

Abstract
Cryptosporidium parvum is one of the major species causing mild to severe cryptosporidiosis in humans and animals. We have previously observed that 2-deoxy-D-glucose (2DG) could inhibit both the enzyme activity of C. parvum hexokinase (CpHK) and the parasite growth in vitro. However, the action and fate of 2DG in C. parvum was not fully investigated. In the present study, we showed that, although 2DG could be phosphorylated by CpHK to form 2DG-6-phosphate (2DG6P), the anti-cryptosporidial activity of 2DG was mainly attributed to the action of 2DG on CpHK, rather than the action of 2DG or 2DG6P on the downstream enzyme glucose-6-phosphate isomerase (CpGPI) nor 2DG6P on CpHK. These observations further supported the hypothesis that CpHK could serve as a drug target in the parasite. We also screened 1200 small molecules consisting of marketed drugs against CpHK, from which four drugs were identified as CpHK inhibitors with micromolar level of anti-cryptospordial activities at concentrations nontoxic to the host cells (i.e., hexachlorphene, thimerosal, alexidine dihydrochloride and ebselen with EC50 = 0.53, 1.77, 8.1 and 165 μM, respectively). The anti-CpHK activity of the four existing drugs provided us new reagents for studying the enzyme properties of the parasite hexokinase.
Keywords: Apicomplexan, Cryptosporidium parvum, hexokinase (HK), drug target, 2-deoxy-D-glucose (2DG)
Cryptosporidium parvum is zoonotic protozoan parasite causing mild to severe watery diarrhea in humans and some other mammals (Chen et al. 2002). Cryptosporidial infection can be prolonged and deadly in immunocompromised patients. It is also one of the four pathogens responsible for the most cases of moderate to severe diarrhea in children (0 – 5 years old) and increased risk of death in toddlers (1 – 2 years old) in low-income countries in sub-Saharan Africa and south Asia (Collaborators 2017, Kotloff et al. 2013). However, fully effective drugs are yet unavailable to treat cryptosporidiosis in humans and animals. In fact, nitazoxanide (NTZ) is the only drug approved by Food and Drug Administration (FDA) in the United States to treat cryptosporidial infection in human patients with a healthy immune system (Chen et al. 2002, Fox and Saravolatz 2005, Kelly 2011, Rossignol 2010).
Unlike other apicomplexans, Cryptosporidium has a compact genome that encodes a highly streamlined metabolism. In energy metabolism, C. parvum lacks Krebs cycle and cytochrome-based respiration, but relies on glycolysis to synthesize ATP (Abrahamsen et al. 2004, Rider and Zhu 2010, Zhu and Guo 2014). Its glycolytic pathway may start with a number of mono-, di-, and poly-saccharides (e.g., glucose, mannose, sucrose, amylose and amylopectin), and exits with lactate, ethanol or acetate as the organic end products or provides substrates for the synthesis of trehalose and N-glycans, the elongation of very long chain fatty acids, and the formation of complex lipids. Several unique glycolytic and fermentative enzymes are present in Cryptosporidium, such as the hexokinase (HK) that is more closely related to those from prokaryotes than to those from fungi, animals and plants, two pyrophosphate-dependent phosphofructokinases (PPi-PFKs) that differ from the ATP-dependent PFK in humans and animals, a bifunctional pyruvate:NADP+ oxidoreductase (PNO), and a bacterial-type lactate dehydrogenase (LDH) (Rotte et al. 2001, Thompson et al. 2005, Yu et al. 2014, Zhang et al. 2015). Therefore, the glycolytic and fermentative enzymes are an attractive subject of research not only for the understanding of the parasite metabolism and evolution, but also for the exploration as novel drug targets in the parasite.
Cryptosporidium possesses a single hexokinase [EC: 2.7.1.1] that catalyzes the first committed reaction in the glycolysis of glucose and other hexoses that may be obtained from the host through sugar transporters or by the degradation of amylopectin. We have previously characterized the C. parvum hexokinase (CpHK) that showed a number of unique molecular and biochemical features (Yu et al. 2014). We also observed that 2-deoxy-D-glucose (2DG), a glucose analog and classic HK inhibitor, was able to inhibit both the enzyme activity of CpHK and the growth of C. parvum in vitro. The inhibition of the parasite growth was more effective in the absence or lower concentrations of glucose and could rescued by adding higher levels of glucose, confirming that “C. parvum relies heavily on glucose as a carbon source and 2DG likely acted on the glycolytic pathway in the parasite” (Yu et al. 2014).
However, the action and fate of 2DG in C. parvum was not fully investigated. It is known that 2DG can be phosphorylated by human and animal HK to form 2DG-6-phosphate (2DG6P) that cannot be further metabolized. However, it was unclear whether 2DG could also be phosphorylated by CpHK. The anti-cryptosporidial activity of 2DG could be attributed mostly to the action of 2DG on CpHK, but the action of 2DG on the parasite glucose-6P isomerase (CpGPI) catalyzing the second reaction and/or the action of 2DG6P on CpHK or CpGPI might potentially contribute to the observed anti-parasitic activity as well (Fig. 1).
Fig. 1. Potential action of 2DG in C. parvum cell leading to the inhibition of parasite growth by blocking glycolysis.
