

Abstract
Ubiquinol cytochrome c reductase core protein I (UQCRC1) is a component of the complex III in the respiratory chain. Its biological functions are unknown. Here, we showed that knockout of UQCRC1 led to embryonic lethality. Disrupting one UQCRC1 allele in mice (heterozygous mice) of both sexes did not affect their growth but reduced UQCRC1 mRNA and protein in the brain. These mice had decreased complex III formation, complex III activity and ATP content in the brain at baseline. They developed worsened neurological outcome after brain ischemia/hypoxia or focal brain ischemia compared with wild-type mice. The ischemic cerebral cortex of the heterozygous mice had decreased mitochondrial membrane potential and ATP content as well as increased free radicals. Also, the heterozygous mice performed poorly in the Barnes maze and novel object recognition tests. Finally, UQCRC1 was expressed abundantly in neurons and astrocytes. These results suggest a critical role of UQCRC1 in embryo survival. UQCRC1 may also be important by forming the complex III to maintain normal brain ischemic tolerance, learning and memory.
Keywords: Brain ischemia, Complex III, Learning and memory, Ubiquinol cytochrome c reductase core protein I
Introduction
Human ubiquinol cytochrome c reductase core protein 1 (UQCRC1) is a nuclear DNA encoded protein consisting of 480 amino acids and is a subunit of the complex III (also known as complex bc1) in the respiratory chain [1–3]. Human complex III consists of 11 subunits encoded by nuclear and mitochondrial DNA [4, 5]. Only three of these subunits (cytochrome b, cytochrome c1 and Rieske iron-sulfur protein) contain redox prosthetic groups that may be directly involved in electron transport [4]. The functions of other subunits in the complex III are not clear. Two large proteins, UQCRC1 and ubiquinol cytochrome c reductase core protein 2 (UQCRC2), are found to be critical for the assembly of reductase and, thus, the integrity of the complex III in yeast [5]. However, the biological functions of UQCRC1 in mammalian cells are unknown.
It has been shown that UQCRC1 is increased in a mouse model of Rett syndrome [6]. UQCRC1 overexpression increases complex III activity in cell cultures [6]. The expression of UQCRC1 is decreased after ischemia/reperfusion in rat heart [7]. Its expression is increased in the heart by a protective agent [8]. These results indicate the association of UQCRC1 expression change with pathological processes but are insufficient to imply its biological functions. We have shown in recent studies that UQCRC1 overexpression reduces oxygen–glucose deprivation-induced injury of rat embryonic ventricular myocytes and that silencing UQCRC1 has the opposite effects [9, 10]. These results indicate a protective effect of UQCRC1 on cardiomyocyte cultures.
Ischemic brain injury is the underlying pathophysiology for stroke, a common cause of mortality and morbidity in human [11]. Impairment of the respiratory chain occurs during ischemia [12], which interrupts energy supply to induce cell injury [13]. The respiratory chain including UQCRC1 is in the mitochondria. It has been shown that mitochondria play a vital role in brain ischemia pathology via various mechanisms, such as reactive oxygen species (ROS) generation and scavenging [11, 14], electron transporting [15], mitochondrial dynamic changes [16–18] and involvement in inflammation [19–21]. Mitochondria are the major source of ROS and are also a target of ROS [22, 23]. Accumulating evidence shows that excessive ROS cause functional and structural impairment of brain tissues and play a critical role in the pathogenesis of cerebral ischemia [11]. If UQCRC1 is a necessary component in the mitochondrial respiratory chain, it is conceivable that UQCRC1 may play a role in determining brain ischemic tolerance. However, the role of UQCRC1 in brain ischemic injury has not been reported.
This study was designed to determine the biological functions of UQCRC1. We hypothesized that UQCRC1 was an important component of complex III in mammalian cells and that disruption of its basic cellular function might impair cell survival/organ function, such as learning, memory and brain ischemic tolerance. Here, we showed that knockout of UQCRC1 in mice led to embryonic lethality. Mice with disruption of one Uqcrc1 allele (heterozygous mice) had decreased UQCRC1 in the brain. These mice had decreased brain ischemic tolerance and poor learning and memory.
Results
Critical role of UQCRC1 in embryo survival
The ES cell from 129/OlaHsd sub-strain contained the inserted Neo cassette that substituted the gene sequence between exon 3 and exon 4. It was predicted to result in 104 base-pair loss of the coding sequence and early truncated UQCRC1 protein (Fig. 1). With the use of the designed primers, the following products were anticipated: one band of 493 base-pairs for the PCR product of the homozygous UQCRC1−/− mice, one band of 223 base-pairs for wild-type UQCRC1+/+ mice, and two bands of 223 and 493 base-pairs for heterozygous UQCRC1+/− mice (Fig. 1). A total of 121 25-day-old mice born from heterozygous parents were genotyped. Homozygous UQCRC1−/− mice were not found (Fig. 1), suggesting that the homozygous UQCRC1−/− mutant mice (mice with UQCRC1 knockout) were not viable.
Fig. 1.

Generation of the heterozygous UQCRC1+/− mice. a Construction of the mutant UQCRC1. b Representative images of the PCR products from embryos. C Results of genotypes of embryos and mice. ND non-determined
To determine the role of UQCRC1 in regulating mouse survival, the embryos at E15.5, E9.5 and E7.5 stages were isolated from 6th or 7th generation of heterozygous mothers for genotyping. There were no homozygous UQCRC1−/− embryos among 28 embryos at E15.5. One and two homozygous UQCRC1−/− embryos were found at E9.5 (39 total) and E7.5 (23 total), respectively. All the homozygous embryos appeared abnormal and hypogenetic under microscope. Six homozygous UQCRC1−/− blastocysts at E3.5 were identified among 46 samples; two of these homozygous UQCRC1−/− blastocysts had obviously abnormal morphology. There were 8 undetermined blastocysts, probably due to an insufficient amount of DNA. These results indicate that knockout of the gene UQCRC1 causes early embryo death and that the disruption of UQCRC1 expression results in embryonic hypogenesis.
Expression of UQCRC1 and UQCRC2 in the brain
In the cerebral cortex, a significant amount of UQCRC1 was expressed in the cells expressing microtubule-associated protein 2 (MAP-2), suggesting that UQCRC1 is expressed in neurons. The immunostaining of UQCRC1 was also co-localized with glial fibrillary acidic protein (GFAP), a marker for astrocytes [24], indicating that UQCRC1 is expressed in astrocytes. However, UQCRC1 staining was not co-localized with ionized calcium-binding adaptor molecule 1 (Iba-1) or transmembrane protein 119 (TMEM119) (Fig. 2), two markers for microglia [25, 26], suggesting that UQCRC1 is not detected in microglia. To determine the co-localization of UQCUC protein with various cell-type markers, microscopic fields with strong staining of one marker were selected for photographing. These fields may not contain many cells of other types, which makes it appear that UQCRC protein is located mostly in the focused type of cells in the microscopic field. Interestingly, UQCRC1 staining was circular surrounding the nuclei in neurons but was diffuse in astrocytes. The pattern of the staining of UQCRC2, the only other core protein in the complex III, was similar to that of UQCRC1 (Fig. 2). These results suggest that UQCRC1 and UQCRC2 are expressed in neurons and astrocytes but may not be expressed in a detectable amount in microglia. Of note, some cells expressed UQCRC1 but did not appear to express UQCRC2. All cells that expressed UQCRC2 also had UQCRC1 (Fig. 3). These results suggest that some cells may express only UQCRC1.
