

ABSTRACT
Immunotherapeutic treatments in head and neck cancer clinical trials include cancer vaccines targeting foreign viral antigens or mutational neoantigens derived from cancer-expressed proteins. Anti-tumor immune responses place cancer cells under selective pressure to lose or downregulate target antigens; therefore, vaccination against virus- or host- “driver” oncogenes are proposed as a strategy to overcome immune escape. Herein, we demonstrate the impact of immunogenic viral antigens on anti-tumor response and immune editing in MOC2-E6E7, a syngeneic murine oral cancer cell line expressing HPV-16 E6 and E7 oncoproteins. Using orthotopic syngeneic models, we observed in vivo tumor growth kinetics of MOC2-E6E7 is delayed in immunocompetent mice compared to parental MOC2 tumors. In contrast, tumor growth remained similar in Rag1-/- mice lacking adaptive immunity. MOC2-E6E7 tumors demonstrated an “inflamed” or immune-activated tumor microenvironment and greater infiltration of CD8+ T cells compared to MOC2. By real-time PCR, we detected downregulation of E6 and E7 genes in MOC2-E6E7 tumors only in immunocompetent mice, suggesting the loss of ectopic viral antigen expression due to immune editing. We then assessed the efficacy of a biomaterials-based mesoporous silica rod (MSR) cancer vaccine targeting HPV-16 E7 in our model. Vaccination induced robust infiltration of antigen-specific CD8+ T cells, which led to tumor growth delay and modestly prolonged survival in MOC2-E6E7 tumors. Increased efficacy was seen in a separate head and neck cancer tumor model, mEER, which obligately expresses E7 antigen. Collectively, our data highlight the need for both immunogenicity and ‘driver’ status of target antigens to be considered in cancer vaccine design.
KEYWORDS: Immune editing, immunotherapy, biomaterials, mesoporous silica rods, cancer vaccine, head and neck cancer, HPV16 E6 and E7
Introduction
More than 65,000 men and women will develop head and neck squamous cell carcinoma (HNSCC) this year and currently, it represents 4% of all cancers in the United States.1 The most common causes of HNSCC include exposure to chemical carcinogens such as tobacco and alcohol and viral etiologies such as high-risk forms of human papillomavirus (HPV). While carcinogen- and viral-associated HNSCC appear similar histologically, their demographic profile, anatomic location, mutational landscape, and response to conventional therapies differ.2,3 Despite advances in surgery, radiation- and chemotherapy, the 5-year overall survival (OS) has remained at 40–60% for the last 50 years.4 Given the well-known co-morbidities and recurrence rates associated with conventional treatments, there is a real need for innovative new approaches to treating this disease.
The ability to target immune responses against malignancies has surged to the forefront of cancer research and treatment in recent years, demonstrating the potential to generate specific and durable anti-tumor responses. Significant progress has been made in cancer immunotherapy, generating encouraging results in the treatment of cancers such as metastatic melanoma, for which the 5-year overall survival rate has increased from less than 10% to almost 40%.5-7 Immune checkpoint inhibitors have been the most successful cancer immunotherapy approach thus far in solid tumors and are currently FDA approved as first-line therapy for several advanced-stage cancers.8-10 Nonetheless, HNSCC remains a challenge since the majority of HNSCC tumors remain resistant to immune checkpoint blockade, with only 15–20% of the patients benefiting from this treatment modality. Thus, there is a need to overcome the challenges and limitations of current immunotherapeutic strategies used to treat HNSCC.11 A multitude of approaches including other immunomodulatory antibodies, chimeric antigen receptor (CAR)-T cell therapies, and cancer vaccines have been explored.12 Therapeutic cancer vaccines, in particular, have been used in combination with other immunotherapies or as monotherapy.12
One of the primary determinants of successful cancer immunotherapy is the generation of a functional tumor-specific cytotoxic lymphocyte pool.13,14 Therapeutic cancer vaccines aim to activate host T-cells and generate a potent, specific, anti-tumor response. Dendritic cells (DCs) are critical to this process because they act as efficient antigen-presenting cells and inducers of T cell immunity.15 Current DC vaccination approaches isolate and activate DCs against tumor antigens ex vivo, and then re-introduce them to the patient. However, more than 90% of the transplanted DCs die, few homes to the lymph nodes, and the ex vivo activated DCs may lose effectiveness after replantation has occurred.16
In response to this challenge, we have been investigating a biomaterial-based cancer vaccine system. Biomaterial platforms can provide spatiotemporal control over the delivery of multiple bioactive molecules and/or cells to direct cell behavior and drive functional tissue formation.17 With our in situ DC vaccination approach, the implanted biomaterial scaffold is designed for controlled release of a recruitment factor which promotes the trafficking of immune cells to the implantation site. Once there, recruited cells such as immature DCs infiltrate the scaffold and are simultaneously presented with tumor antigen and pro-inflammatory “danger signals” in the form of a pattern recognition receptor (PRR) ligand adjuvant. The mature, antigen-loaded DCs which are generated then traffic out of the scaffold towards draining lymph nodes where they can facilitate anti-cancer immunity through T cell priming and activation.18-22 Not only does this bypass the need for ex vivo DC manipulation and transplantation, but also allows for the generation of anti-tumor immune cells in a controlled setting, away from the immunosuppressive milieu of the tumor microenvironment. Thus, the ability of these engineered scaffolds to perform in situ cell programming has made them an attractive technology to improve DC and T cell function in the context of therapeutic cancer vaccines.16,18-21
The tumor microenvironment and the immunogenic nature of the tumor cells are critical factors which can affect the efficacy of biomaterial-based vaccines. Tumor specific-T cell responses are induced by three classes of antigens: antigens from viral proteins (e.g. HPV), somatic mutations, and those encoded by cancer-germline genes.23 Tumor antigens, recognized by, tumor antigen-specific CD8+ T cells induce tumor-specific T-cell responses because they display a tumor-specific pattern.24 In viral-associated cancers such as HPV-related HNSCC, antigens produced within the tumor cells are detected by T cells. Therefore, vaccines containing long HPV peptides have emerged as promising therapeutic modalities for HPV-related cancers, since these long peptides can increase the number and activity of HPV-16-specific CD4 and CD8 T cells.25-27 Critical for recognition of tumor antigens by tumor antigen-specific CD8+ T cells is the major histocompatibility complex – Class I (MHC-I) and antigen processing machinery (APM), which presents processed tumor antigenic peptides to T lymphocytes.
Most HPV-associated non-cancerous lesions undergo clearance by HPV-specific circulating CD4+ and CD8+ T cells. However, HPV infection occasionally persists, and the eventual transformation to malignancy is associated with the viral proteins E5, E6 and E7 which are critical for immune escape.28,29 Therefore, designing therapies that target these antigens to enhance clearance of HPV-associated cancer may be effective.30 For example, vaccination with HPV-E7 linked to dendritic cells using a fusion protein containing an extra domain from fibronectin regressed tumor growth and increased antitumor CD8+ T-cell responses in a murine model of cervical cancer.31 In addition, previous studies have shown that a biomaterial-based cancer vaccine combining granulocyte-macrophage colony stimulating factor (GM-CSF), cytosine-guanine oligonucleotide (CpG-ODN), and tumor lysates in a mouse melanoma model were able to increase animal survival by up to 90%.16 More recently, mesoporous silica rod (MSR)-based biomaterial vaccines have demonstrated the ability to form structures which provide a microenvironment that can support and modulate immune cells in vivo. MSR-based vaccines have also been shown to confer long-term immunity and protect against tumor re-challenge in multiple preclinical models21,32 and reviewed in.22,33 These studies illustrate the potential of MSR-based cancer vaccines to generate a potent anti-tumor effector T cell response in situ.