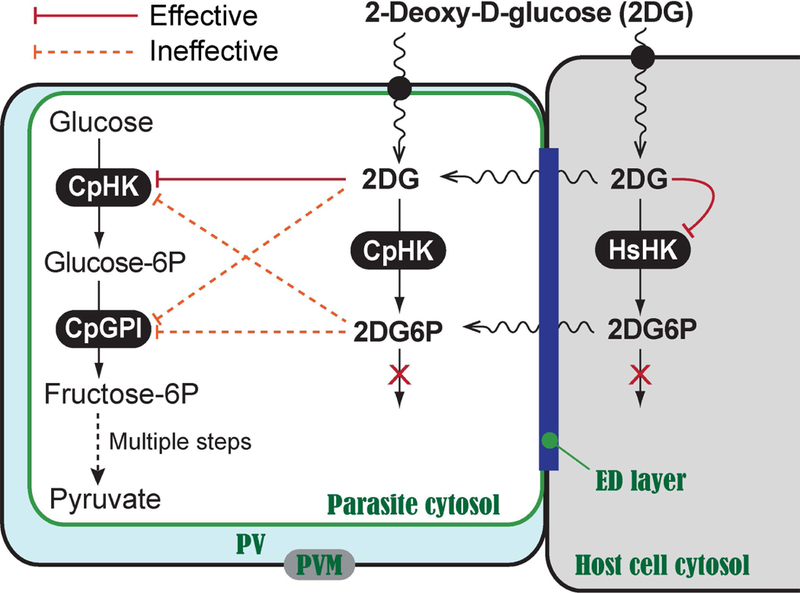
2DG inhibited CpHK and HsHK1 (solid lines), but was ineffective on CpGPI up to 50 mM (dotted lines). 2DG6P was ineffective (dotted lines) against CpGPI, CpHK and HsHK up to 28 mM. ED layer: electron-dense layer. PVM: parasitophorous vacuole membrane. Sugar transporters across the PVM and plasma membranes are indicated by solid circles.
The present study aimed to address the questions by determining the inhibitory effects of compounds 2DG and 2DG6P on the enzymes CpHK and CpGPI, and comparing the anti-cryptosporidial activity and cytotoxicity between 2DG and 2DG6P, which would help us to better understand the action of 2DG in the parasite. Additionally, although 2DG can be employed to probe the effects of inhibition of HK, the chance for being developed into anti-cryptosporidial drug is low due to the little selectivity between CpHK and host HKs and its micromolar level of in vitro anti-cryptosporidial activity (i.e., EC50 = 0.54 mM) (Yu et al. 2014). Therefore, we also initiated a drug screening campaign aimed to discover more selective and more efficacious anti-CpHK inhibitors. As a first step of the campaign, we screened 1200 existing drugs for potential anti-CpHK activities, and identified 4 existing drugs with not only lower micromolar level anti-CpHK enzyme, but also lower micromolar anti-cryptosporidial growth activities as described below. While the screening of larger compound libraries is ongoing for discovering more selective and efficacious anti-CpHK inhibitors, the identification of the four hits from existing drugs has provided us new anti-CpHK reagents with diversified chemical structures.
MATERIALS AND METHODS
Chemical and other reagents
Molecular kits and reagents were purchased from various resources as specified. Chemicals including substrates and inhibitors at analytical grade or higher purity were purchased from Sigma-Aldrich (St. Louis, MO) or as specified. The compound library was purchased in 2012 from Prestwick Chemical (San Diego, CA) (http://prestwickchemical.com) and used in primary screening by HK/G6PDH-coupled assay and validating hit specificity by G6PDH assay. The 2012 collection of the Prestwick Chemical Library contained 1200 small molecules that were 100% being marketed drugs, which differed slightly from the current collection of 1280 small molecules (see Supplemental Material). Compounds representing top hits were purchased from Sigma-Aldrich for subsequent studies of inhibitory kinetics on enzymes and anti-cryptosporidial efficacy in vitro.
Oocysts of C. parvum (subtype IIaA17G2R1 at gp60 locus) were purchased from Brunch Grass Farm (Deary, ID) and only those less than 3 months old were used in the in vitro drug efficacy assay. For clarity, the original strain propagated in Brunch Grass Farm was the Iowa-1 strain (subtype IIaA15G2R1), but has been replaced by a local isolate with a subtype IIaA17G2R1. Oocysts were cleaned by a treatment of 10% Clorox in water for 10 min on ice, followed by extensive washes to completely remove the chlorine and suspension in PBS that were used immediately or stored at 4 °C before use.
Preparation of recombinant proteins
The CpHK gene (CryptoDB gene ID: cgd6_3800; GenBank:XM_627719) was previously cloned into an MBPHT-mCherry2 vector for the expression of CpHK as a recombinant protein fused with maltose-binding protein (MBP)-His-mCherry tags (Yu et al. 2014). The CpGPI gene (Gene ID: cgd2_3200; GenBank:XP_626511) was previously cloned into a pMAL-c2E-TEV-His vector for the expression of CpGPI as an MBP-fusion protein (Eltahan et al. 2018). The expression of recombinant CpHK and CpGPI proteins in Escherichia coli and the amylose resin-based affinity purification of recombinant proteins followed standard protocols as described (Guo and Zhu 2012).
For determining the selectivity of inhibitors, we also expressed the human (Homo sapiens) HK-I (HsHK1) as an MBP-fusion protein. HsHK1 was selected from the four HKs presented in mammals because it is the house-keeping enzyme found in all mammalian tissues. For cloning, a plasmid containing the cDNA of the open reading frame (ORF) of HsHK1 gene (GenBank:NM_000188.2) was purchased from GeneCopoeia (Rockville, MD). The plasmid was used as a template for amplifying the 2754-bp HsHK1 ORF by a high-fidelity PCR using a HiFi PCR Kit (KAPA Biosystems, Wilmington, Massachusetts) and subcloned into pMAL-c2E-TEV-His vector at the EcoRI and HindIII restriction sites using NEB assembly kits (New England BioLabs Inc.). Primers used for HsHK assembly are HsHK_assemlby_F (att ttc agg gca tgg tac cgA TGA TCG CCG CGC AGC TC′) and HsHK_assembly_R (tgg tgg tga tga tgg tgg tgg tgG CTG CTT GCC TCT GTG C) (lower cases indicate linker sequences). Clones were screened by PCR for the presence of inserts in plasmids, and a selected number of plasmids containing correctly oriented HsHK1 inserts were sequenced to confirm the accuracy of the inserts. The expression of recombinant MBP-HsHK1 protein was carried out in a Rosetta-2 strain of E. coli cells. The induction of expression and purification of MBP-HsHK1 protein followed standard protocols (Eltahan et al. 2018, Guo and Zhu 2012, Yu et al. 2014).