Fig. 2.
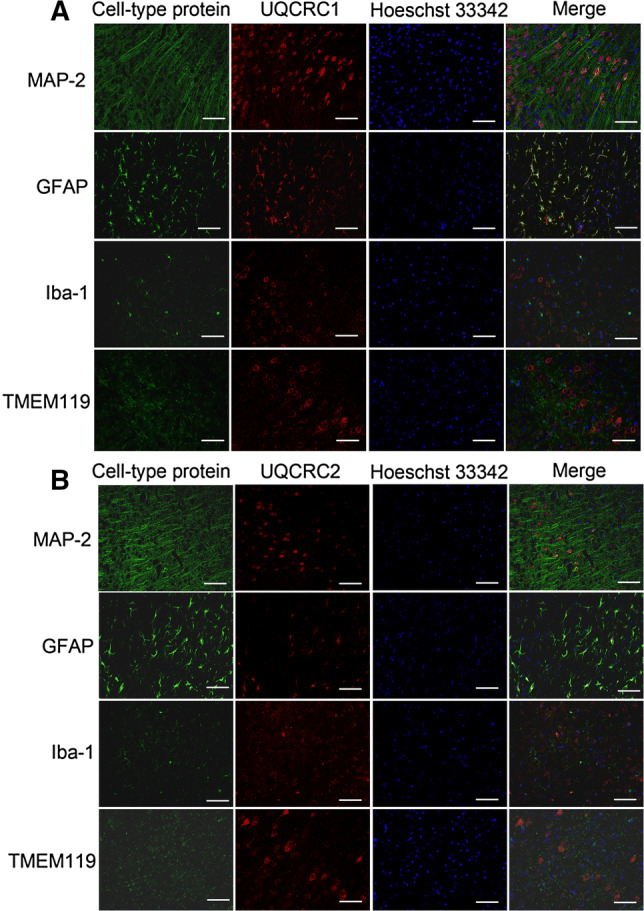
Double immunofluorescent staining of a cell-type specific protein and UQCRC1 (a) or UQCRC2 (b) in mouse cerebral cortex. Scale bar 200 µm
Fig. 3.
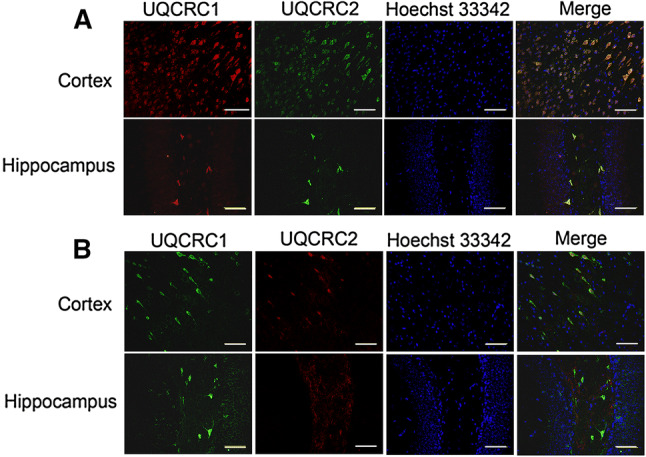
Double immunofluorescent staining of UQCRC1 and UQCRC2. a Rabbit polyclonal anti-UQCRC1 antibody and mouse monoclonal anti-UQCRC2 antibody were used. b Mouse monoclonal anti-UQCRC1 antibody and rabbit monoclonal anti-UQCRC2 antibody were used. Scale bar 200 µm
The heterozygous UQCRC1+/− mice had reduced UQCRC1 mRNA and protein in their brain (parametric results are presented as mean ± SD in this paper). Although the mRNA of UQCRC2 was increased in the cerebral cortex, a compensatory increase in the expression of UQCRC2 protein was not present in the brain (Fig. 4).
Fig. 4.

Decreased UQCRC1 expression in the heterozygous UQCRC1+/− mice. a UQCRC1 mRNA expression analyzed by real-time PCR. b UQCRC2 mRNA expression analyzed by real-time PCR. c UQCRC1 protein expression analyzed by Western blotting. Representative images of Western blotting are at top and quantitative results are at bottom. d UQCRC2 protein expression analyzed by Western blotting. Representative images of Western blotting are at top and quantitative results are at bottom. Results are mean ± SD (n = 5 for a and b, = 10 for c and = 6 for d). *P < 0.05 compared with wild-type mice. W wild-type, H heterozygous
Physiological characteristics of heterozygous UQCRC1+/− mice
The heterozygous UQCRC1+/− mice appeared normal and reproduced normally. Their body weights were similar to those of wild-type mice at ~ 3 months of age no matter whether they were male (26.7 ± 2.7 g of wild-type mice vs. 25.5 ± 2.6 g of the heterozygous mice, n = 20, P = 0.175) or female (19.3 ± 2.5 g of wild-type mice vs. 20.3 ± 1.8 g of the heterozygous mice, n = 20–27, P = 0.170). Their body weight that increased with age was similar to that of wild-type [F(1, 27) = 0.432, P = 0.517]. However, sex was a significant factor to affect the body weight increase with age [F(1, 27] = 20.564, P < 0.001]. Similarly, male mice had a slower respiratory rate than female mice [F(1, 1) = 10.959, P = 0.003] but there was no difference between wild-type mice and heterozygous UQCRC1+/− mice [F(1, 1) = 0.0085, P = 0.927]. There was no difference in the heart rate and mean arterial blood pressure between these two types of mice and between male and female mice (Fig. 5).
Fig. 5.

Physiological characteristics of the heterozygous UQCRC1+/− mice. a Growth curve. b Respiratory rates. c Heart rates. d Mean arterial pressure. Results are mean ± SD (n = 6–9)
Role of UQCRC1 in brain ischemic tolerance, learning and memory
To determine whether the reduced UQCRC1 expression in the heterozygous UQCRC1+/− mice had any functional consequence, we subjected these mice to brain ischemia/hypoxia. Six mice in 30 wild-type mice and 7 mice in 18 heterozygous UQCRC1+/− mice died before the end of the observation period (3 days) after this brain ischemia/hypoxia. There was no difference in the mortality between these two types of mice (P = 0.19). The heterozygous UQCRC1+/− mice had a larger infarct brain volume and poorer neurological deficit scores than the wild-type mice (Fig. 6a–c). Only mice that survived to the end of the observation period contributed data to the infarct volume analysis but the neurological deficit scores from all mice were included in analysis. Neurological deficit scores were evaluated just before the animals were killed for brain infarction assessment for those animals that survived throughout the observation period. The scores were 7 for those animals that died within the observation period. The heterozygous UQCRC1+/− mice also had worsened neurological outcome after they were subjected to the middle cerebral arterial occlusion (MCAO) (Fig. 6d–f), a commonly used focal brain ischemia model [27]. No animal died before the end of observation period (24 h) after MCAO. The heterozygous UQCRC1+/− mice after a Sham surgery did not have brain infarction or neurological deficits (Fig. 6d–f). Mice in the Sham surgery group had an identical surgical procedure to that of MCAO group but without receiving a suture to achieve MCAO. Two ischemia models, brain ischemia/hypoxia and MCAO models, were used to complement each other. These results suggest that the heterozygous mice have a decreased brain ischemic tolerance.