Importantly, while targeting clinically relevant antigens such as HPV E6 or E7 with a vaccine strategy may lead to successfully targeted immunotherapy against HPV-associated HNSCC, the host’s anti-tumor immune response can play a role in both tumor suppression and promotion. The host immune system shapes tumor fate by “cancer immunoediting”.28,34
In order to study the effect of vaccine-induced immunologic targeting on the progression of viral-associated HNSCC, we utilized MOC2-E6E7, a preclinical model of HNSCC which expresses a clinically relevant model antigen.35 MOC2-E6E7 is derived from MOC2, a murine model of carcinogen-associated HNSCC successfully used as a preclinical tool to study HNSCC, mimicking several aspects of its human counterpart such as lymph node metastases and showing cross-species conservation of its genomic landscape with human HNSCC.36-39 Here we demonstrate that ectopic expression of the clinically relevant, tumor-specific antigens HPV-16 E6 and E7 in the MOC2-E6E7 tumor model leads to an initial robust immune response, manifested in an “inflamed tumor” phenotype and delayed tumor growth. However, the loss of these tumor-specific antigens through the immunoediting process40-42 in MOC2-E6E7 tumors leads to immune escape, contributing to only modest efficacy of an E7-targeted MSR vaccine. In contrast, we observed enhanced vaccine efficacy against the HPV-driven mEER tumor model which requires the presence of HPV oncogenes for continued growth.43 Given the current widespread interest in neoantigen identification and therapeutic cancer vaccine development, these findings suggest that the selection of driver mutations for immunologic targeting may be important.
Results
MOC2-E6E7 tumor growth kinetics
Previous work demonstrated that MOC cell lines have different growth phenotypes in vivo.38 MOC2 cells showed no difference in tumor growth when implanted into immunocompromised Rag2-/- and wild type (WT) mice.38 To determine if MOC2-E6E7 cells displayed the same growth characteristics as parental MOC2, they were compared both in vitro and in vivo. Growth rates of these two cell lines were similar in vitro (Supplemental Figure 1). However, when MOC2 and MOC2-E6E7 cells were inoculated in the oral cavity of C57BL/6J WT mice and their tumor growths compared, we found that MOC2-E6E7 displayed different tumor growth kinetics from parental MOC2 in these immunocompetent mice. MOC2-E6E7 tumors showed a significant growth delay versus MOC2 tumors. Interestingly, MOC2-E6E7 and MOC2 tumor growth kinetics were similar in Rag1-/- mice, indicating the delay in growth of MOC2-E6E7 tumors in immunocompetent mice is T-cell-mediated (Figure 1).
Figure 1.
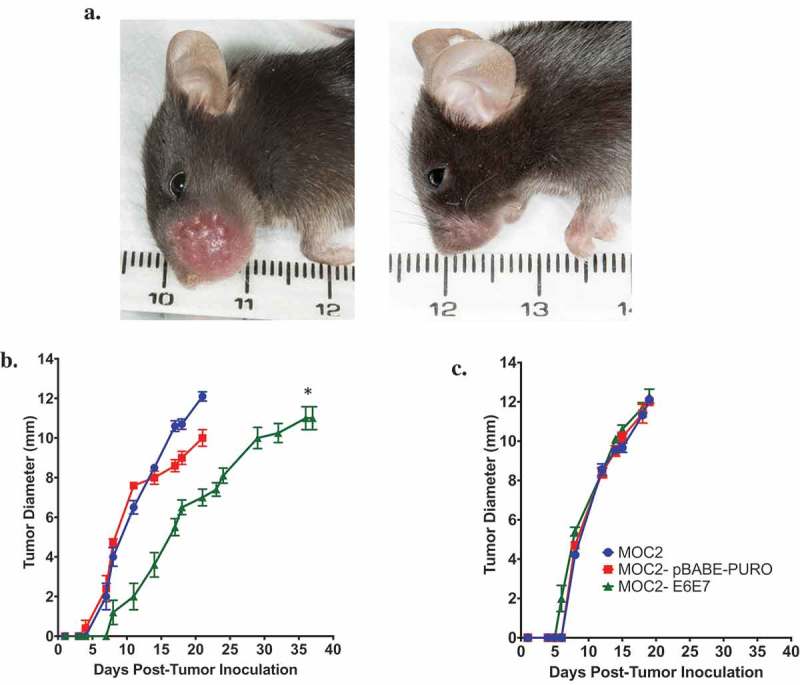
Growth of MOC2-E6E7 in immunocompetent mice is delayed vs. growth of MOC2.
Wild type or Rag1-/- C57BL/6J mice were injected with MOC2, MOC2-pBABE-PURO or MOC2-E6E7 cells in the oral cavity and tumor growth monitored. Representative image of tumor growth in wild type mice injected with MOC2 (left) and MOC2-E6E7 (right) 15 days after oral inoculation (a). Tumor growth was delayed in MOC2-E6E7 vs. MOC2 in immunocompetent mice (*, p = 0.0001 vs MOC2 and MOC2-pBABE-PURO), (b) whereas in Rag1-/- immunocompromised mice tumor growth rates were similar (c) (n = 10/group, except for Rag1-/- MOC2, n = 9). Error bars, as mean ± SEM.
E6 and E7 mRNA expression in MOC2-E6E7 tumors
To determine if the delay in the growth of MOC2-E6E7 tumors compared to parental MOC2 tumors was attributed to immune surveillance and editing, we assessed E6 and E7 mRNA expression in MOC2-E6E7 early-stage (6 mm) and late-stage (10 mm) tumors depleted of CD45+ cells in WT and Rag1-/- mice (Figure 2). For controls, E6 and E7 mRNA expression at similar time-points in CD45 depleted-mEER tumors were determined. As expected, E6 and E7 mRNA expression were maintained in the HPV-driven mEER tumors. Interestingly, E6 and E7 mRNA expression were reduced in MOC2-E6E7 tumors (as compared to MOC2-E6E7 cells grown in vitro) while expression of E6 and E7 was consistently higher in MOC2-E6E7 early- and late-stage tumors grown in Rag1-/- mice. We observed that the levels of E6 and E7 expression in early-stage MOC2-E6E7 tumors appeared to be low. In vitro E6 and E7 expression was maintained over a period of two weeks both in the presence/absence of puromycin selection media (Supplemental Figure 2). Collectively, our data showed reduced E6 and E7 expression in MOC2-E6E7 tumors when grown in immunocompetent mice whereas expression remained unchanged in immunocompromised mice over time, suggesting that immunoediting was taking place.
Figure 2.
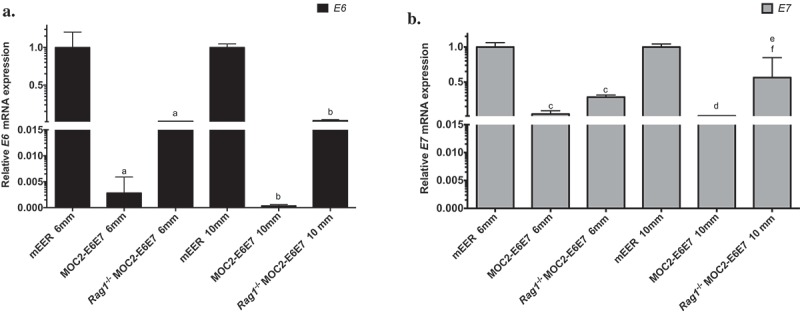
E6 and E7 expression in early and late MOC2-E6E7 tumors in immunocompetent and immunocompromised mice.
E6 (a) and E7 (b) mRNA expression were determined relative to that of ß-actin in CD45-depleted MOC2-E6E7 tumors from C57BL/6J wild type or Rag1-/- mice. As controls, E6 and E7 expression in CD45-depleted mEER tumors were determined. E6 and E7 expression are maintained in mEER and MOC2-E6E7 tumors grown in Rag1-/- mice while a loss of E6 and E7 expression is observed in MOC2-E6E7 tumors grown in wild type mice. a, p < 0.0001 vs. E6 mRNA expression in mEER 6 mm tumors, b, p < 0.0001 vs. E6 mRNA expression in mEER 10 mm tumors p = 0.0032; c, p < 0.0001 vs. E7 mRNA expression in mEER 6 mm tumors. d, p < 0.0001 vs. E7 mRNA expression in mEER 10 mm tumors, e, p = 0.0102 vs. E7 mRNA expression in mEER 10 mm tumors, f, p = 0.0002 vs. E7 mRNA expression in MOC2-E6E7 10 mm tumors. Error bars, as mean ± SD.