The MBP-tag was similarly expressed and purified from the blank pMAL-c2E-TEV-His plasmid for use as a negative control in biochemical analysis of recombinant proteins. The quality and quantity of MBP-tag and recombinant proteins including MBP-CpHK, CpGPI and MBP-HsHK1 were evaluated by SDS-PAGE analysis and Bradford protein assay using bovine serum albumin (BSA) as the standard.
Biochemical analyses
HK assays.
The CpHK and HsHK1 enzyme activities were evaluated by two assays as previously described (Yu et al. 2014). The first one was an HK/glucose-6-phosphate dehydrogenase (G6PDH)-coupled assay that detected the product glucose-6P. Briefly, the assay was performed in 200 μL reactions containing D-glucose (2 mM or as specified), ATP (2.5 mM or as specified), NAD+ (0.3 mM), G6PDH (2 U), MgCl2 (5 mM) in Tris.HCl buffer (50 mM, pH 7.5), and MBP-CpHK or HsHK1 (500 ng) in the absence or the presence of an inhibitor at specified concentrations. In this assay, glucose was phosphorylated to form glucose-6P by HK, and then glucose-6P was converted into glucono-1′5-lactone-6P by G6PDH. The consumption of NAD+ was monitored spectrophotometrically at a 340 nm every minute for up to 30 min using Multiscan Spectrum spectrophotometer (Thermo Scientific, Waltham, MA). When an inhibitor was tested by the HK/G6PDH-coupled assay, the observed inhibitory effect might possibly be attributed to the action of the inhibitor on G6PDH. This possibility was tested by G6PDH assay, which was performed similarly to the G6PDH-coupled assay in the absence of HK and glucose, but using glucose-6P (0.2 mM) as the substrate. A lower concentration of G6PDH (0.04 U) was also used to avoid the potential masking of the inhibitory effect on G6PDH (Yu et al. 2014).
The second one was a pyruvate kinase/lactate dehydrogenase (PK/LDH)-coupled assay. This assay detected the formation of ADP, thus allowing the evaluation of HK activity towards various substrates in addition to glucose, such 2DG in this study. It was performed in 200 μL reactions containing D-glucose (5 mM) or other substrates (e.g., 2DG) at specified concentrations, ATP (2.5 mM), phosphoenolpyruvate (PEP; 1 mM), NADH (0.15 mM), PK (5 U), rabbit muscle LDH (10 U), and MBP-CpHK, MBP-HsHK1 or MBP (500 ng). In this assay, ADP was converted back to ATP to produce pyruvate from PEP by PK. Pyruvate was then converted to lactate and the consumption of NADH was monitored spectrophotometrically at a 340 nm every minute for up to 30 min. G6PDH, PK and LDH used in these assays were purchases from Sigma Aldrich (St. Louis, MO).
GPI assay.
The potential inhibitory effect of 2DG and 2DG6P on CpGPI was evaluated by a GPI/G6PDH-coupled assay as previously described (Eltahan et al. 2018). It was performed similarly to the HK/G6PDH-coupled assay, but in a reverse reaction direction that converted fructose-6P into glucose-6P by CpGPI. Glucose-6P was then converted into glucono-1,5-lactone-6-phosphate by G6PDH using NAD+ as a cofactor. A typical assay was performed using 200 μL reactions containing D-fructose-6P (2 mM), NAD+ (0.2 mM), G6PDH (2 U), MgCl2 (5 mM) in Tris.HCl buffer (50 mM, pH8.5), and MBP-CpGPI or MBP (50 ng).
All assays were performed at least twice in triplicated reactions at room temperature (23 °C) except for the reactions involving HsHK1 enzyme that was assayed at 37 °C due to its low activity at room temperature. Enzyme kinetics were analyzed using GraphPad Prism version 5.0f or higher (http://www.graphpad.com). MBP-tag was used as the negative control in all assays for background subtraction.
High-throughput screening (HTS) of existing drugs
The Prestwick chemical library containing 1200 existing drugs were screened in 96-well plates for potential anti-CpHK activities using the HK/G6PDH-coupled assay as described above. All drugs were screened at of 20 μM in primary screening. Negative controls using MBP-tag (2 wells/plate) and positive controls using 2DG (5 mM; 2 wells/plate) were included in each plate. Each reaction was performed at least twice independently at room temperature.
Top hits were then evaluated by G6PDH assay to exclude those showing significant anti-G6PDH activities from further analysis as described above. Hits with confirmed anti-CpHK activities were repurchased from Sigma-Aldrich and subjected to further analysis to determine their inhibitory kinetic parameters on CpHK in comparison with those of HsHK1. Because of the low activity of HsHK1 at room temperature, detailed comparison on the effects of inhibitors on CpHK and HsHK1 were performed at 37 °C. All reactions contained 1% DMSO that was used to increase the solubility of compounds and performed at least twice in triplicates. All assays were performed independently for at least three times.