Fig. 6.

Decreased brain ischemic tolerance of the heterozygous UQCRC1+/− mice. a Representative images of TTC-stained brain slices of mice after brain ischemia/hypoxia. b Quantitative results of infarct volume of mice after brain ischemia/hypoxia. Results are mean ± SD (n = 12–18). c Neurological deficit scores of mice after brain ischemia/hypoxia. Results are in box plot (n = 18–30). d Representative images of TTC-stained brain slices of mice after MCAO or without MCAO (Sham surgery group). e Quantitative results of infarct volume of mice after MCAO or without MCAO (Sham surgery group). Results are mean ± SD (n = 7–8). f Neurological deficit scores of mice after MCAO or without MCAO (Sham surgery group). Results are in box plot (n = 7–8). *P < 0.05 compared with wild-type mice
The mice that were not exposed to anesthesia, brain ischemia and hypoxia were subjected to Barnes maze to test their spatial learning and memory. The time for mice to identify the target box was decreased with increased training sessions. This time on day 4 for both wild-type and heterozygous UQCRC1+/− mice was shorter than the time on day 1. Also, the heterozygous nature is a significant factor to influence the performance of these mice during the training sessions of Barnes maze test [F(1, 17) = 5.766, P = 0.028]. In addition, the heterozygous UQCRC1+/− mice took a longer time than wild-type mice to identify the target box at 1 day and 8 days after the training sessions (Fig. 7a, b). These results suggest that the heterozygous UQCRC1+/− mice have poorer spatial learning and memory than wild-type mice. The heterozygous UQCRC1+/− mice also performed poorly in novel object recognition test compared with wild-type mice (Fig. 7c), providing additional evidence that these heterozygous mice have poor learning and memory.
Fig. 7.

Impaired performance of the heterozygous UQCRC1+/− mice without brain ischemia and hypoxia in Barnes maze and novel object recognition test. a Results of the training sessions of Barnes maze. b Results of the memory phase of Barnes maze. Results are mean ± SD (n = 8–11). ^P < 0.05 compare with the corresponding data on day 1. *P < 0.05 compared with wild-type mice. c Novel object recognition. Results are mean ± SD (n = 9–14). *P < 0.05 compared with wild-type mice. d Total travel distance in open field. e Performance in open field. Results are mean ± SD (n = 13–16)
The amount of time spent in the corner, center and border areas was not different between the heterozygous UQCRC1+/− mice and wild-type mice. They traveled the same distances in the open field test (Fig. 7d, e). These results suggest that the heterozygous UQCRC1+/− mice and wild-type mice have similar exploratory behavior. These two types of mice also have a similar anxiety level.
Biochemical changes induced by UQCRC1 down-regulation
To determine possible mechanisms for the decreased brain ischemic tolerance and impaired learning and memory, biochemical analysis was performed. First, there was a decreased amount of complex III in the brain of the UQCRC1+/− mice as shown in the native page (Fig. 8a, b). Consistent with this finding, the complex III activity at baseline in the heterozygous UQCRC1+/− mice was lower than that of wild-type mice. The mitochondrial membrane potential (MMP) and cellular ATP content in the heterozygous UQCRC1+/− mice were also lower than those in wild-type mice (Fig. 8c–e). The activity of the complex III, MMP and ATP content in the ischemic brain tissues was reduced and the free radical levels in the ischemic brain tissues were increased compared with the corresponding non-ischemic brain tissues of the wild-type mice or the heterozygous UQCRC1+/− mice (Fig. 8c–f). These results suggest that the heterozygous UQCRC1+/− mice have a disturbed energy supply and MMP at baseline and that brain ischemia further disturbs these basic biochemical processes and increases oxidative stress.
Fig. 8.

Biochemical changes in the ischemic and non-ischemic tissues of wild-type and heterozygous UQCRC1+/− mice. a Representative images of native page. MW molecular weight markers, WT wild-type mice, HZ heterozygous mice, WB western blotting. Western blotting was performed with an anti-UQCRC2 antibody to identify the location of complex III. b Quantification of complex III in native pages. Results are mean ± SD (n = 7–9). #P < 0.05 compared with wild-type mice. c Complex III activity. d Mitochondrial membrane potential. e ATP content. f Free radicals. Results are mean ± SD (n = 6 for c, 11 for d, 12 for e and 6 for f). *P < 0.05 compared with the corresponding non-ischemic tissues of the wild-type mice. ^P < 0.05 compared with the corresponding non-ischemic tissues of the heterozygous UQCRC1+/− mice. $P < 0.05 compared with the corresponding ischemic tissues of wild-type mice
Discussion
UQCRC1 is a core protein of the respiratory chain complex III [1–3, 5]. We attempted to get homozygous UQCRC1−/− offspring by crossing the heterozygous UQCRC1+/− F1 mice. Unfortunately, although a large number of postnatal mice had been collected, no homozygous UQCRC1−/− mice were found. Only one homozygous UQCRC1−/− embryo was detected among 39 embryos at E9.5 and no homozygous UQCRC1−/− embryo was observed at E15.5, implying that the UQCRC1 plays an essential role during embryo development and for embryo survival and that disruption of this gene causes early embryonic lethality.
The incidence of mitochondria disorders in human is 1:10,000 live births. Deficiencies in complex I and IV are the two most common causes of mitochondria disorders. The deficiency of other components of the respiratory chain is comparatively scarce. Combined complex I and complex III deficiency was found in a patient due to the mutation of NADH:ubiquinone oxidoreductase subunit S4 gene of complex I. This patient died at the age of 3 months [28]. Another patient that has deficiency of complex I and complex III suffers from benign congenital myopathy [28, 29]. Mutations of the complex III cause respiratory enzyme dysfunction in human [30]. Among complex III deficiency-caused diseases, the mitochondrial encoded cytochrome b mutation is the commonest form. Several mutations of cytochrome b have been found, which lead to diseases, such as Leber hereditary optic neuropathy [31], progressive exercise muscle intolerance [32–34], histiocytoid cardiomyopathy [35] and ischemic cardiomyopathy [36]. Of note, mutations of the UQCRC1 gene in human have not been reported.
In our study, the heterozygous UQCRC1+/− mice have reduced UQCRC1 mRNA and protein expression, suggesting a partially dominant mutation in our heterozygous mice. These mice had a decreased brain ischemic tolerance as shown in two different brain ischemia models. In addition, these mice had impaired spatial and vision-dependent learning and memory. These results suggest a critical role of UQCRC1 in maintaining brain functions.
UQCRC1 is a mitochondrial protein [1–3]. It combines with other 10 proteins to form the complex III, which catalyzes the reduction of cytochrome c [4, 5]. This process pumps four protons from the mitochondrial matrix to the intermembrane space to maintain MMP [37]. UQCRC1 is not an electron transport protein but may act as a core protein to facilitate the interaction between cytochrome c (complex IV component) and cytochrome c1 (Complex III component) [4].
As the electron is passing through the five complexes of the electron transport chain, proton is moved from the matrix of the mitochondria to the space between the inner and outer membranes of the mitochondria. This process produces energy that is often stored as ATP [37–39]. Block of normal electron transport can decrease energy production and MMP. In addition, leaked electron can react with oxygen to produce ROS [40, 41]. These pathophysiological changes may harm cells, especially when detrimental insults, such as ischemia, are present [11, 40, 41].