MOC2-E6E7 tumors are characterized by an inflamed phenotype
In order to determine if the immunophenotype of the MOC2-E6E7 tumor microenvironment differed from MOC2, we first comprehensively profiled immune-related gene expression in MOC2-E6E7 and MOC2 tumors, using cell lines and normal mouse oral mucosa as controls in nanoString™’s PanCancer Immune Profiling Panel (mouse) (Figure 3(a–d)). Multiplex gene analysis including 770 genes was performed that included up to 40 reference genes. Several genes associated with a T-cell inflamed status such as CD4, CD8, IL-12, Gzmb, Ido1, Prf1, Stat1, PD-L1, Pdcd1, Ifng, and Tnf were upregulated in MOC2-E6E7 tumors. CD4 and IL-12 expression increase were not significant (Figure 3(c)). Collectively, our data confirms a T-cell inflamed microenvironment in MOC2-E6E7 tumors, characterized by cytotoxic effector molecules, antigen presentation functions, IFN-γ signaling and T-cell functions (Figure 3 (b–d)). We then sought to compare the immune cell infiltrate in the tumor microenvironment, draining lymph nodes and blood of MOC2 and MOC2-E67 tumor-bearing mice on day 16 post-tumor inoculation (Figure 4(a–f), Supplemental Figure 3) by analytical flow cytometry. We observed a significantly increased quantity of CD8+ tumor-infiltrating lymphocytes (TILs) (12.5%) in the Day 16 MOC2-E6E7 tumors (Figure 4(a)) compared to MOC2 tumors. CD4+ TILs were similar in both MOC2-E6E7 and MOC2 tumors (Figure 4(b)). We observed similar differences in proportions of CD8+ and CD4+ cells amongst CD45+ and CD3+ cells (Figure 4(a,b)). FoxP3 and myeloid-derived suppressor cells accumulated in both MOC2-E6E7 and MOC2 tumors at a similar rate (Figure 4(c,d)). An increase in the ratio of CD8+/regulatory T cells (Treg) was also observed in MOC2-E6E7 tumors (Figure 4(e)). Collectively, we observed that MOC2-E6E7 tumors are characterized by an increase in CD8+ TILs using flow cytometry.
Figure 3.
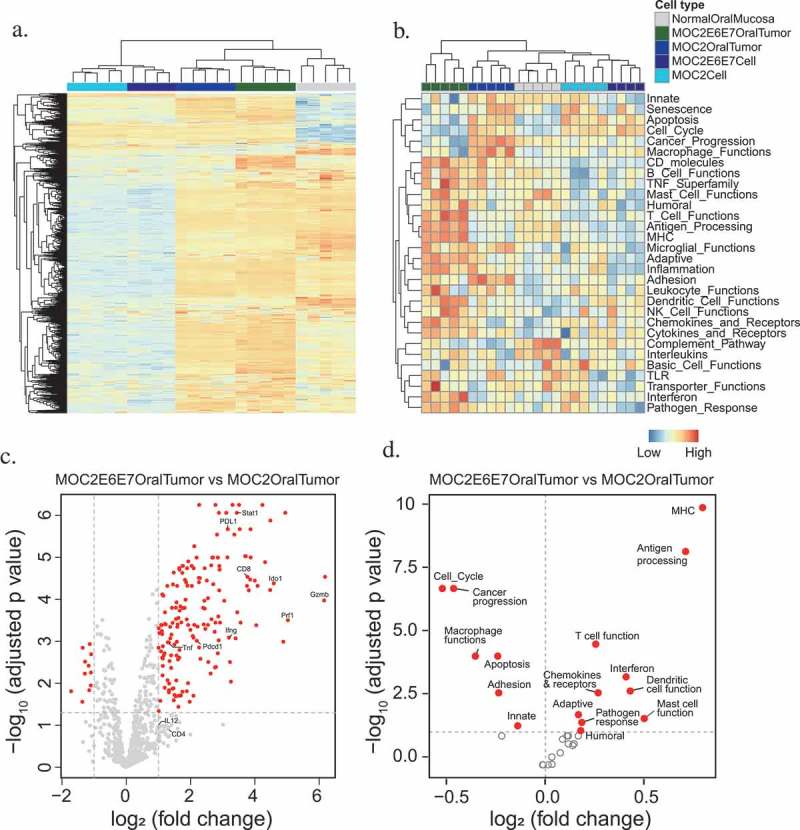
Differential gene expression analysis and pathway activity of MOC2-E6E7 tumors show the inflamed phenotype.
Multiplex gene expression analysis using nanoString™’s Pan Cancer Immune Profiling panel revealed that MOC2-E6E7 tumors show greater upregulation of immune-related genes compared to MOC2 tumors. Heat map representation of gene clustering of MOC2, MOC2-E6E7 cell line and tumors in replicates with blue indicating gene downregulation and orange indicating upregulation (a). Heat map representation of genes from the different groups clustered according to pathway activity (b). Volcano plots showing differential gene expression (c) and pathway activity (d) show the inflamed phenotype of MOC2-E6E7 tumors in comparison with MOC2 tumors.
Figure 4.
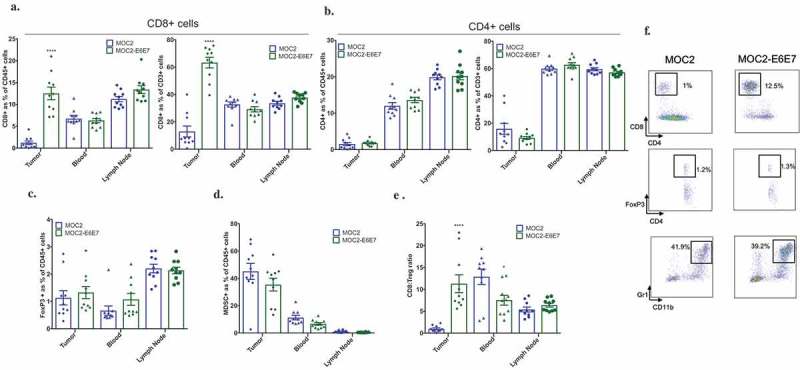
MOC2-E6E7 tumors have increased T-cell immune infiltrate.
Tumors, lymph nodes and blood from MOC2 and MOC2-E6E7 tumor-bearing mice were harvested at day 16 post-tumor inoculation and analyzed by flow cytometry (n = 10 mice/group). Increased CD8+ cells were observed in MOC2-E6E7 generated tumors. Quantification for CD8+ and CD4+ cells was done among CD45+ and CD3+ cells (a and b). Quantification of TILs (a-e) and representative flow cytometry dot plots are shown (f). ****, p < 0.0001; *, p = 0.0342. Error bars, as mean ± SEM.
Conventional H&E examination of MOC2-E6E7 tumors showed abundant lymphocyte infiltration (Supplemental Figure 4). Additional immunophenotyping of the MOC2-E6E7 tumor microenvironment on day 15 was performed using multiplex immunohistochemistry staining (Figure 5(a)) and quantification (Figure 5(b)). Altogether, our data indicate that MOC2-E6E7 tumors display an inflamed tumor phenotype with an increased CD8+ T cell infiltrate and markers of cytolytic activity.
Figure 5.
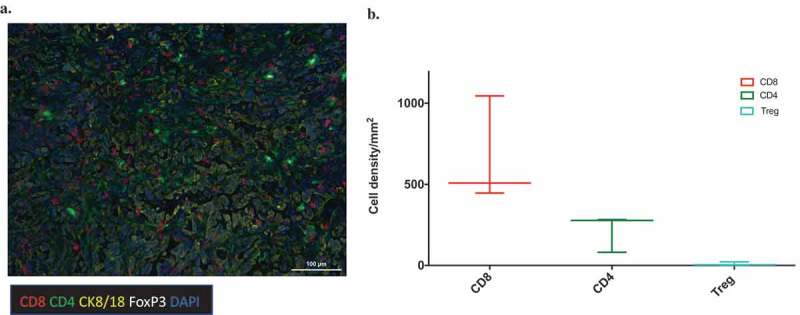
Multiplex fluorescent immunohistochemistry of MOC2-E6E7 tumors at day 15 shows T-cell infiltration.