Drug efficacy against the parasite growth in vitro
The efficacies of 2DG, 2DG6P and hits identified from existing drugs were evaluated against the growth of C. parvum in vitro using a qRT-PCR assay as previously described (Cai et al. 2005, Guo et al. 2018, Zhang and Zhu 2015). Briefly, HCT-8 cells (ATCC # CCL-225) were cultured overnight in 96-well plates in RPMI-1640 medium containing 10% fetal bovine serum (FBS) at 37 °C under 5% CO2 atmosphere until they reached to ~80% confluence. Oocysts were then added to the host cells monolayers (oocyst:host cell ratio = 1:2; i.e., ~2×104 oocysts with ~4×104 host cells/well) and incubated for 3 h at 37 °C, followed by a medium exchange to remove uninfected parasites and the addition of compounds at specified concentrations. Infected cells were incubated for an additional 41 h (total 44 h infection time). Cell lysates were prepared, diluted, and used directly to evaluate the parasite loads by qRT-PCR in 384-well plates as described (Zhang and Zhu 2015). Paromomycin (0.14 mM) and 2DG (0.5 mM) were used as positive controls and diluent (1% DMSO) is used as negative control in the assay. Cytotoxicity of drugs was evaluated by an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. In addition, the relative levels of 18S rRNA in uninfected HCT-8 cells was also evaluated for cytotoxicity. All assays were performed for at least three times independently in triplicates. Data was analyzed with Microsoft Excel and GraphPad Prism version 5.0f or higher.
RESULTS
The anti-cryptosporidial activity of 2-deoxy-D-glucose (2DG) was mainly attributed to its action on the parasite hexokinase (CpHK)
We have previously shown that 2DG was a competitive inhibitor of CpHK (IC50 = 5.75 mM; Ki = 0.34 mM) and could inhibit the growth of C. parvum (EC50 = 0.54 mM) at concentrations non-toxic to host cells (Fig. 1) (Yu et al. 2014). To fully understand the action of 2DG in the parasite, we wanted to determine whether CpHK could catalyze 2DG to form 2DG6P and whether 2DG6P could act on the downstream enzyme CpGPI to contribute to the anti-cryptosporidial activity.
We first confirmed that, similar to HsHK1, CpHK could utilize 2DG as a substrate to form 2DG6P (Km = 0.204 mM), but with ~4.5-fold lower affinity in comparison to its activity towards glucose (Km = 0.046 mM) (Table 1). The activities of CpHK on glucose and 2DG were comparable to HsHK1 (i.e., Km = 0.265 mM on 2DG and Km = 0.027 mM on glucose) (Table 1). The data also agreed with our previous observation that the action of 2DG on CpHK and HsHK1 was due to its direct competition with glucose as a slow substrate of HKs (Yu et al. 2014).
Table 1.
CpHK and HsHK1 enzyme parameters towards D-glucose and 2-deoxy-D-glucose (2DG) as determined by HK/G6PDH-coupled assay
| Substrate | CpHK | HsHK1 | ||
|---|---|---|---|---|
| Km (mM) | Vmax (U)1 | Km (mM) | Vmax (U)1 | |
| D-glucose | 0.046 | 155.2 | 0.027 | 53.3 |
| 2DG | 0.204 | 111.5 | 0.265 | 68.4 |
| Ratio (2DG/glucose) | 4.43 | 0.72 | 9.81 | 1.28 |
U = nmol/μg/min.
However, 2DG6P was ineffective on both CpHK and CpGPI at concentrations up to 28 mM (the maximal concentration achievable in the assay). While 2DG and its analog fluorodeoxyglucose (FDG) inhibited CpHK activity at low millimolar levels (IC50 = 5.75 mM and 3.7 mM, respectively), both 2DG and FDG were ineffective on CpGPI at concentrations up to 50 mM and 150 mM respectively (Table 2). For comparison, 2DG6P was also ineffective on HsHK1, whereas 2DG was effective on HsHK1 (IC50 = 5.75 mM) (Table 2).
Table 2.
Effects of 2-deoxy-D-glucose (2DG), 2-deoxy-D-glucose-6-phosphate (2DG6P) and fluorodeoxyglucose (FDG) on CpHK, HsHK1 and CpGPI enzyme activities
| Inhibitor | Effects of compounds on enzymes | ||
|---|---|---|---|
| CpGPI | CpHK | HsHK1 | |
| 2DG | Ineffective at ≤50 mM | IC50 = 5.75 mM | IC50 = 10.1 mM |
| 2DG6P | Ineffective at ≤28 mM | Ineffective at ≤28 mM | Ineffective at ≤28 mM |
| FDG | Ineffective at ≤150 mM | IC50 = 3.7 mM | IC50 = 15.2 mM |
These observations were correlated well with the in vitro efficacy data on the parasite, in which 2DG and FDG inhibited the parasite growth at lower millimolar levels (EC50 = 0.54 mM and 0.30 mM, respectively), whereas 2DG6P could inhibit the parasite growth at concentrations of more than 33-fold higher than 2DG (i.e., EC50 = 18.0 mM) (Table 3). In contrast, host cells were much less sensitive to the inhibition of 2DG (TC50 = 36.0 mM; in vitro safety interval (SI) = 66.7), 2DG6P (TC50 >72.0 mM; SI >4.0) and FDG (TC50 >72 mM; SI >240) (Table 3).
Table 3.
In vitro efficacy of 2-deoxy-D-glucose (2DG), 2-deoxy-D-glucose-6-phosphate (2DG6P) against the growth of C. parvum (44 h infection assay) and cytotoxicity on the HCT-8 cells (44 h treatment)
| Inhibitor | In vitro efficacy (EC50, mM) | Cytotoxicity (TC50, mM) |
Safety Interval (TC50/EC50) | |||
|---|---|---|---|---|---|---|
| 2DG | 0.54 | 36.0 | 66.7 | |||
| 2DG6P | 18.0 | >72.0 | >4.0 | |||
| 2FDG | 0.30 | >72.0 | >240 | |||
On the other hand, it remained to be determined whether 2DG6P was permeable to the PVM and parasite plasma membrane. While reduced permeability of 2DG6P may explain the observed ineffectiveness of the compound, our in vitro data was in congruence with the enzyme kinetic data, or at least did not reject our working hypothesis that the anti-cryptosporidial activity of 2DG was mainly attributed to the action of 2DG on CpHK, rather than the action of 2DG6P or 2DG on CpGPI, nor 2DG6P on CpHK (Fig. 1).