Very little is known about the biological function of UQCRC1. It is not known whether UQCRC1 is necessary for maintaining complex III activity. If it is necessary, the decrease of UQCRC1 expression may lead to a decreased complex III activity, which can result in disrupted MMP, reduced ATP production and increased ROS. These pathophysiological changes may make tissues or organs very vulnerable to injury [11]. Consistent with this possibility, the heterozygous UQCRC1+/− mice had decreased MMP and ATP content and increased ROS compared to the wild-type mice in the ischemic brain tissues, which may explain the decreased brain ischemic tolerance of the heterozygous UQCRC1+/− mice. Interestingly, the heterozygous UQCRC1+/− mice had decreased complex III formation, complex III activity, MMP and ATP content in their brain at baseline, which may be a mechanism for the impaired learning and memory in these mice because ATP supply impairment and oxidative stress can impair learning and memory [42]. In addition, the heterozygous UQCRC1+/− mice had decreased UQCRC1 expression and complex III formation and activity. These findings, along with the finding that UQCRC1 knockout is lethal, indicate that UQCRC1 is an important component of complex III.
Of note, cell-type distribution of UQCRC1 and UQCRC2 has not been determined. Our results showed that UQCRC1 and UQCRC2 were expressed in neurons and astrocytes. However, neither UQCRC1 nor UQCRC2 appeared to be co-localized with the staining of Iba-1 or TMEM119, two microglial markers [25, 26], suggesting that microglia may not express a detectable amount of UQCRC1 and UQCRC2. These findings are consistent with that in a previous study in which a mitochondrial protein was not found in microglia [43]. Interestingly, some cells that expressed UQCRC1 did not appear to express UQCRC2, suggesting that UQCRC1 has a wider expression pattern than UQCRC2. Together, these findings suggest that not all respiratory chain components are expressed in all cells. Further studies are required to test this provocative suggestion. Nevertheless, the wider expression pattern of UQCRC1 may explain why UQCRC2 cannot compensate the functions of UQCRC1.
We did not see any difference between the wild-type and heterozygous UQCRC1+/− mice in the travel distance in the open field test. This result suggests that the difference observed in the Barnes maze and novel object recognition tests may not be caused by decreased motor functions in the heterozygous UQCRC1+/− mice. Also, the exploratory and anxious behaviors of the heterozygous mice are similar to those of wild-type mice, suggesting that these heterozygous mice do not have a generalized behavioral impairment. Interestingly, being heterozygous in the UQCRC1 gene did not affect the general physiological characteristics including growth curve, respiratory rate, heart rate and mean arterial blood pressure, suggesting ineffectiveness of mild impairment of the complex III on these basic life parameters.
In conclusion, our results suggest that UQCRC1 is critical for the development and survival of embryos. UQCRC1 is also important for the proper function of the complex III, which may affect the brain ischemic tolerance and normal learning and memory.
Materials and methods
The animal protocol was approved by the institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 2011.
Eighteen pregnant female mice were used to isolate embryos to generate data presented in Fig. 1c. About 475 mice were used to generate the data presented in the other figures.
Generation of Uqcrc1 knockout/knockdown mice
The F1 heterozygous Uqcrc1 mice with partial gene sequence replaced by Lac-Neo cassette in one of the Uqcrc1 alleles were generated using 129/OlaHsd targeted ES cells obtained from Deltagen. The targeted ES cells harboring a LacO-SA-IRES-lacZ-wtNEO/Kan cassette insert that spanned part of exon 3, exon 4 and the intervening intronic sequence were used to generate chimeras by blastocyst injection. The chimeras were then crossed with wild-type C57BL/6N to get heterozygous F1 offspring. F2 offspring mice were produced by intercrossing F1 heterozygous males and females. The F2 heterozygous mice were intercrossed again to get the F3 offspring as we described before [44]. All these mice were genotyped by PCR. The wild-type and heterozygous mice generated from this cross-breeding were used in the experiments described in the following sections.
Isolation and in vitro culture of mouse embryos
As we described before [44], female heterozygous mice at 4 weeks old received intraperitoneal injection of pregnant mare serum gonadotropin (National Hormone and Peptide Program, Torrance, CA, USA) followed 48 h later by an injection of human chorionic gonadotropin (National Hormone and peptide program, Torrance, CA, USA). The female mice were then mated with sexually mature male heterozygous mice. Fertilized eggs were collected in EmbryoMax® flushing and holding medium (FHM; Merck Millipore, Billerica, MA, USA), treated with hyaluronidase (Sigma, St. Louis, MO, USA) and rinsed with FHM. The embryos were then cultured in EmbryoMax® potassium-supplemented simplex optimized medium (MerckMillipore) overlaid with mineral oil in a humidified air containing 5% CO2 at 37 °C. Embryos were lyzed at various times in a lysis buffer consisting of 50 mM NaOH at 95 °C for 5 min. The solution was cooled to room temperature and then neutralized with 50 mM Tris–HCl (pH 8.0). The lysate was then used for PCR.
Genotyping of the Uqcrc1 knockout/knockdown mice
The genomic DNA from post-implantation embryos and mouse tails was extracted using the DNeasy blood and tissue kit (QIAGEN, USA). The primers of F1 (5′-GGGATGTGGAGCCAGAATCAACAAC-3′) and R1 (5′-CCAGAGCTACCCAACAGCCTATCTT-3′) were designed to identify the gene sequence of Uqcrc1 that should be substituted by the Neo cassette in the heterozygous and homozygous mice. The primers of F2 (5′-AGGATCTCCTGTCATCTCACCTTGCTCCTG-3′) and R2 (5′-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-3′) were used to verify the inserted Neo cassette (Fig. 1). The PCR program was set as 94 °C for 5 min, 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 35 s per cycle for 35 cycles and then 72 °C for 10 min.
Physiological measurement
The body weights of male and female mice were recorded once every week for 8 weeks with the first measurement at an age of 1 week.
Heart rate and blood pressure were measured at an age of about 8 weeks with a non-invasive CODA monitor (Kent Scientific Corporation, Connecticut, USA) as we did before [45]. Briefly, the mouse was kept in a mouse restrainer and wrapped with warming pad for 5 min. Heart rate and blood pressure were then measured via a tail cuff by a CODA monitor. Mean value from a total of five measurements of each mouse was used for statistical analysis. Respiratory rate was counted three times manually and their mean value was used for statistical analysis.
Total RNA extraction and real-time PCR
Cortex and hippocampus were harvested from wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) mice under deep anesthesia and after cardiac perfusion with normal saline. RNeasy mini kit (catalog number: 74104, QIAGEN) was used for RNA isolation. Reverse transcription PCR was conducted by 5 × All-In-One RT MasterMix (catalog number: G486, Applied Biological Materials Inc., Richmond, Canada) for cDNA synthesis. A total of 125 ng cDNA was used for real-time PCR procedure by the CFX Connect system (Bio-Rad). Primers were as follows: Uqcrc1, F3 (5′-CAGTGTCTCCCGAGTGTATG-3′) and R3 (5′-GGTCACGTTGTCTGGGTTAG-3′); Uqcrc2, F4 (5′-CCAAATACCGTGGAGGTG-3′) and R4 (5′-ACACTCTGGCTCAGGAGG-3′). The house keeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal reference [F5 (5′-AATGTGTCCGTCGTGGATCT-3′) and R5 (5′-GGTCCTCAGTGTAGCCCAAG-3′)]. The relative amount of mRNA was determined using the comparative threshold cycle method.