Representative image after multispectral imaging for CD8, CD4, FoxP3, cytokeratin-8/18 together with a nuclear marker, DAPI in MOC2-E6E7 tumor at Day 15 (a). Tumor sections from day 15 were sequentially stained using Opal fluorophores for Ck8 (yellow), FoxP3 (white), CD8 (red), CD4 (green) and DAPI (blue). Images were taken on the Vectra platform and analyzed using InForm for quantitation of immune cells. The median value is represented by the line drawn at the center. The above and below whiskers show the maximum and minimum values, respectively (b).
MSR-vaccines generate a tumor-specific cytotoxic T-lymphocyte response in MOC2-E6E7 tumor-bearing mice
Previous work from the Mooney laboratory has investigated the potential of MSR-based vaccines to induce adaptive immune responses and building tumor immunity.21 We first sought to characterize MSRs in our laboratory (Supplemental Figure 5) and formulated injectable MSR vaccines (Supplemental Figure 6) for a therapeutic vaccination study in MOC2-E6E7 tumor-bearing mice. Prior to the efficacy study, we sought to examine if injectable MSR-vaccines were able to generate an E7-specific response in MOC2-E6E7 tumor-bearing mice treated with PBS alone or vaccinated with the E7 peptide-loaded MSR vaccine (Figure 6(a–g), See supplemental Figure 7). Blood, tumors, spleen, inguinal and cervical lymph nodes were harvested from both groups on day 13 post-tumor inoculation and processed by flow cytometry to investigate antigen-specific T-cell responses. While both PBS-treated and MSR vaccine-treated MOC2-E6E7-bearing mice showed comparable TILs, Tregs, MDSCs and other inflammatory myeloid cell infiltration in the tumor microenvironment and other harvested tissues, E7-tetramer-positive CD8+ lymphocytes (2%) were only observed in the tumors of the MSR-vaccinated group (Figure 6(a–e)). Collectively, this data indicates that treatment with an E7 peptide-loaded MSR-vaccine was able to generate an E7-specific CD8 + T cell response that could result in anti-tumor immunity.
Figure 6.
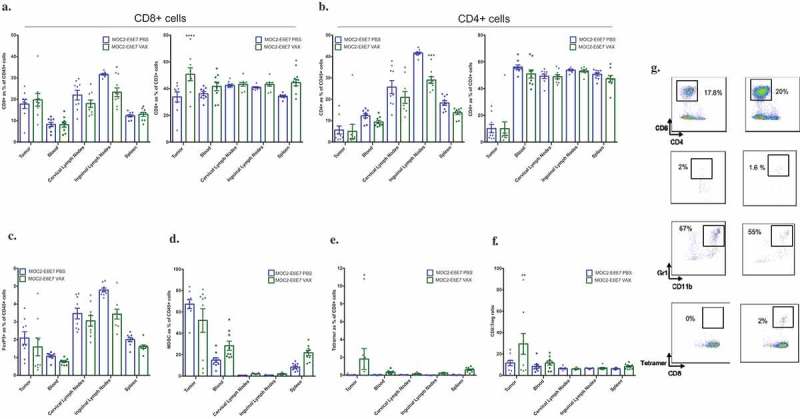
Mesoporous silica rod (MSR)-based vaccine formulations induces a tumor antigen-specific-CD8+ T-cell response in MOC2-E6E7 tumors.
Tumors, blood, spleen, inguinal and cervical lymph nodes from PBS (MOC2-E6E7-PBS) and MSR-vaccinated (MOC2-E6E7-VAX) MOC2-E6E7 tumor-bearing mice were harvested at day 13 post-tumor inoculation and analyzed by flow cytometry (n = 10 mice/group). Increased E7-specific CD8+ cells were observed in MOC2-E6E7 generated tumors (*, p = 0.034). Quantification for CD8+ and CD4+ cells was done among CD45+ and CD3+ cells (a and b). Quantification of data (a-f) and representative flow cytometry dot plots are shown (g). Error bars, as mean ± SEM.
MSR-vaccines prolong survival and delay tumor growth in MOC2-E6E7 and mEER tumors
We next determined the efficacy of E7 peptide-loaded MSR-vaccines targeted against MOC2-E6E7 tumors. MSRs loaded with E7 antigen in addition to GM-CSF and CpG-ODN promoted a statistically significant prolongation of survival in MOC2-E6E7 tumor-bearing mice (Figure 7(a)) when compared with PBS control treated groups. Tumor growth curves for the MSR vaccine-treated group showed a delay in tumor growth kinetics as well (Figure 7(b)). Interestingly, HPV-driven mEER tumor-bearing mice that were treated with the E7 peptide-loaded MSR vaccine showed even more robust prolongation of survival versus mice treated with PBS controls (Figure 7(d)). mEER tumor growth kinetics were also significantly slowed in vaccine-treated animals (Figure 7(e)). In mEER flank-tumor bearing mice, we also observed similar effects (Supplemental Figure 8). Collectively, we determined that MSR-based vaccines loaded with E7 in combination with GM-CSF and CpG-ODN enabled a modest prolongation of survival and delayed tumor growth in carcinogen-driven MOC2-E6E7 tumor-bearing mice, with improved efficacy seen in HPV-driven mEER tumor-bearing mice (Figure 7(g)).
Figure 7.
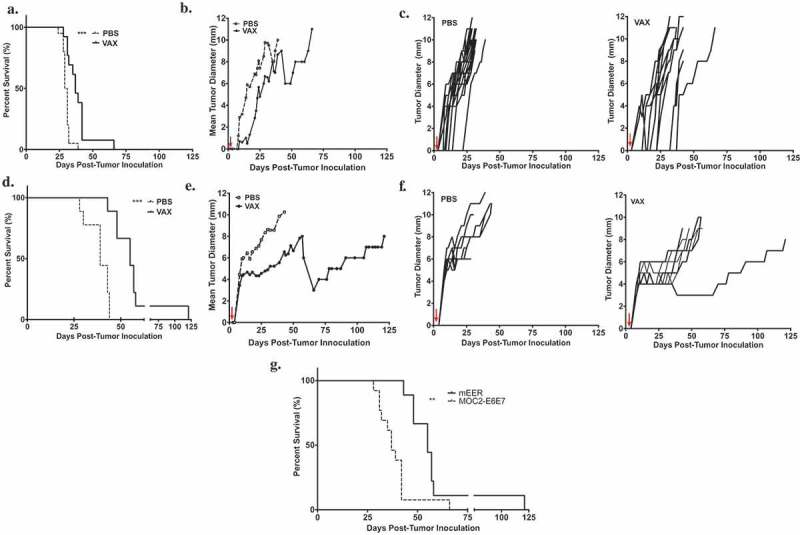
MSR-based vaccine prolongs survival and delays growth of MOC2-E6E7 tumors.
Vaccine efficacy study using injectable MSRs loaded with GM-CSF, CpG-ODN and antigen (E7 synthetic long peptide) in MOC2-E6E7 (a-c) and mEER (d-f) tumors, administered 3 days after tumor inoculation. Kaplan-Meier survival curves of tumor-bearing mice given E7-loaded MSR vaccines and PBS controls (a) and (d). Log-rank test was employed to measure significance (***, p = 0.0002 for MOC2-E6E7; ***, p = 0.0001 for mEER). Individual tumor growth curves in PBS controls and MSR vaccine-treated groups (n = 20 mice for PBS; n = 13 for vaccine group for MOC2-E6E7 and n = 9 for mEER challenged-tumors) (c) and (f). Comparison of tumor growth rates show delayed MOC2-E6E7 (b) and mEER (e) tumor growth in MSR vaccine-treated mice versus PBS controls. Prolongation of survival is enhanced in vaccine-treated mEER tumors as compared to MOC2-E6E7 tumors (**, p = 0.0039) (g).