Four existing drugs were discovered to possess anti-CpHK activities
As a first step to discover new CpHK inhibitors, we screened 1200 small molecules (2012 collection) in the Prestwick Chemical Library using an HK/G6PDH-coupled assay. The assay was validated by the NIH-recommended intra-plate, inter-plate, and inter-day uniformity tests (Iversen et al. 2012), in which the signal window (SW) and Z′ factor values were ranging from 7.1 to 37.59 and from 0.70 to 0.92, respectively. These values were all better than the recommended acceptance criterions (i.e., SW ≥ 2 and Z′ ≥ 0.4).
Our primary screening identified 21 compounds with ≥ 50% inhibition of enzyme activity at 20 μM (Fig 2a). The 21 compounds were subjected to a G6PDH assay using lower concentrations of G6PDH (0.04 U) in the absence of CpHK and glucose, by which 12 compounds were confirmed to be specific. Among them, 8 hits were excluded for further assays due to cytotoxicity or insolubility at applicable concentrations. The remaining 4 specific CpHK inhibitors are thimerosal, alexidine•2HCl, hexachlorophene and ebselen (Fig. 2b).
Fig. 2. Identification of four known compounds as inhibitors of CpHK.
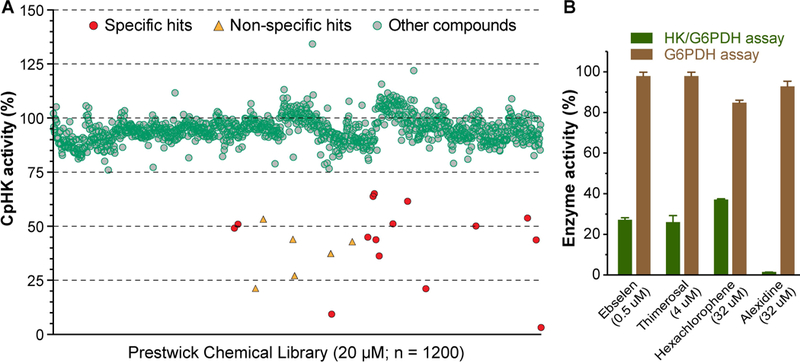
(A) Screening of the Prestwick chemical library containing 1200 marketed drugs at 20 μM (green round circles) showing specific hits (red round circles) and non-specific hits (triangles). 2-deoxy-D-glucose at 0.5 mM was used as a positive control. (B) Hit validation assay showing that the four drugs were authentic hits because they were ineffective on G6PDH used in the CpHK/G6PDH-coupled assay. Bars represent standard errors of the mean (SEMs) derived from at least three replicated reactions.
Among the 4 hits, ebselen was previously found to also inhibit CpGPI (Eltahan et al. 2018). However, other studies had showed that ebselen could act on as a reversible inhibitor of Plasmodium falciparum HK (PfHK) or an irreversible inhibitor for Trypanosoma brucei HK (TbHK) (Harris et al. 2013, Joice et al. 2013), so that it was not a surprise to also see its inhibition on CpHK. We observed that, similar to its action on CpGPI and TbHK, ebselen was an irreversible inhibitor of CpHK, as the inhibition could not be reversed by up to 50-fold dilution of the inhibitor (i.e., from 100 to 2 μM) (Eltahan et al. 2018, Joice et al. 2013) (Fig. 3A). Additionally, the reducing agent dithiothreitol (DTT) at 100 mM could prevent the inhibition when incubated with the enzyme prior to the reaction and partially reversed the inhibition when added after incubation of ebselen with enzyme (Fig. 3B). These observations suggest that ebselen might covalently bind to CpHK, likely via the modification of Cys residues of CpHK, while the direct contact between DTT and ebselen could interfere the enzyme-inhibitor association at certain levels.
Fig. 3. Effects of dilution and DTT on the inhibition of ebselen (Ebs) on CpHK.
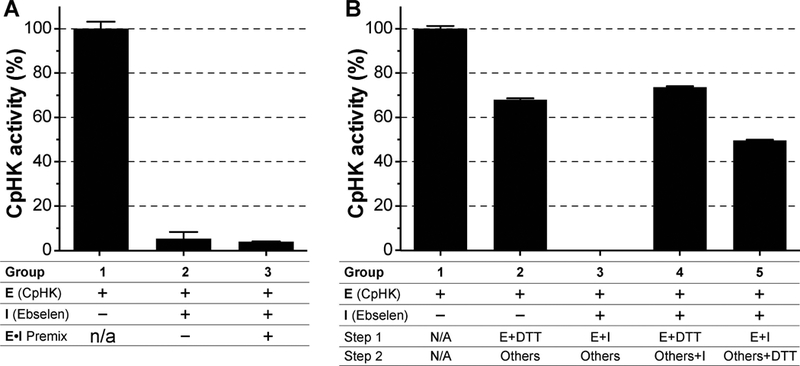
(A) In the inhibitor dilution assay, pre-incubation of ebselen (100 μM) with CpHK on ice for 10 min followed by 50-fold dilution with the addition of other reaction components had no effect on the inhibition of ebselen on CpHK (experimental group 3; vs. group 2 without premix). Group 1 was the positive control without ebselen. (B) In the DTT assay, pre-incubation of DTT (100 mM) and CpHK only slightly affected the enzyme activity (group 2) compared with group 3 in which ebselen inhibited CpHK in the absence of DTT. DTT blocked the inhibition of ebselen on CpHK (group 4) and partially reversed ebselen inhibition without its prior incubation with CpHK (group 5). Group 1 was the positive control without ebselen. Bars represent standard errors of the mean (SEMs) derived from at least three replicated reactions.