Native-PAGE
Samples prepared from freshly isolated mitochondria of wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) mice were applied to native PAGE to evaluate the quantity of complex III according to a method described before [46]. Gel was scanned by a Bio-Rad GS 800 densitometer and quantified by Image J. The band corresponding to the complex III was confirmed by Western blotting with an anti-UQCRC2 antibody.
Western blotting
The expression level of UQCRC1 and UQCRC2 in wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) mice was assessed by Western blotting. Cortex and hippocampus of wild-type and heterozygous mice without any interventions were harvested and homogenized in RIPA buffer (Thermo Fisher Scientific, Waltham, MA) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Twenty microgram protein was loaded per lane and separated by 12% gels (Bio-Rad Laboratories, Hercules, CA, USA). After transferred to a polyvinylidine difluoride membrane (catalogue number: 162-0177, Bio-Rad), the membrane was blocked for 1 h in blocking buffer (Pierce Protein-free T20, catalogue number: PI207535, Thermo Fisher Scientific) and then incubated with a primary antibody overnight at 4 °C. The primary antibodies were rabbit polyclonal anti-UQCRC1 antibody (1:1000 dilution, catalogue number: ab96333, Abcam, Cambridge, United Kingdom), rabbit monoclonal anti-UQCRC2 antibody (1:1000 dilution, catalogue number: ab203832, Abcam) and rabbit polyclonal anti-GAPDH (1:5000 dilution, catalogue number: G9545, Sigma). Secondary antibodies were goat anti-rabbit antibody and goat anti-mouse antibody conjugated with horse radish peroxidease (1:5000 dilution, Santa Cruz Biotechnology, Dallas, TX). Protein bands were quantified by Genetools version 4.01. The relative protein expression of UQCRC1 and UQCRC2 was normalized to the level of GAPDH in the same sample.
Immunofluorescent staining
Immunofluorescent staining of UQCRC1 and UQCRC2 in the brain was conducted as described before [47]. Wild-type male and female mice were perfused with normal saline. Brain slices at Bregma − 2 to − 5 mm were fixed in 4% paraformaldehyde for 2 days at 4 °C and then embedded in paraffin. Five-micrometer thick coronal sections were cut and mounted on slides. Antigen retrieval was performed in sodium citrate buffer (10 mM sodium citrate, 0.1% Tween 20, pH 6.0) for 20 min. Primary antibodies used were mouse monoclonal anti-UQCRC1 (1:100 dilution, catalogue number: ab110252, Abcam), rabbit polyclonal anti-UQCRC1 (1:100 dilution, catalogue number: ab96333, Abcam), mouse monoclonal anti-UQCRC2 (1:500 dilution, catalogue number: ab14745, Abcam), rabbit monoclonal anti-UQCRC2 (1:200 dilution, catalogue number: ab203832, Abcam), mouse monoclonal anti-MAP2 (1:200 dilution, catalogue number: ab7756, Abcam), goat polyclonal anti-GFAP (1:200 dilution, catalogue number: ab53554, Abcam), rabbit monoclonal anti-TMEM119 (1:100 dilution, catalogue number: ab209064, Abcam), and rabbit polyclonal anti-Iba-1 (1:200 dilution, catalogue number: 019-19741, Wako Chemicals USA, Richmond, VA). Secondary antibodies were donkey anti-rabbit IgG conjugated with Alexa Fluor 488/594 (1:200 dilution, catalogue number: A-21206/A-21207, Invitrogen, Eugene, ON); donkey anti-goat IgG conjugated with Alexa Fluor 488 (1:200 dilution, catalogue number: A-11055, Invitrogen) and donkey anti-mouse IgG conjugated with Alexa Fluor 488/594 (1:200 dilution, catalogue number: A21202/A-21203, Invitrogen). They were applied for 1 h at room temperature in the dark. Sections were counterstained with Hoechst 33342 (1:1000 dilution, catalogue number: 62249, Thermo Scientific, Pittsburgh, PA) for 5 min, then rinsed and mounted with Vectashield mounting medium (catalogue number: H-1000, Vector Laboratories, Burlingame, CA, USA). Images were acquired with a fluorescent microscope (Olympus DP70, Olympus Corporation, Tokyo, Japan).
Brain hypoxia–ischemia model and middle cerebral arterial occlusion model
Ten-week-old wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) C57BL/6N male and female mice were subjected to brain ischemia/hypoxia. These mice had a left common carotid arterial ligation [48–50]. To achieve this, mice were anesthetized with 2.0% isoflurane that were delivered by an anesthetic vaporizer (Vaporizer Sale & Service, Inc., Rockmart, GA) gassed with oxygen. The concentration of isoflurane was monitored by an anesthetic analyzer (Datex-Engstrom Helsinki, Finland). A 1.5 cm incision at the midline of neck was made. The left common carotid artery was dissected carefully without damaging the vagus nerve. The artery was cut after both sides were ligated with a 6-0 silk. The wound was closed with a clip and the mouse was placed in a warm container until recovery. A servo-controlled warming blanket (TCAT-2LV, Physitemp instruments Inc., Clifton, NJ) was used to maintain the rectal temperature at 37 °C. The mouse’s heart rate and pulse oxygen saturation were monitored by a MouseOX Murine Plus Oximeter System (Starr Life Sciences Corporation, Oakmont, PA). All animals received a subcutaneous injection of 3 mg/kg bupivacaine after surgery. The surgery lasted for 8 min for each mouse. Two hours after the carotid artery ligation and cut, wild-type and heterozygous mice were put in a chamber filled with humidified 8% oxygen in nitrogen for 60 min at 37 °C. This hypoxia chamber was placed in a cell culture incubator whose temperature was set at 37 °C. The oxygen concentration and temperature in the hypoxia chamber were continuously monitored. This brain ischemia/hypoxia model is adopted from a neonatal hypoxic–ischemic encephalopathy model as we described before [48–50] and has been used in adult mice [51].
To induce focal brain ischemia, 10-week-old wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) C57BL/6N male and female mice were subjected to left MCAO as we described before [27]. Briefly, mice were anesthetized with 2.0% isoflurane that was delivered by an anesthetic vaporizer (Vaporizer Sale & Service, Inc., Rockmart, GA, USA) gassed with oxygen. The concentration of isoflurane was monitored by an anesthetic analyzer (Datex-Engstrom Helsinki, Finland). The MCAO was achieved by inserting a monofilament nylon suture (Beijing CiNontech Co. Ltd., Beijing, China) with a rounded end to the left internal carotid artery via the left external carotid artery until slight resistance was felt. The ipsilateral common carotid artery was occluded temporarily during the placement of the suture. Mice were re-anesthetized by isoflurane to remove the suture 90 min later. The wound was closed with a clip and the mouse was placed in a warm container until recovery. Mice in the Sham surgery group had an identical surgical procedure to that of MCAO group but without the insertion of the suture. During surgery, a servo-controlled warming blanket (TCAT-2LV, Physitemp instruments Inc., Clifton, NJ) was used to maintain the rectal temperature at 37 °C. Their heart rate and pulse oxygen saturation were monitored by a MouseOX Murine Plus Oximeter System (Starr Life Sciences Corporation, Oakmont, PA). All animals received a subcutaneous injection of 3 mg/kg bupivacaine after surgery for pain control.