Discussion
Despite the encouraging efficacy of immunotherapy in several types of cancer, the treatment of HNSCC with this modality remains a challenge. Survival rates of HNSCC patients receiving conventional multi-modality treatment have been relatively stagnant for decades and even those treated with FDA-approved immunotherapies such as checkpoint inhibition show an overall survival rate of only about 20%. This underscores a need for innovative approaches to treat this disease.44,45 In particular, biomaterials-based cancer immunotherapy platforms have arisen as a way for investigators to enhance conventional immunotherapeutic strategies.21-23,27,46 The parallel development of immunocompetent preclinical models of HNSCC has also been important. Uppaluri and colleagues36,38 previously established a syngeneic murine model of HNSCC that displays high fidelity with human HNSCC39 and identified key immune components to delineate therapeutic targets in immunocompetent C57BL/6J mice.36 While these MOC tumor models have shown utility both in modeling the “inflamed” tumor microenvironment36,47 and use as a testbed for various immunotherapeutic strategies,37,48,49 the absence of a clearly defined tumor-specific antigen makes it a challenge to study therapeutic cancer vaccines, the generation of specific anti-tumor immune responses, and immunoediting. High-risk HPV 16 and 18 are well-known etiologies of HNSCC50,51 and the immunogenic nature of their E6 and E7 oncoproteins identify them as ideal tumor-specific antigens for immunotherapeutic targeting. We, therefore, generated MOC2-E6E7, a syngeneic murine model of HNSCC which allowed us to study the in vivo effects of expressing clinically relevant model antigens (HPV-16 E6 and E7) on a previously immunologically “cold” MOC2 tumor, while simultaneously giving us the ability to target a tumor-specific antigen with a biomaterials-based cancer vaccine.
Although there were no differences in the in vitro growth kinetics of parental MOC2 and MOC2-E6E7, MOC2-E6E7 had a delayed growth phenotype in vivo in immunocompetent C57BL/6J mice. The reverse was observed in Rag1-/- immunocompromised mice with tumor growth rates similar to MOC2 and MOC2-E6E7 in vivo. Our investigations revealed that the in vitro expression levels of the target antigens, E6 and E7 is maintained at low levels. However, in vivo examination of E6 and E7 expression at early and later time-points of tumor growth in wild type and immunodeficient mice differed. Interestingly, tumors depleted of immune cells (CD45 depletion) after harvest had reduced E6 and E7 expression levels in immunocompetent mice whereas levels of E6 and E7 expression were maintained in Rag1-/- immunocompromised mice emphasizing that the loss of expression is not a ‘dilution’ effect due to the presence of immune cells. Reduced levels of E6 and E7 expression in MOC2-E6E7 bearing tumors resulted from ‘cancer immunoediting’. In this paradigm, the host immune system plays a protective role by destroying tumor cells in the elimination phase, and if all tumor cells are destroyed, then the endpoint of the immunoediting process is reached. However, rare tumor cell mutants may escape elimination and subsequently enter an equilibrium phase where overall tumor growth is controlled, but the immunogenicity of the tumor is being actively sculpted (and reduced) by host immune cells. As the “fittest” residual tumor cells with low antigenicity and MHC-I expression survive, eventually the tumor cell population acquires the most immunoevasive mutations and escapes immune control, leading to tumor growth. Similar to an earlier study, it seems that cancer immunoediting is a T-cell dependent immune-selection process leading to the growth of tumor cells lacking immunodominant rejection antigens (in this case ectopically-expressed E6 and E7 antigens) that display weak immunogenicity.52 The delayed, but eventual growth of MOC2-E6E7 tumors in immunocompetent mice is best described by this phenomenon.
Multiplex gene expression analyses and flow cytometric analyses of MOC2 and MOC2-E6E7-generated tumors established that MOC2-E6E7 displays an inflamed tumor phenotype. By nanoString™ analyses, we observed increases in expression of genes involved in T cell functions (CD8+ T cells), MHC functions, antigen-processing machinery, and interferon regulatory genes in MOC2-E6E7 tumors when compared to MOC2 tumors. Flow cytometry analyses revealed the presence of Treg cells, MDSCs and other inflammatory myeloid cells in MOC2-E6E7 bearing tumors. The percentage of CD8+ T cells was higher than that of the Tregs. Multiplex immunohistochemical staining of MOC2-E6E7 tumors also confirmed the presence of a robust – CD8+ T cell infiltrate. The presence of both CD8+ T cells and Tregs was similar to results obtained from flow cytometry and gene analysis. The efficacy of immunotherapy has been shown to be dependent upon the cancer being an immunogenic ‘hot’ tumor and not a hypoimmunogenic ‘cold’ tumor.53 Given our flow cytometry results, gene analysis and multiplex immunohistochemistry, we defined that the presence of the model antigens in MOC2-E6E7 altered the immunophenotype typically seen in the parental MOC2 tumor microenvironment. MOC2-derived tumors have a less inflamed phenotype and display aggressive and rapid tumor growth kinetics.36 Indeed, we observed increased CD8+ T cell infiltration into the tumor microenvironment in MOC2-E6E7 tumors.
Having established that MOC2-E6E7 tumors display an inflamed tumor phenotype with evidence of robust anti-tumor immunity, we subsequently investigated whether the efficacy of an MSR-E7 peptide cancer vaccine would be affected by targeting a non-essential model antigen in the context of tumor immunoediting. Treatment with the biomaterials-based vaccine generated an E7-specific CD8+ T cell response within the tumor microenvironment, although the mean percentage of CD8+ cells, Tregs, MDSCs and other inflammatory myeloid cells did not differ between treated and untreated tumors. Furthermore, Li et al. demonstrated that CD8+ T-cell depletion using anti-CD8a monoclonal antibody abrogates the effectiveness of the MSR vaccine system, indicating the anti-tumor effect of this biomaterial-based vaccine approach is mediated by CD8+ T cells.32 These findings suggest that injectable MSR-based biomaterials may serve as a multifunctional vaccine platform to modulate host immune cell function and provoke tumor-specific adaptive immune responses.21
Not surprisingly, the loss of E7 antigen via immunoediting in MOC2-E6E7 affected the efficacy of the MSR-based vaccine. Treatments with the E7-loaded MSR-vaccine resulted in improved survival and reduction of MOC2-E6E7 tumor growth rate compared to treatments with PBS alone. Seeing that treatment of MOC2-E6E7 tumors with the MSR-based E7 vaccine had only modest survival benefit (increase in median survival from 30 days to 37 days), we next tested the same vaccination approach in mEER tumors, which represent an HPV-driven HNSCC model (as opposed to MOC2-E6E7 whose model antigens are merely passengers in the carcinogen-induced, mutation-driven MOC2 model). Previous work has shown that mEER tumors require both E6 and E7 to maintain their tumorigenicity43 and continued E6 and E7 antigen expression within mEER tumors over time was also verified. The MSR-based E7 vaccine was capable of evoking better efficacy in mEER tumor-bearing mice with prolonged survival and a decrease in tumor growth rate as compared to MOC2-E6E7 tumor-bearing mice. Thus, while an inflamed tumor phenotype has traditionally been seen as a favorable prognostic marker for immunotherapy approaches, our findings suggest that this paradigm may be dependent on the modality of immunotherapy chosen, i.e., checkpoint inhibitors vs. vaccination approaches. This data has general implications for cancer vaccine design, whereby investigators should not only consider the immunogenicity of a tumor-specific antigen, but also whether the target is a “driver” mutation essential to tumor survival or merely an immunogenic “passenger” that can be eliminated without significant implications for the tumor cell population.
Modifications to MSR surfaces have been reported to regulate immune cell infiltration.54 Results from the Mooney lab utilizing the MSR-polyethylenemine (PEI)-based vaccine approach using E7 peptide elicited a higher vaccine efficacy in TC-1 tumors.32 The differences in our present study include methods employed for MSR formulations (i.e. without PEI), type of tumor (a highly immunoedited challenging oral tumor model), site of tumor inoculation (orthotopic inoculation) and the distance from the vaccine site (flank) to the tumor site (oral cavity). In addition, the MSR-vaccine formulation differed by using double the dose and then dividing into two flank sites unlike previously reported.21,54 Nevertheless, our results exhibit a delay in tumor growth in the challenging syngeneic MOC2-E6E7 tumor model.
