arrays of annexin V on phosphatidylserine-containing liposomes. J. Mol. Biol. 236, 199–208 10.1006/jmbi.1994.1129 [DOI] [PubMed] [Google Scholar]
- 23. Bouter A., Gounou C., Bérat R., Tan S., Gallois B., Granier T., D'Estaintot B. L., Pöschl E., Brachvogel B., and Brisson A. R. (2011) Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2, 270 10.1038/ncomms1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerke V., and Moss S. E. (2002) Annexins: from structure to function. Physiol. Rev. 82, 331–371 10.1152/physrev.00030.2001 [DOI] [PubMed] [Google Scholar]
- 25. Dempsey B. R., Rezvanpour A., Lee T. W., Barber K. R., Junop M. S., and Shaw G. S. (2012) Structure of an asymmetric ternary protein complex provides insight for membrane interaction. Structure 20, 1737–1745 10.1016/j.str.2012.08.004 [DOI] [PubMed] [Google Scholar]
- 26. Rezvanpour A., Santamaria-Kisiel L., and Shaw G. S. (2011) The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 286, 40174–40183 10.1074/jbc.M111.244038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ozorowski G., Milton S., and Luecke H. (2013) Structure of a C-terminal AHNAK peptide in a 1:2:2 complex with S100A10 and an acetylated N-terminal peptide of annexin A2. Acta Crystallogr. D Biol. Crystallogr. 69, 92–104 10.1107/S0907444912043429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y., Myrvang H. K., and Dekker L. V. (2015) Annexin A2 complexes with S100 proteins: structure, function and pharmacological manipulation. Br. J. Pharmacol. 172, 1664–1676 10.1111/bph.12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ecsédi P., Kiss B., Gógl G., Radnai L., Buday L., Koprivanacz K., Liliom K., Leveles I., Vértessy B., Jeszenöi N., Hetényi C., Schlosser G., Katona G., and Nyitray L. (2017) Regulation of the equilibrium between closed and open conformations of annexin A2 by N-terminal phosphorylation and S100A4-binding. Structure 25, 1195–1207.e5 10.1016/j.str.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 30. Pollard H. B., Burns A. L., Rojas E., Schlaepfer D. D., Haigler H., and Brocklehurst K. (1989) Purification and biochemical assay of synexin and of the homologous calcium-dependent membrane-binding proteins, endonexin II and lipocortin I. Methods Cell Biol. 31, 207–227 10.1016/S0091-679X(08)61611-8 [DOI] [PubMed] [Google Scholar]
- 31. Pollard H. B., and Rojas E. (1988) Ca2+-activated synexin forms highly selective, voltage-gated Ca2+ channels in phosphatidylserine bilayer membranes. Proc. Natl. Acad. Sci. U.S.A. 85, 2974–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rojas E., and Pollard H. B. (1987) Membrane capacity, measurements suggest a calcium-dependent insertion of synexin into phosphatidylserine bilayers. FEBS Lett. 217, 25–31 10.1016/0014-5793(87)81235-8 [DOI] [PubMed] [Google Scholar]
- 33. Rojas E., Pollard H. B., Haigler H. T., Parra C., and Burns A. L. (1990) Calcium-activated endonexin II forms calcium channels across acidic phospholipid bilayer membranes. J. Biol. Chem. 265, 21207–21215 [PubMed] [Google Scholar]
- 34. Langen R., Isas J. M., Hubbell W. L., and Haigler H. T. (1998) A transmembrane form of annexin XII detected by site-directed spin labeling. Proc. Natl. Acad. Sci. U.S.A. 95, 14060–14065 10.1073/pnas.95.24.14060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burmann B. M., Knauer S. H., Sevostyanova A., Schweimer K., Mooney R. A., Landick R., Artsimovitch I., and Rösch P. (2012) An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell 150, 291–303 10.1016/j.cell.2012.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 37. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 10.1107/S0021889897006766 [DOI] [Google Scholar]
- 39. Réty S., Sopková-de Oliveira Santos J., Dreyfuss L., Blondeau K., Hofbauerová K., Raguénès-Nicol C., Kerboeuf D., Renouard M., Russo-Marie F., and Lewit-Bentley A. (2005) The crystal structure of annexin A8 is similar to that of annexin A3. J. Mol. Biol. 345, 1131–1139 10.1016/j.jmb.2004.11.015 [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brunger A. T. (2007) Version 1.2 of the crystallography and nmr system. Nat. Protoc. 2, 2728–2733 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 44. Grega-Larson N. E., Crawley S. W., Erwin A. L., and Tyska M. J. (2015) Cordon bleu promotes the assembly of brush border microvilli. Mol. Biol. Cell 26, 3803–3815 10.1091/mbc.E15-06-0443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mazerik J. N., and Tyska M. J. (2012) Myosin-1A targets to microvilli using multiple membrane binding motifs in the tail homology 1 (TH1) domain. J. Biol. Chem. 287, 13104–13115 10.1074/jbc.M111.336313 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.