Evaluation of neurological deficit scores and infarct volumes
Neurological deficit scores were evaluated based on an eight-point scale [52]. This evaluation was performed just before the animals were used for brain infarction assessment. Mice were scored as follows: 0, no apparent deficits; 1, failure to extend right forepaw fully; 2, decreased grip of the left forelimb; 3, spontaneous movement in all directions, contralateral circling only if pulled by the tail; 4, circling or walking to the left; 5, walking only if stimulated; 6, unresponsiveness to stimulation and with depressed level of consciousness; and 7, dead. The score was evaluated by a person who is blinded to the mouse types.
The assessment of infarct volumes at 72 h after brain ischemia/hypoxia or 24 h after MCAO was performed after the staining of brain slices with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (catalog number: T8877, Sigma, St. Louis, MO) as we described before [53, 54]. Briefly, the mouse was deeply anesthetized by isoflurane and perfused with normal saline. The brain was harvested and sliced with a microtome into 6 pieces at 2 mm thickness. Slices were incubated in 2% TTC for 30 min at 37 °C. After fixation in 4% paraformaldehyde overnight, the slices were pictured and analyzed by image J. The percentage of infarct volume in the contralateral hemisphere volume was calculated [55]. The quantification was performed by a person who is blinded to the mouse types. Animals in the MCAO group were eliminated from the study if there was no brain infarction as assessed by TTC staining.
Barnes maze
Eight-week-old wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) mice without hypoxia and brain ischemia were assessed by Barnes maze test as we did before [56]. The mouse was placed in the center of a circle platform that has 20 equally spaced holes with one of them connected to a dark chamber (target box) (SD Instruments, San Diego, CA, USA). Aversive noise (85 dB) and bright light (200 W) shed on the platform to encourage the mouse to find the target box. All mice were trained for 4 consecutive days with 3 min per trial, 4 trials per day. Their reference memory was tested on day 5 (short-term retention) and day 12 (long-term retention). Each mouse had only one trial on each of these two test days. No test was performed during the period from day 5 to day 12. The latency to find the target box during each trial was recorded with ANY-Maze video tracking system (Stoelting Co., IL).
Open-field and novel object recognition tests
As described before [57], 8-week-old mice were put in the open-field box for 10 min. The time of mice spent in the corner, border and center areas and the travel distance were recorded by ANY-maze behavioral tracking software.
One day after open field test, mice were subjected to novel object recognition test. As described before [58], two identical objects were placed in opposite sides of the box on the training day. A mouse was placed in the center and allowed to explore the box for 5 min. An animal was eliminated from the experiment if the total exploration time on two objects was less than 5 s. Twenty-four hours later, a novel object and a familiar object were placed in the same locations again as in the training phase. The mouse was put in the middle of the box and allowed to explore for 5 min. Animal behavior (e.g., the time and frequency of exploring novel/familiar object) was recorded by ANY-maze behavioral tracking software. The ratio of time spent with the novel object to the total exploring time was calculated.
Assessment of reactive oxygen species
The wild-type (Uqcrc1+/+) and heterozygous (Uqcrc1+/−) mice were randomly assigned to the control and MCAO groups. The control group of mice was not subjected to anesthesia or surgical procedure. The OxiSelect™ In Vitro ROS/RNS Assay Kit (catalog number: STA-347, Cell Biolabs Inc., San Diego, CA, USA) was chosen to detect ROS as we did before [59]. Briefly, the left frontal cortex area 1 (Fr1) at Bregma 1 to − 1 was harvested 24 h after MCAO or from control mice. Tissue was homogenized on ice in cold phosphate-buffered saline (10 mg/100 μL), and centrifuged at 10,000g for 5 min at 4 °C. Fifty microliter supernatant was mixed with 50 μL catalyst in a well of 96-well plates and incubated at room temperature for 5 min. Subsequently, 2′,7′-dichlorodihydrofluorescin diacetate solution (100 μL) was added to each well and was reacted for 45 min in the dark. Fluorescence at 486 nm excitation and 538 nm emission was read on a fluorescence plate reader (Gemini EM Microplate Reader, Molecular Devices, Sunnyvale, CA, USA). Measurements were performed in triplicate and then averaged as the value for each mouse.
Measurement of mitochondrial transmembrane potential
Fresh Fr1 at 24 h after MCAO was prepared into single cell suspension in Neurobasal-A medium (20 mg/400 μL, catalog number: A24775-01, Thermo Fisher Scientific) by passing the tissues through a 70 µm mesh in a way similar to what we described before [60, 61]. Tetramethylrhodamine ethyl ester (catalog number: K238-200, BioVision) was used to detect the transmembrane potential changes per the manufacture’s instruction. Measurements were run in duplicate and then averaged as the value for each mouse.
Mitochondrial isolation
Mitochondria of control animals and animals with MCAO were isolated by the Mitochondria Isolation Kit for Tissue and Cultured Cells (catalog number: K288-50, BioVision, Milpitas, CA, USA) according to the instructions and similar to the protocol we used before [62]. Briefly, the mouse was deeply anesthetized 24 h after MCAO. Left cerebral cortex at Bregma 1.5 to − 1 was collected and rinse in 1 mL cold isolation buffer. Tissue was homogenized with Dunce Tissue Homogenizer in isolation buffer (10 mg/100 μL) for 15 strokes. The homogenate was then transferred to a tube and centrifuged at 600g for 10 min at 4 °C. Supernatant was collected to a new tube and centrifuged at 7000g for 10 min at 4 °C. The formed pellet was re-suspended in 100 μL storage buffer. Protein concentration was measured by Bradford method. Isolated mitochondria were immediately used for mitochondrial complex III activity assay.
Assessing mitochondrial respiratory enzyme complex III activity
Mitochondrial Complex III Activity Assay Kit (catalog number: K520-100, BioVision) was used to measure the mitochondrial complex III activity according to the manufacturer’s instruction. The absorbance of reduced cytochrome c was measured at 550 nm by an end point model. A total of 10 μg mitochondria from each group were used for this assay. Measurements were run in triplicate. The mean value of the triplicates was used to represent the value of each mouse.
ATP assay
ATP content was measured by an ADP/ATP ratio Assay Kit (catalog number: ab65313, Abcam) with some modifications. Briefly, freshly harvested cortex tissue at Bregma − 1 to − 2.5 was prepared into a single-cell suspension with cold PBS (20 mg/400 μL). The sample was diluted at a ratio of 1:10 with nuclear releasing buffer, incubated for 5 min at room temperature and then added to 100 μL ATP monitoring enzyme mix. The bioluminescent intensity of the sample was read by a luminometer (GloMax 20/20, Promega). Measurements were conducted in duplicate. The mean value of the duplicates was used to represent the value of each mouse.
Statistical analysis
Parametric data with normal distribution are presented as mean ± SD (n ≥ 5). The other types of data are in box plot in the figures. One-way or two-way repeated measures analysis of variance followed by Tukey test was used to analyze the data from the training sessions of Barnes maze test within the same group or between groups, respectively. Other data were analyzed by t test, or rank sum test, one-way analysis of variance followed by the Tukey test or two-way analysis of variance followed by the Tukey test. Differences were considered statistically significant at P < 0.05 based on two-tailed hypothesis testing. All statistical analyses were performed with SigmaStat version 3.5 (Systat Software, Inc., Point Richmond, CA, USA). Sample size estimate was not performed for each study parameter before the experiments because there were no data to base on for performing the estimate. In addition, it is difficult to select a primary outcome for sample size estimate in a complex basic science study. Thus, we aimed to have a sample size at least five for biochemical analysis and eight for behavior study based on our experience and sample size for these types of studies in the literature [63, 64].