In summary, we have described a syngeneic model of HNSCC, MOC2-E6E7 that expresses well-defined tumor-specific antigens and allows for the study of immunoediting and the downstream implications on targeting these ectopic immunogens in the context of therapeutic cancer vaccines. While the initial expression of the model antigens E6 and E7 led to a robust immune response resulting in an inflamed tumor phenotype and delayed tumor growth, the eventual loss of these targets via immunoediting rendered these tumors resistant to cancer vaccines targeting these epitopes. In contrast, the mEER model of HNSCC which is HPV-driven, appeared to be more amenable to E7-vaccine treatment, implying both immunogenicity and the “driver” status of targets should be given consideration in cancer vaccine design.
Materials and methods
Materials
All chemicals used were reagent grade or better. All reagents used for the experiments were purchased from VWR (Radnor, PA), Fisher Scientific (Pittsburgh, PA) or Sigma Aldrich (St. Louis, MO) unless otherwise indicated.
Plasmids
pBABE-puro was a gift from Hartmut Land & Jay Morgenstern & Bob Weinberg (Addgene plasmid # 1764).55 p1321 HPV-16 E6/E7 was a gift from Peter Howley (Addgene plasmid # 8641).56
Animals
For tumor experiments, 6–8-week old wild-type C57BL/6J or Rag1-/- female mice were obtained from Jackson Laboratory. Mice were maintained in standard housing conditions for the duration of the study. All protocols were in accordance with the guidelines for humane treatment of laboratory animals by the National Institutes of Health, the Animal Welfare Committee and the Center for Laboratory Animal Medicine and Care (CLAMC) at the University of Texas Health Science Center at Houston. mEER flank tumor experiments in 6–8 week old male C57BL/6J mice and animal care procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee and were conducted at Baylor College of Medicine.
Murine tumor cell lines
The murine oral cancer cell line, MOC2 was generously provided by Dr. Ravindra Uppaluri (Dana-Farber Cancer Institute, Harvard University, Boston) and maintained as previously described.36 The MOC2-E6E7 cell line was generated by retroviral transduction of HPV-16 E6 and E7 in parental MOC2 cells. Briefly, the E6 and E7 coding region was cloned from p1321 HPV-16/E6/E7 and inserted between the BamHI and SalI sites of pBABE-puro and confirmed by sequencing. The vector was sequenced to confirm the presence of the transgenes and then used to produce VSV-G pseudotyped retrovirus using the Retro-X™ Universal Packaging System (Clontech, 631530). Parental MOC2 cells were treated with a retrovirus in the presence of 8 μg/ml polybrene (EMD Millipore, TR-1003-G) overnight. After culturing for two days, the cells were selected for transgene expression by culturing in medium containing 4 μg/mL puromycin dihydrochloride (Sigma, P8833) for 7 days. MOC2-E6E7 cells were maintained in medium in the presence of 4 μg/mL puromycin. The MOC2-pBABE-puro (empty vector) cell line was generated using a similar protocol. Both MOC2 and MOC2-E6E7 cell lines were routinely tested for mycoplasma. E6 and E7 expression in the MOC2-E6E7 cell line was confirmed by qRT-PCR (Supplemental Figure 2). Cells were harvested in log-phase growth before injection into mice. The HPV-driven mEER murine HNSCC cell line was maintained and provided by Dr. Andrew Sikora (BCM).43,57
In vitro growth rate
MOC2, MOC2-pBABE-PURO and MOC2-E6E7 cells were seeded in 6-well plates at 10,000 cells/well in medium free of puromycin. Cells were harvested at days 3, 4 and 5 post-seeding. Cell count and viability was performed and analyzed using a Muse® Cell Analyzer Muse® Cell Analyzer (MilliporeSigma, 0500-3115) (Supplemental Figure 1).
In vivo tumor growth rate
MOC2, MOC2-pBABE-PURO, and MOC2-E6E7 tumors were established by injecting 30,000 cells into the maxillary vestibule of the left oral cavity in C57BL/6J or Rag1-/- mice. mEER tumors were established by injecting 500,000 cells into the maxillary vestibule of the left oral cavity in C57BL/6J mice. Tumor size measurements (in diameter) were taken two to three times per week to monitor tumor growth using digital calipers to measure the longest dimension (single dimension).
Characterization of mesoporous silica rods, preparation of vaccine and subcutaneous injection
Mesoporous silica rods (MSRs) were synthesized as previously described.21 Imaging was performed at the NanoCharacterization Core at Baylor College of Medicine. Scanning electron microscopy (SEM) characterization of MSRs was performed with a Hitachi FE-SEM SU8230. The low magnification image was taken by sprinkling MSRs on carbon tape and imaging at 5.0 kV with 600X magnification. The high-resolution image utilized a scanning transmission electron microscopy (STEM) detector, by dipping a 200-mesh carbon TEM grid with Cu support. The image was taken at high resolution with a low accelerated voltage of 1.0 kV at 220,000X magnification (Supplemental Figure 5). Preparation of the MSR vaccine was done as previously described.21 Briefly, 5 mg of MSRs loaded with bioactive reagents including 1 μg recombinant murine GM-CSF (Peprotech, 315-03) + 50 μg CpG-ODN (Invivogen vac-1826-1) + 50 μg E7 synthetic long peptide (Peptide2.0, GQAEPDRAHYNIVTFCCKCDSTLRLCVQSTHVDIR), suspended in cold PBS (150 μl) were injected subcutaneously in the bilateral flanks of mice using an 18-gauge needle (Supplemental Figure 6).
Tissue processing, cell separation and flow cytometry
Blood, tumors, spleen, cervical and inguinal lymph nodes were harvested from tumor-bearing mice for flow cytometric analysis. Preparation of single-cell suspensions from harvested tissues and blood was performed according to a standard protocol (see supplementary material and methods for details). To enrich these suspensions for tumor-infiltrating lymphocytes (TIL), the digested tumor single cell suspensions were first enriched for lymphocytes using Lymphoprep™ (Stemcell, 07801), prior to staining. A lymphoid staining panel of antibodies was then utilized including: CD45-PE-Cy7 (Clone30-F11, 60-0451-U100), CD19-FITC (clone 1D3, 35-0193-U100), CD3e-PE (clone 145-2C11, 50-0031-U100), CD4-PerCP-Cy5.5 (clone RM4-5, 65-0042-U100), CD8-APC (clone 53-6.7, 20-0081-U100), all from Tonbo Biosciences. E7 tetramer-BV421 was obtained from the NIH Tetramer Core Facility, Emory University (H-2Db RAHYNIVTF). For detection of myeloid and Treg cell subsets, an antibody panel including CD45-PE-Cy7, Gr-1-FITC (Clone RB-6-8C5; ThermoFisher, 50-133-29); CD11b-PE (Clone M1/70; ThermoFisher, 12-0112-81), CD4-PerCP-Cy5.5, CD25-BV421 (Clone 3C7; BD, Biosciences, 566228), and FoxP3-APC (Clone FJK-16s; ThermoFisher, 17-5773-80) was used. Dead cells were excluded via Near IR live/dead staining (ThermoFisher, L10119). Specific staining was validated using the ‛fluorescence minus one’ method as a control. Data were acquired on a LSRFortessa X-20 Cell Analyzer using FACSDiva software (BD Biosciences) and analyzed on FlowJo software vX10.0.7r2. See Supplemental Figures 9 and 10 for flow gating strategy.
E6 and E7 expression by qRT-PCR
Freshly dissected MOC2-E6E7 tumors (6 mm, early time point or 10 mm, late time-point) from C57BL/6J or Rag1-/- mice or mEER (6 mm or 10 mm) tumors from C57BL/6J were processed into single-cell suspensions by mincing and chemical dissociation (Tumor Dissociation Kit, Miltenyi Biotec, 130-096-730) as per the manufacturer’s protocol. Isolated cell suspensions were filtered through 70 μM filters and enrichment of the tumor cell population was performed on an AutoMACS Pro (Miltenyi Biotec) separator using murine anti-CD45 microbeads (Miltenyi Biotec, 130-052-301) to deplete leukocytes. The enriched tumor cell population was spun down and resuspended in TRIzol™ (ThermoFisher, 15596026) for RNA extraction. cDNA was synthesized using qScript™ cDNA SuperMix (QuantaBio, 95048-100). Gene expression was determined relative to β-actin using the indicated primers (IDT) on a CFX iCycler (Bio-rad).