All four hits were further analyzed to determine their inhibitory kinetics and IC50 values on CpHK and HsHK. Ebselen (IC50 = 0.241 μM) and alexidine•2HCl (IC50 = 22.1 μM) acted as noncompetitive inhibitor of CpHK, while the action of thimerosal (IC50 = 2.3 μM) and hexachlorophene (IC50 = 8.8 μM) on CpHK fit with the mixed model of inhibition (Table 4; Supplemental Fig. S1). They also showed very similar IC50 values on HsHK1 (Table 4), suggesting no selectivity of the four compounds between CpHK and HsHK1.
Table 4.
Inhibition of the CpHK and HsHK1 enzyme activity by the top four hits identified from 1200 existing drugs and their efficacies against the growth of C. parvum in vitro
| Compound | Inhibition on CpHK (IC50, μM) |
Inhibition on HsHK1 (IC50, μM) |
Selectivity on enzyme | Efficacy on C. parvum (EC50, μM) | Cytotoxicity (TC50, μM) | Safety interval (SI) | Compound description |
|---|---|---|---|---|---|---|---|
| Alexidine•2HCl | 22.1 | 22.7 | 1.03 | 8.1 | 32.6 | 4.02 | Biguanide compound; Antibacterial, detergent, oral disinfectant, apoptosis promotic agent |
| Ebselen | 0.241 | 0.265 | 1.10 | 165* | 700* | 4.24 | Organoselenium compound; Anti-inflammatory, anti-thrombotic, anti-atherosclerotic, anti-oxidant |
| Hexachlorophene | 8.8 | 6.5 | 0.74 | 0.53 | 100 | 188.7 | Organochlorine compound; Antiseptic, bacteriostatic, anti-helminthic |
| Thimerosal | 2.3 | 0.35 | 1.52 | 1.77 | 18.5 | 10.5 | Organomercury compound; Antiseptic, antifungal, vaccine preservative |
Data adapted from a previous study (Eltahan et al, 2018).
Despite of the lack of selectivity between the parasite and host HK enzymes, the four compounds affect the growth of C. parvum in vitro more that of the host cells. The in vitro safety intervals (SI = TC50/EC50) ranged from ~4 for alexidine•2HCl and ebselen to 10.5 for thimerosal and 188.7 for hexachlorophene (Table 4), indicating that the parasite was much more sensitive than host cells to the inhibition by the four compounds, particularly by hexachlorophene.
DISCUSSION
In the present study, we first confirmed that 2DG could be phosphorylated by CpHK to form 2DG6P, but the phosphorylated form of 2DG was ineffective on CpGPI, CpHK and HsHK1 at concentrations comparable to or higher than the effective concentrations of 2DG (Table 2). 2DG6P was much less effective than 2DG on the parasite grown in vitro and host cells, albeit with an alternative explanation that 2DG6P is impermeable or less permeable to the host and/or parasite plasma membranes (Table 3). These observations are in consistent with our working hypothesis that the anti-cryptosporidial action of 2DG was attributed to its action on CpHK and further support the notion that CpHK is a valid drug target in the parasite (Fig. 1).
As discussed earlier, the chance to develop 2DG into an anti-cryptosporidial therapeutic might be low, but further investigation including in vivo assays might be needed to make a firm conclusion (Yu et al. 2014). However, 2DG can serve as a useful reagent to study the function of CpHK in the parasite. Our data implies 2DG can be converted to 2DG6P, leading to the loss of inhibitory effect on the parasite enzymes. Therefore it would be highly appealing to design/identify 2DG analogs that cannot be phosphorylated by CpHK to extend its action on CpHK and increase its efficacy on the parasite. This notion is partly supported by the data on FDG that displayed slightly better anti-CpHK activity (IC50 = 3.7 mM on CpHK, vs. 5.75 mM by 2DG) (Table 2), and slightly improved in vitro anti-C. parvum efficacy (EC50 = 0.3 mM, vs. 0.54 mM by 2DG) and safety interval (>240-fold, vs. 66.7 for 2DG) (Table 3).
Secondly, we initialized a drug screening campaign for identifying new anti-CpHK compounds by first screening 1200 marketed drugs with the greatest possible degree of drug-likeliness to validate the HTS assay before starting to screen large compound libraries and to identify potential anti-CpHK activity from marketed drugs. Among the four anti-CpHK hits, only ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one; CAS # 60940–34-3) can be orally administrated for medical use (Chew et al. 2010, Dawson et al. 1995, Imai et al. 2001, Saito et al. 1998, Takasago et al. 1997). Ebselen is a synthetic organoselenium known to have multiple targets enzymes in many species including HKs from several protozoan parasites (Azad and Tomar 2014, Harris et al. 2013, Joice et al. 2013, Koizumi et al. 2011, Terentis et al. 2009, Ullrich et al. 1996, Zhao et al. 2002). It was also recently identified as the only inhibitor of CpGPI among marketed drugs (Eltahan et al. 2018). Therefore, it is likely that ebselen might also affect other Cys-containing enzymes in the parasite. Although the efficacy of ebselen at micromolar levels was not impressive (EC50 = 0.165 mM), it might be worth to evaluate its anti-cryptosporidial efficacy in vivo. There could also be a chance to investigate its analogs to identify more efficacious and selective inhibitors.
The other three hits were antiseptic biguanide (alexidine•2HCl), organochlorine (hexochlorophene) and organomercury (thimerosal) compounds (Gołoś and Lutyńska 2015, Spolsky and Forsythe 1977). These compounds were also reported to possess activities against some other parasites, such as the anti-Acanthamoeba activity of alexidine (Tarun et al. 2008), anti-Plasmodium and anti-trypanosome activities of thimerosal (Mackey et al. 2006, Zidovetzki et al. 1994), and anti-nematode and anti-protozoan activities of hexochlorophene (Allen et al. 1967, Keiser et al. 2016, Takeuchi et al. 1985, Thong and Coombs 1987). On the other hand, these compounds are only labeled for use as vaccine preservative or disinfectants, rather than for oral medication, for which they are unlikely to be developed into anti-cryptosporidial therapeutics.