Significance statement
Ubiquinol cytochrome c reductase core protein I is important by forming complex III to maintain normal brain ischemic tolerance and cognition and for embryo survival.
Acknowledgements
This study was supported by Grants (GM098308, AG056995, HD089999 and NS099118) from the National Institutes of General Medical Sciences and National Institutes of Health, Bethesda, MD, the Robert M. Epstein Professorship endowment, University of Virginia, Charlottesville, VA.
Abbreviations
- UQCRC1
Ubiquinol cytochrome c reductase core protein I
- UQCRC2
Ubiquinol cytochrome c reductase core protein 2
- MAP-2
Microtubule associated protein 2
- GFAP
Glial fibrillary acidic protein
- Iba-1
Ionized calcium-binding adaptor molecule 1
- TMEM119
Transmembrane protein 119
- MCAO
Middle cerebral arterial occlusion
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- TTC
2,3,5-Triphenyltetrazolium chloride
- ROS
Reactive oxygen species
Author contributions
ZZ conceived the project. WS, JL, WX, HL and ZZ designed the study, WS, JL and WX performed the experiments. WS did the initial data analysis and drafted Methods section. ZZ performed the final data analysis and wrote the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare no competing financial interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schulte U, Arretz M, Schneider H, Tropschug M, Wachter E, Neupert W, Weiss H. A family of mitochondrial proteins involved in bioenergetICS and biogenesis. Nature. 1989;339:147–149. doi: 10.1038/339147a0. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman GG, Lee S, Christiano AM, Chung-Honet LC, Cheng W, Katchman S, Uitto J, Greenspan DS. Complete coding sequence, intron/exon organization, and chromosomal location of the gene for the core I protein of human ubiquinol-cytochrome c reductase. J Biol Chem. 1993;268:21113–21119. [PubMed] [Google Scholar]
- 3.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CA, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 4.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 5.Fu W, Beattie DS. Assembly of the iron-sulfur protein into the cytochrome b–c1, complex of yeast mitochondria. J Biol Chem. 1991;266:16212–16218. [PubMed] [Google Scholar]
- 6.Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin HB, Cadete VJ, Sawicka J, Wozniak M, Sawicki G. Effect of the myosin light chain kinase inhibitor ML-7 on the proteome of hearts subjected to ischemia–reperfusion injury. J Proteom. 2012;75:5386–5395. doi: 10.1016/j.jprot.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 8.Wong R, Aponte AM, Steenbergen C, Murphy E. Cardioprotection leads to novel changes in the mitochondrial proteome. Am J Physiol Heart Circ Physiol. 2010;298:H75–H91. doi: 10.1152/ajpheart.00515.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi T, Wu X, Long Z, Duan G, Wu Z, Li H, Chen H, Zhou X. Overexpression of ubiquinol-cytochrome c reductase core protein 1 may protect H9c2 cardiac cells by binding with zinc. Biomed Res Int. 2017;2017:1314297. doi: 10.1155/2017/1314297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long Z, Duan G, Li H, Yi T, Wu X, Chen F, Wu Z, Gao Y. Ubiquinol-cytochrome c reductase core protein 1 may be involved in delayed cardioprotection from preconditioning induced by diazoxide. PLoS One. 2017;12:e0181903. doi: 10.1371/journal.pone.0181903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 12.Davis M, Whitely T, Turnbull DM, Mendelow AD. Selective impairments of mitochondrial respiratory chain activity during aging and ischemic brain damage. Acta Neurochir Suppl. 1997;70:56–58. doi: 10.1007/978-3-7091-6837-0_17. [DOI] [PubMed] [Google Scholar]
- 13.Moro MA, Almeida A, Bolanos JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005;39:1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Chen SD, Wu HY, Yang DI, Lee SY, Shaw FZ, Lin TK, Liou CW, Chuang YC. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem Biophys Res Commun. 2006;351:198–203. doi: 10.1016/j.bbrc.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Ames A., 3rd CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000;34:42–68. doi: 10.1016/S0165-0173(00)00038-2. [DOI] [PubMed] [Google Scholar]
- 16.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao YX, Cui M, Chen SF, Dong Q, Liu XY. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci Ther. 2014;20:528–538. doi: 10.1111/cns.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, Gagnon E, McBride HM, Desjardins M. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- 20.Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. 2016;46:13–21. doi: 10.1002/eji.201445427. [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Mou Y, Bernstock JD, Klimanis D, Wang S, Spatz M, Maric D, Johnson K, Klinman DM, Li X, Li X, Hallenbeck JM. Synthetic oligodeoxynucleotides containing multiple telemeric TTAGGG motifs suppress inflammasome activity in macrophages subjected to oxygen and glucose deprivation and reduce ischemic brain injury in stroke-prone spontaneously hypertensive rats. PLoS One. 2015;10:e0140772. doi: 10.1371/journal.pone.0140772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal. 2011;14:1437–1448. doi: 10.1089/ars.2010.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niizuma K, Yoshioka H, Chen H, Kim GS, Jung JE, Katsu M, Okami N, Chan PH. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta. 2010;1802:92–99. doi: 10.1016/j.bbadis.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacque CM, Vinner C, Kujas M, Raoul M, Racadot J, Baumann NA. Determination of glial fibrillary acidic protein (GFAP) in human brain tumors. J Neurol Sci. 1978;35:147–155. doi: 10.1016/0022-510X(78)90107-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Tan H, Jiang W, Zuo Z. Amantadine alleviates postoperative cognitive dysfunction possibly by increasing glial cell line-derived neurotrophic factor in rats. Anesthesiology. 2014;121:773–785. doi: 10.1097/ALN.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology. 2016;36:39–49. doi: 10.1111/neup.12235. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Shan W, Zuo Z. Age-related upregulation of carboxyl terminal modulator protein contributes to the decreased brain ischemic tolerance in older rats. Mol Neurobiol. 2018;55(7):6145–6154. doi: 10.1007/s12035-017-0826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Budde SM, van den Heuvel LP, Janssen AJ, Smeets RJ, Buskens CA, DeMeirleir L, Van Coster R, Baethmann M, Voit T, Trijbels JM, Smeitink JA. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem Biophys Res Commun. 2000;275:63–68. doi: 10.1006/bbrc.2000.3257. [DOI] [PubMed] [Google Scholar]
- 29.Castro-Gago M, Eiris J, Pintos E, Rodrigo E, Blanco-Barca O, Campos Y, Arenas J. Benign congenital myopathy associated with a partial deficiency of complexes I and III of the mitochondrial respiratory chain. Rev Neurol. 2000;31:838–841. [PubMed] [Google Scholar]
- 30.Haut S, Brivet M, Touati G, Rustin P, Lebon S, Garcia-Cazorla A, Saudubray JM, Boutron A, Legrand A, Slama A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum Genet. 2003;113:118–122. doi: 10.1007/s00439-003-0946-0. [DOI] [PubMed] [Google Scholar]