Immune profile expression
Differences in the immune profile expression between MOC2 and MOC2-E6E7 tumors at day 14 were determined by performing multiplex gene expression analysis with the nCounter PanCancer Immune Profiling Panel (nanoString™, Seattle, Washington). Raw read counts from the nanoString™ PanCancer Immune Profiling Panel were normalized by nanoString™ nSolver (version 3.0) following the manufacturer’s instructions. Briefly, the normalization subtracted the background measured by a negative control probe set, and the geometric mean of a set of housekeeping genes was then used as the normalization factor to scale the read count for each sample. The normalized read counts were log2-transformed and used for differential gene expression analysis by the R package “limma”.58 We defined genes as differentially expressed if they displayed a fold change larger than 2 and an adjusted P value (Benjamini-Hochberg procedure) less than 0.05 in a pairwise comparison. To calculate the combined activity for a functional gene group, we downloaded the gene set annotation from nanoString™
(https://www.nanostring.com/download_file/view/436/3808). (Note that like all pathway databases, such gene sets are subjected to some arbitrary definition. Nonetheless, the majority of the gene sets are consistent with our understanding in cancer immunology and are unbiased from our perspective). We then used the single sample GSEA algorithm (ssGSEA)59 to project the gene expression into pathway activity scores. The ssGSEA we used was implemented in the R package “GSVA”.60 Pairwise activity scores were compared with the “limma” R package and an adjusted P value (Benjamini-Hochberg procedure) less than 0.05 was set to define differential activated pathways in the comparison. All differential analysis was performed under the R computation environment (3.4.0).
Multiplex immunohistochemistry
Day 15 tumors were collected, frozen in Tissue Tek Optimal Cutting Temperature (Sakura Finetechnical, 6255001) and preserved at -20°C until cryosectioning for multiplex immunohistochemistry. 5 μm tumor sections were prepared on a Leica CM3050 S Cryostat (Leica Microsystems Inc.) mounted on Superfrost blue slides (Fisher Scientific) and tumor histology was confirmed by H&E staining. Multiparametric immune profiling was performed with certain modifications to protocols as mentioned in references61-64 and the Opal™Assay Development guide (PerkinElmer). Sections were sequentially stained for four different antigens. Each staining step involved blocking with bovine serum albumin (BSA), application of primary antibody and corresponding secondary horseradish peroxidase-conjugated polymer antibodies, followed by the covalent binding of a different fluorophore using tyramide signal amplification. Briefly, frozen sections were allowed to dry for 30 min at room temperature (RT), fixed in 10% neutral buffered formalin (ThermoScientific) for 15 min at RT, rinsed in distilled water for 2 min, followed by rehydration in 1X TBS-T for 2 min. Antigen retrieval was performed with AR6 or AR9 buffers (PerkinElmer, AR600250ML; AR900250ML) in an EZ-Retriever System (Biogenex, MW014-MO), at 95°C for 15 min. Slides were allowed to cool before processing for staining using the Opal™ 5-color fIHC kits (PerkinElmer, NEL795001KT). Antibodies were used in the following order to detect cytokeratin 8/18 (TROMA-1, TROMA-I was deposited to the Developmental Studies Hybridoma Bank, the University of Iowa by Brulet, P./Kemler, R), Technology), FoxP3 (clone FJK-16s, ThermoFisher, 14-5773-80), CD8a (clone D4W2Z, Cell Signaling Technology, 98941), CD4 (clone 4SM95, ThermoFisher, 14–9766-80). TSA-Opal 520, TSA-Opal 540, TSA-Opal 620 and TSA-Opal 690 were applied to each antibody in the order stated. All staining from blocking to TSA-signal amplification was performed on an intelliPATH FLX automated slide stainer (Biocare Medical, IPS0001). Sections were then counterstained with Spectral DAPI (PerkinElmer, NEL795001KT) and mounted with Fluoromount-G™ (Electron Microscopy Services, 17984–25), manually. Slides were imaged using the PerkinElmer Vectra platform and analyzed using inForm image analysis software at the University of Texas MD Anderson Cancer Center North Campus Flow Cytometry and Cellular Imaging Core Facility (Houston, TX).
Statistical analysis
Growth spline modeling analysis was performed to test significances for tumor growth kinetics. Tests of significance between pairs of data are reported as p-values, derived using a student’s t-test with a two-tailed distribution and calculated at 95% confidence. Comparison of multiple sets of data was achieved with an analysis of variance with Tukey’s multiple comparisons. Survival analysis was determined by Log-Rank (Mantel-Cox) analysis. All error bars indicate standard error of measurement unless indicated otherwise. The analysis was performed using GraphPad Prism v7 or SPSS statistical analysis software.
Funding Statement
NIDCR-R00-DE023577 and UTHealth Rising STARs grants to SY. NIGMS T32 (T32GM088129) and NIDCR (F31DE026682) both of the NIH to JMN. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH; National Institute of Dental and Craniofacial Research [F31DE026682]; National Institute of Dental and Craniofacial Research [R00-DE-023577]; National Institute of General Medical Sciences [T32GM088129]; University of Texas Health Science Center at Houston [Rising STARs];
Acknowledgments
This research was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award R00-DE023577 and a University of Texas System Rising STAR grants to SY. This research was performed in the Flow Cytometry and Cellular Imaging Facility, which is supported in part by the National Institutes of Health through M.D. Anderson’s Cancer Center Support Grant CA016672. AGS acknowledges financial support from Baylor College of Medicine Weiss Law Endowment for Academic Excellence. JMN acknowledges financial support from NIGMS T32 (T32GM088129) and the NIDCR F31 NRSA training grants (F31DE026682). This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors would also like to thank Drs. Vlad Sandulache, Mitch Frederick and Yohannes Ghebre, Baylor College of Medicine for helpful scientific discussions and Dr. Liang Zhu (UTHealth) for statistical support.