Modern drug discovery mainly relies on two complementary approaches to identify leads, i.e., phenotypic drug screening and target-based drug screening assays. In the anti-cryptosporidial drug discovery, two phenotypic HTS assays have been recently reported to screen chemicals against the growth of C. parvum in vitro, including a high-content imaging analysis using wild-type or transgenic parasites (Bessoff et al. 2013, Vinayak et al. 2015), and a qRT-PCR-based assay using wild-type parasites (Guo et al. 2018, Zhang and Zhu 2015). These assays have been employed to identified several leads with outstanding anti-cryptosporidial potency in vitro and in vivo. Target-based approach has also been employed to identify several leads, such as the fatty acyl-CoA synthetase (ACS) inhibitor triacsin C with excellent anti-cryptosporidial potency in vitro and in vivo (Guo et al. 2014). Our serial genomic and biochemical analyses, including this study, indicate that key enzymes in the glycolytic pathway in Cryptosporidium including CpHK could be targeted for developing therapeutics, and CpHK is worth to be pursued for identifying more selective and efficacious inhibitors.
In summary, our study on the action of 2DG further supported that CpHK could serve as a drug target in the parasite, and the drug screening identified four existing drugs as CpHK inhibitors that could be used as new reagents for studying the CpHK enzyme properties. We are currently screening large compound libraries including the TimTec Diversity Screening Set of 10,000 compounds (http://www.timtec.net) aimed to identify more efficacious and selective anti-CpHK leads for developing anti-cryptosporidial therapeutics, and will report our findings in the near future.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by National Institute of Infectious and Allergic Diseases (NIAID) of the National Institutes of Health (NIH) (R21 AI103668 to G.Z.), the United State Department of Agriculture (USDA) Formula Animal Health grant (TEX09591 to G.Z.), and the Department of Veterinary Pathobiology and the Texas Engineering Experiment Station (TEES), Texas A&M University (TAMU) to R.E. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH, USDA or TAMU.
LITERATURE CITED
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L & Kapur V 2004. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science, 304:441–5. [DOI] [PubMed] [Google Scholar]
- Allen RW, Enzie FD & Samson KS 1967. Trials with Yomesan and other selected chemicals against Thysanosoma actinioides, the fringed tapeworm of sheep. Proc. Helminthol. Society, 34:195–199. [Google Scholar]
- Azad GK & Tomar RS 2014. Ebselen, a promising antioxidant drug: mechanisms of action and targets of biological pathways. Mol. Biol. Rep, 41:4865–4879. [DOI] [PubMed] [Google Scholar]
- Bessoff K, Sateriale A, Lee KK & Huston CD 2013. Drug repurposing screen reveals FDA-approved inhibitors of human HMG-CoA reductase and isoprenoid synthesis that block Cryptosporidium parvum growth. Antimicrob. Agents Chemother, 57:1804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Woods KM, Upton SJ & Zhu G 2005. Application of quantitative real-time reverse transcription-PCR in assessing drug efficacy against the intracellular pathogen Cryptosporidium parvum in vitro. Antimicrob. Agents Chemother, 49:4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-M, Keithly JS, Paya CV & LaRusso NF 2002. Cryptosporidiosis. N. Engl. J. Med, 346:1723–1731. [DOI] [PubMed] [Google Scholar]
- Chew P, Yuen DY, Stefanovic N, Pete J, Coughlan MT, Jandeleit-Dahm KA, Thomas MC, Rosenfeldt F, Cooper ME & de Haan JB 2010. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes, 59:3198–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators GDD 2017. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis, 17:909–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson D, Masayasu H, Graham D & Macrae I 1995. The neuroprotective efficacy of ebselen (a glutathione peroxidase mimic) on brain damage induced by transient focal cerebral ischaemia in the rat. Neurosci. Lett, 185:65–69. [DOI] [PubMed] [Google Scholar]
- Eltahan R, Guo F, Zhang H, Xiang L & Zhu G 2018. Discovery of ebselen as an inhibitor of Cryptosporidium parvum glucose-6-phosphate isomerase (CpGPI) by high-throughput screening of existing drugs. Int. J. Parasitol. Drug Drug Resist, 8:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LM & Saravolatz LD 2005. Nitazoxanide: a new thiazolide antiparasitic agent. Clin. Infect. Dis, 40:1173–1180. [DOI] [PubMed] [Google Scholar]
- Gołoś A & Lutyńska A 2015. Thiomersal-containing vaccines–a review of the current state of knowledge. Przegl. Epidemiol, 69:59–64. [PubMed] [Google Scholar]
- Guo F & Zhu G 2012. Presence and removal of a contaminating NADH oxidation activity in recombinant maltose-binding protein fusion proteins expressed in Escherichia coli. BioTechniques, 52:247. [DOI] [PubMed] [Google Scholar]
- Guo F, Zhang H, McNair NN, Mead JR & Zhu G 2018. The existing drug vorinostat as a new lead against cryptosporidiosis by targeting the parasite histone deacetylases. J. Infect. Dis, 217:1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Zhang H, Fritzler JM, Rider SD Jr., Xiang L, McNair NN, Mead JR & Zhu G 2014. Amelioration of Cryptosporidium parvum infection in vitro and in vivo by targeting parasite fatty acyl-coenzyme A synthetases. J. Infect. Dis, 209:1279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MT, Walker DM, Drew ME, Mitchell WG, Dao K, Schroeder CE, Flaherty DP, Weiner WS, Golden JE & Morris JC 2013. Interrogating a hexokinase-selected small-molecule library for inhibitors of Plasmodium falciparum hexokinase. Antimicrob. Agents Chemother, 57:3731–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Masayasu H, Dewar D, Graham D & Macrae I 2001. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke, 32:2149–2154. [DOI] [PubMed] [Google Scholar]