- 31.Johns DR, Neufeld MJ. Cytochrome b mutations in Leber hereditary optic neuropathy. Biochem Biophys Res Commun. 1991;181:1358–1364. doi: 10.1016/0006-291X(91)92088-2. [DOI] [PubMed] [Google Scholar]
- 32.Andreu AL, Bruno C, Shanske S, Shtilbans A, Hirano M, Krishna S, Hayward L, Systrom DS, Brown RH, Jr, DiMauro S. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology. 1998;51:1444–1447. doi: 10.1212/WNL.51.5.1444. [DOI] [PubMed] [Google Scholar]
- 33.Bouzidi MF, Carrier H, Godinot C. Antimycin resistance and ubiquinol cytochrome c reductase instability associated with a human cytochrome b mutation. Biochim Biophys Acta. 1996;1317:199–209. doi: 10.1016/S0925-4439(96)00055-5. [DOI] [PubMed] [Google Scholar]
- 34.Dumoulin R, Sagnol I, Ferlin T, Bozon D, Stepien G, Mousson B. A novel gly290asp mitochondrial cytochrome b mutation linked to a complex III deficiency in progressive exercise intolerance. Mol Cell Probes. 1996;10:389–391. doi: 10.1006/mcpr.1996.0053. [DOI] [PubMed] [Google Scholar]
- 35.Andreu AL, Checcarelli N, Iwata S, Shanske S, DiMauro S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr Res. 2000;48:311–314. doi: 10.1203/00006450-200009000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Marin-Garcia J, Hu Y, Ananthakrishnan R, Pierpont ME, Pierpont GL, Goldenthal MJ. A point mutation in the cytb gene of cardiac mtDNA associated with complex III deficiency in ischemic cardiomyopathy. Biochem Mol Biol Int. 1996;40:487–495. doi: 10.1080/15216549600201053. [DOI] [PubMed] [Google Scholar]
- 37.Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu Rev Biophys Biomol Struct. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–139. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- 39.Dimroth P, Kaim G, Matthey U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol. 2000;203:51–59. doi: 10.1242/jeb.203.1.51. [DOI] [PubMed] [Google Scholar]
- 40.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–508. doi: 10.1016/S0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 41.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 42.Yamagami S, Tamura M, Hayashi M, Endo N, Tanabe H, Katsuura Y, Komoriya K. Differential production of MCP-1 and cytokine-induced neutrophil chemoattractant in the ischemic brain after transient focal ischemia in rats. J Leukoc Biol. 1999;65:744–749. doi: 10.1002/jlb.65.6.744. [DOI] [PubMed] [Google Scholar]
- 43.Hu WH, Hausmann ON, Yan MS, Walters WM, Wong PK, Bethea JR. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP) J Neurochem. 2002;81:36–45. doi: 10.1046/j.1471-4159.2002.00788.x. [DOI] [PubMed] [Google Scholar]
- 44.Feng C, Xu W, Zuo Z. Knockout of the regulatory factor X1 gene leads to early embryonic lethality. Biochem Biophys Res Commun. 2009;386:715–717. doi: 10.1016/j.bbrc.2009.06.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng J, Li J, Li L, Feng C, Xiong L, Zuo Z. Glutamate transporter type 3 knockout leads to decreased heart rate possibly via parasympathetic mechanism. Transgenic Res. 2013;22:757–766. doi: 10.1007/s11248-012-9680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz F, Barrientos A, Fontanesi F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using blue native gel electrophoresis. Curr Protoc Hum Genet Chapter. 2009;19(Unit19):14. doi: 10.1002/0471142905.hg1904s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi J, Shan W, Luo A, Zuo Z. Critical role of matrix metallopeptidase 9 in postoperative cognitive dysfunction and age-dependent cognitive decline. Oncotarget. 2017;8(31):51817–51829. doi: 10.18632/oncotarget.15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao P, Peng L, Li L, Xu X, Zuo Z. Isoflurane preconditioning improves long-term neurologic outcome after hypoxic-ischemic brain injury in neonatal rats. Anesthesiology. 2007;107:963–970. doi: 10.1097/01.anes.0000291447.21046.4d. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Zhao H, Peng S, Zuo Z. Intranasal pyrrolidine dithiocarbamate decreases brain inflammatory mediators and provides neuroprotection after brain hypoxia-ischemia in neonatal rats. Exp Neurol. 2013;249:74–82. doi: 10.1016/j.expneurol.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren X, Ma H, Zuo Z. Dexmedetomidine postconditioning reduces brain injury after brain hypoxia-ischemia in neonatal rats. J Neuroimmune Pharmacol. 2016;11:238–247. doi: 10.1007/s11481-016-9658-9. [DOI] [PubMed] [Google Scholar]
- 51.Kossatz E, Maldonado R, Robledo P. CB2 cannabinoid receptors modulate HIF-1alpha and TIM-3 expression in a hypoxia-ischemia mouse model. Eur Neuropsychopharmacol. 2016;26:1972–1988. doi: 10.1016/j.euroneuro.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke. 1997;28:2060–2065. doi: 10.1161/01.STR.28.10.2060. [DOI] [PubMed] [Google Scholar]
- 53.Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- 54.Li L, Zuo Z. Isoflurane preconditioning improves short-term and long-term neurological outcome after focal brain ischemia in adult rats. Neuroscience. 2009;164:497–506. doi: 10.1016/j.neuroscience.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume [see comments] J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, Jiang W, Zuo Z. Pyrrolidine dithiocarbamate attenuates surgery-induced neuroinflammation and cognitive dysfunction possibly via inhibition of nuclear factor kappaB. Neuroscience. 2014;261:1–10. doi: 10.1016/j.neuroscience.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seibenhener ML, Wooten MC. Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp. 2015;96:e52434. doi: 10.3791/52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, Freret T. Object recognition test in mice. Nat Protoc. 2013;8:2531–2537. doi: 10.1038/nprot.2013.155. [DOI] [PubMed] [Google Scholar]
- 59.Li L, Wang Z, Zuo Z. Chronic intermittent fasting improves cognitive functions and brain structures in mice. PLoS One. 2013;8:e66069. doi: 10.1371/journal.pone.0066069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yun J, Li J, Zuo Z. Transferred inter-cell ischemic preconditioning-induced neuroprotection may be mediated by adenosine A1 receptors. Brain Res Bull. 2014;103:66–71. doi: 10.1016/j.brainresbull.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McMurtrey RJ, Zuo Z. Isoflurane preconditioning and postconditioning in rat hippocampal neurons. Brain Res. 2010;1358:184–190. doi: 10.1016/j.brainres.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li L, Peng L, Zuo Z. Isoflurane preconditioning increases B-cell lymphoma-2 expression and reduces cytochrome c release from the mitochondria in the ischemic penumbra of rat brain. Eur J Pharmacol. 2008;586:106–113. doi: 10.1016/j.ejphar.2008.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xing W, Huang P, Lu Y, Zeng W, Zuo Z. Amantadine attenuates sepsis-induced cognitive dysfunction possibly not through inhibiting toll-like receptor 2. J Mol Med (Berl) 2018;96:391–402. doi: 10.1007/s00109-018-1631-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Al-Ghobashy MA, ElMeshad AN, Abdelsalam RM, Nooh MM, Al-Shorbagy M, Laible G. Development and pre-clinical evaluation of recombinant human myelin basic protein nano therapeutic vaccine in experimental autoimmune encephalomyelitis mice animal model. Sci Rep. 2017;7:46468. doi: 10.1038/srep46468. [DOI] [PMC free article] [PubMed] [Google Scholar]