Disclosure of Potential Conflicts of Interest
SY receives support from Privo Technologies as the PI of a Phase I/II clinical trial. Aspects of vaccine technology (DJM) have been licensed to Novartis for commercialization. AGS receives unrestricted support from Advaxis for Phase II clinical trial and Tessa Therapeutics for serving on DSMB for Phase III clinical trial, and for clinical trial protocol development.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A.. Cancer statistics. CA Cancer J Clin. 2014. Jan-Feb;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011. August 26;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015. February 1;21(3):632–641. doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Costa AM, Young MR.. Immunotherapy for head and neck cancer: advances and deficiencies. Anticancer Drugs. 2011. August;22(7):674–681. doi: 10.1097/CAD.0b013e328340fd18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, Hwu WJ, Kefford R, Weber JS, Daud A, Hamid O, Patnaik A, Ribas A, Robert C, Gangadhar TC, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016. May 1;34(13):1510–1517. doi: 10.1200/JCO.2015.64.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Errico A. Immunotherapy: PD-1-PD-L1 axis: efficient checkpoint blockade against cancer. Nat Rev Clin Oncol. 2015. February;12(2):63. doi: 10.1038/nrclinonc.2014.221. [DOI] [PubMed] [Google Scholar]
- 7.Hauschild A, Mohr P. [Rationale, visions and limits of Immuno-oncology: checkpoint inhibition as a new pillar of tumor therapy]. Oncol Res Treat. 2015;38(Suppl 3):2–5. doi: 10.1159/000381298. [DOI] [PubMed] [Google Scholar]
- 8.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012. March 22;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010. August 19;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016. November 10;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polverini PJ, D’Silva NJ, Lei YL. Precision therapy of head and neck squamous cell carcinoma. J Dent Res. 2018. June;97(6):614–621. doi: 10.1177/0022034518769645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther. 2017. October;178:31–47. doi: 10.1016/j.pharmthera.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumgaertner P, Jandus C, Rivals JP, Derré L, Lövgren T, Baitsch L, Guillaume P, Luescher IF, Berthod G, Matter M, et al. Vaccination-induced functional competence of circulating human tumor-specific CD8 T-cells. Int J Cancer. 2012. June 1;130(11):2607–2617. doi: 10.1002/ijc.26297. [DOI] [PubMed] [Google Scholar]
- 14.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014. November 27;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012. March 22;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat Mater. 2009. February;8(2):151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kearney CJ, Mooney DJ. Macroscale delivery systems for molecular and cellular payloads. Nat Mater. 2013. November;12(11):1004–1017. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 18.Ali OA, Emerich D, Dranoff G, Mooney DJ. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med. 2009. November 25;1(8):8ra19. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bencherif SA, Warren Sands R, Ali OA, Li WA, Lewin SA, Braschler TM, Shih T-Y, Verbeke CS, Bhatta D, Dranoff G, et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun. 2015. August 12;6:7556. doi: 10.1038/ncomms8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chew SA, Danti S. Biomaterial-based implantable devices for cancer therapy. Adv Healthc Mater. 2017. January;6:2. doi: 10.1002/adhm.201600766. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol. 2015. January;33(1):64–72. doi: 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshy ST, Mooney DJ. Biomaterials for enhancing anti-cancer immunity. Curr Opin Biotechnol. 2016. August;40:1–8. doi: 10.1016/j.copbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan YS, Sansanaphongpricha K, Prince MEP, Sun D, Wolf GT, Lei YL. Engineering vaccines to reprogram immunity against head and neck cancer. J Dent Res. 2018. June;97(6):627–634. doi: 10.1177/0022034518764416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed Res Int. 2015:948501. doi: 10.1155/2015/948501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welters MJ, Kenter GG, Piersma SJ, Vloon APG, Löwik MJG, Berends-van der Meer DMA, Drijfhout JW, Valentijn ARPM, Wafelman AR, Oostendorp J, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res. 2008. January 1;14(1):178–187. doi: 10.1158/1078-0432.CCR-07-1880. [DOI] [PubMed] [Google Scholar]
- 26.Kenter GG, Welters MJ, Valentijn AR, Lowik MJG, Berends-van der Meer DMA, Vloon APG, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009. November 5;361(19):1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 27.Tan YS, Sansanaphongpricha K, Xie Y, Donnelly CR, Luo X, Heath BR, Zhao X, Bellile E, Hu H, Chen H, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. 2018. May 16;24:4242–4255. doi: 10.1158/1078-0432.CCR-17-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014. April;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gildener-Leapman N, Lee J, Ferris RL. Tailored immunotherapy for HPV positive head and neck squamous cell cancer. Oral Oncol. 2014. September;50(9):780–784. doi: 10.1016/j.oraloncology.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albers A, Abe K, Hunt J, Wang J, Lopez-Albaitero A, Schaefer C, Gooding W, Whiteside TL, Ferrone S, DeLeo A, et al. Antitumor activity of human papillomavirus type 16 E7-specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res. 2005. December 1;65(23):11146–11155. doi: 10.1158/0008-5472.CAN-05-0772. [DOI] [PubMed] [Google Scholar]
- 31.Mansilla C, Berraondo P, Durantez M, Martínez M, Casares N, Arribillaga L, Rudilla F, Fioravanti J, Lozano T, Villanueva L, et al. Eradication of large tumors expressing human papillomavirus E7 protein by therapeutic vaccination with E7 fused to the extra domain a from fibronectin. Int J Cancer. 2012. August 1;131(3):641–651. doi: 10.1002/ijc.26412. [DOI] [PubMed] [Google Scholar]
- 32.Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, Weaver JC, Dellacherie MO, Shih T-Y, Ali OA, et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater. 2018. June;17(6):528–534. doi: 10.1038/s41563-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TL, Choi Y, Kim J. Mesoporous Silica as a Versatile Platform for Cancer Immunotherapy. Adv Mater. 2018. November;12:e1803953. doi: 10.1002/adma.201803953. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011. March 25;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 35.Leach DG, Dharmaraj N, Piotrowski SL, Lopez-Silva TL, Lei YL, Sikora AG, Young S, Hartgerink JD. STINGel: controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials. 2018. May;163:67–75. doi: 10.1016/j.biomaterials.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Judd NP, Allen CT, Winkler AE, Uppaluri R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol Head Neck Surg. 2012. September;147(3):493–500. doi: 10.1177/0194599812442037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res. 2016. December;4(12):1061–1071. doi: 10.1158/2326-6066.CIR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS, Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012. January 1;72(1):365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014. June 1;20(11):2873–2884. doi: 10.1158/1078-0432.CCR-14-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002. November;3(11):999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004. May-Jun;27(3):184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012. February 8;482(7385):405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoover AC, Spanos WC, Harris GF, Anderson ME, Klingelhutz AJ, Lee JH. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Arch Otolaryngol Head Neck Surg. 2007. May;133(5):495–502. doi: 10.1001/archotol.133.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016. July;17(7):956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 45.Mery B, Rancoule C, Guy JB, Espenel S, Wozny A-S, Battiston-Montagne P, Ardail D, Beuve M, Alphonse G, Rodriguez-Lafrasse C, et al. Preclinical models in HNSCC: A comprehensive review. Oral Oncol. 2017. February;65:51–56. doi: 10.1016/j.oraloncology.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Cheung AS, Mooney DJ. Engineered materials for cancer immunotherapy. Nano Today. 2015. August 01;10(4):511–531. doi: 10.1016/j.nantod.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013. October;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol Res. 2016. July;4(7):611–620. doi: 10.1158/2326-6066.CIR-15-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cash H, Shah S, Moore E, Caruso A, Uppaluri R, Van Waes C, Allen C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget. 2015. November 3;6(34):36400–36417. doi: 10.18632/oncotarget.5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferris RL, Martinez I, Sirianni N, Wang J, López-Albaitero A, Gollin SM, Johnson JT, Khan S. Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): a natural disease model provides insights into viral carcinogenesis. Eur J Cancer. 2005. March;41(5):807–815. doi: 10.1016/j.ejca.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 51.Gillison ML, Shah KV. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr Opin Oncol. 2001. May;13(3):183–188. [DOI] [PubMed] [Google Scholar]
- 52.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y-S, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012. February 8;482(7385):400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wargo JA, Reddy SM, Reuben A, Sharma P. Monitoring immune responses in the tumor microenvironment. Curr Opin Immunol. 2016. August;41:23–31. doi: 10.1016/j.coi.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li WA, Lu BY, Gu L, Choi Y, Kim J, Mooney DJ. The effect of surface modification of mesoporous silica micro-rod scaffold on immune cell activation and infiltration. Biomaterials. 2016. March;83:249–256. doi: 10.1016/j.biomaterials.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990. June 25;18(12):3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989. October;63(10):4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J Immunol. 2012. June 1;188(11):5365–5376. doi: 10.4049/jimmunol.1103553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. April 20;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009. November 5;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013. January 16;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014. November;70(1):46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 62.Feng Z, Puri S, Moudgil T, Wood W, Hoyt CC, Wang C, Urba WJ, Curti BD, Bifulco CB, Fox BA. Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. J Immunother Cancer. 2015;3:47. doi: 10.1186/s40425-015-0091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng Z, Bethmann D, Kappler M, Schaeffer J, Pannetier D, Guedj J, Rives M, Georges N, Garcia-Bonnet N, Sylla AI, et al. Multiparametric immune profiling in HPV- oral squamous cell cancer. JCI Insight. 2017. July 20;2:14. doi: 10.1172/jci.insight.93652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng Z, Jensen SM, Messenheimer DJ, Farhad M, Neuberger M, Bifulco CB, Fox BA. Multispectral Imaging of T and B Cells in Murine Spleen and Tumor. J Immunol. 2016. May 01;196(9):3943–3950. doi: 10.4049/jimmunol.1502635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.