- Iversen PW, Benoit Beck B, Yun-Fei Chen Y, Dere W, Devanarayan V, Eastwood BJ, Farmen MW, Iturria SJ, Montrose C, Moore RA, Weidner JR & Sittampalam GS (2012) Validation. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. [Google Scholar]
- Joice AC, Harris MT, Kahney EW, Dodson HC, Maselli AG, Whitehead DC & Morris JC 2013. Exploring the mode of action of ebselen in Trypanosoma brucei hexokinase inhibition. Int. J. Parasitol. Drug Drug Resist, 3:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser J, Panic G, Adelfio R, Cowan N, Vargas M & Scandale I 2016. Evaluation of an FDA approved library against laboratory models of human intestinal nematode infections. Parasites & vectors, 9:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly P 2011. Treatment and prevention of cryptosporidiosis: what options are there for a country like Zambia? Parasitology, 138:1488. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Fujisawa H, Suehiro E, Shirao S & Suzuki M 2011. Neuroprotective effects of ebselen following forebrain ischemia: involvement of glutamate and nitric oxide. Neurol. Med. Chir. (Tokyo), 51:337–343. [DOI] [PubMed] [Google Scholar]
- Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D & Breiman RF 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet, 382:209–222. [DOI] [PubMed] [Google Scholar]
- Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR & Williams J 2006. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem. Biol. Drug Des, 67:355–363. [DOI] [PubMed] [Google Scholar]
- Rider SD & Zhu G 2010. Cryptosporidium: genomic and biochemical features. Exp. Parasitol, 124:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol J-F 2010. Cryptosporidium and Giardia: treatment options and prospects for new drugs. Exp. Parasitol, 124:45–53. [DOI] [PubMed] [Google Scholar]
- Rotte C, Stejskal F, Zhu G, Keithly JS & Martin W 2001. Pyruvate: NADP oxidoreductase from the mitochondrion of Euglena gracilis and from the apicomplexan Cryptosporidium parvum: a biochemical relic linking pyruvate metabolism in mitochondriate and amitochondriate protists. Mol. Biol. Evol, 18:710–720. [DOI] [PubMed] [Google Scholar]
- Saito I, Asano T, Sano K, Takakura K, Abe H, Yoshimoto T, Kikuchi H, Ohta T & Ishibashi S 1998. Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery, 42:269–277. [DOI] [PubMed] [Google Scholar]
- Spolsky VW & Forsythe AB 1977. Effects of alexidine·2HCl mouthwash on plaque and gingivitis after six months. J. Dent. Res, 56:1349–1358. [DOI] [PubMed] [Google Scholar]
- Takasago T, Peters E, Graham D, Masayasu H & Macrae I 1997. Neuroprotective efficacy of ebselen, an anti-oxidant with anti-inflammatory actions, in a rodent model of permanent middle cerebral artery occlusion. Br. J. Pharmacol, 122:1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Kobayashi S, Tanabe M & Fujiwara T 1985. In vitro inhibition of Giardia lamblia and Trichomonas vaginalis growth by bithionol, dichlorophene, and hexachlorophene. Antimicrob. Agents Chemother, 27:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun AS, Peng X, Dumpit RF, Ogata Y, Silva-Rivera H, Camargo N, Daly TM, Bergman LW & Kappe SH 2008. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. U. S. A, 105:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentis AC, Freewan M, Sempértegui Plaza TS, Raftery MJ, Stocker R & Thomas SR 2009. The selenazal drug ebselen potently inhibits indoleamine 2, 3-dioxygenase by targeting enzyme cysteine residues. Biochemistry, 49:591–600. [DOI] [PubMed] [Google Scholar]
- Thompson RA, Olson M, Zhu G, Enomoto S, Abrahamsen MS & Hijjawi N 2005. Cryptosporidium and cryptosporidiosis. Adv. Parasitol, 59:77–158. [DOI] [PubMed] [Google Scholar]
- Thong K-W & Coombs GH 1987. Trichomonas species: homocysteine desulphurase and serine sulphydrase activities. Exp. Parasitol, 63:143–151. [DOI] [PubMed] [Google Scholar]
- Ullrich V, Weber P, Meisch F & von Appen F 1996. Ebselen-binding equilibria between plasma and target proteins. Biochem. Pharmacol, 52:15–19. [DOI] [PubMed] [Google Scholar]
- Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ & Striepen B 2015. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature, 523:477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhang H, Guo F, Sun M & Zhu G 2014. A unique hexokinase in Cryptosporidium parvum, an apicomplexan pathogen lacking the Krebs cycle and oxidative phosphorylation. Protist, 165:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H & Zhu G 2015. Quantitative RT-PCR assay for high-throughput screening (HTS) of drugs against the growth of Cryptosporidium parvum in vitro. Front. Microbiol, 6:991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Guo F & Zhu G 2015. Cryptosporidium lactate dehydrogenase is associated with the parasitophorous vacuole membrane and is a potential target for developing therapeutics. PLoS Pathog, 11:e1005250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Masayasu H & Holmgren A 2002. Ebselen: a substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc. Natl. Acad. Sci. U. S. A, 99:8579–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G & Guo F 2014. Cryptosporidium metabolism. Cryptosporidium: parasite and disease. Springer.361–379. [Google Scholar]
- Zidovetzki R, Sherman I, Prudhomme J & Crawford J 1994. Inhibition of Plasmodium falciparum lysophospholipase by anti-malarial drugs and sulphydryl reagents. Parasitology, 108:249–255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.