

 A cooperative Lewis acid/photocatalytic reduction of salicylaldehyde-derived arylidene malonates provides access to a versatile, stabilized radical anion enolate. Using these unusual umpolung operators, we have developed a novel route to access densely functionalized carbo- and heterocycles through a radical annulation addition pathway.
A cooperative Lewis acid/photocatalytic reduction of salicylaldehyde-derived arylidene malonates provides access to a versatile, stabilized radical anion enolate. Using these unusual umpolung operators, we have developed a novel route to access densely functionalized carbo- and heterocycles through a radical annulation addition pathway.
Abstract
A cooperative Lewis acid/photocatalytic reduction of salicylaldehyde-derived arylidene malonates provides access to a versatile, stabilized radical anion enolate. Using these unusual umpolung operators, we have developed a novel route to access densely functionalized carbo- and heterocycles through a radical annulation addition pathway.
The development of annulation strategies has proved invaluable in organic synthesis, particularly for the construction of complex natural products.1 In a broad sense, annulation reactions can be divided into two electron and one electron approaches, where two electron annulation tactics include the venerable Diels–Alder, Michael and Dieckmann reactions, as well as general nucleophilic additions and alkylations. Conversely, one-electron annulation methods have focused on using halogenated starting materials and tin reagents, where seminal reports by Curran and Stork were revolutionary for the advent of radical annulations.2 Despite these elegant approaches, the use of stoichiometric tin hydrides is problematic both due to toxicity and purification problems, driving the need for the development of radical annulation reactivity accessed through catalytic methods using non-prefunctionalized starting materials.3 The development of photoredox chemistry has rendered the catalytic generation of open shell intermediates relatively facile due to the natural abundance and ease of use of visible light, as well as the superior chemoselectivity observed compared with traditional methodologies for radical-based approaches.4
Along these lines, the use of photoredox chemistry to access inverse polarity concepts, termed umpolung, have emerged as instrumental.5 Specifically, the generation of ketyl radical species (e.g. d1 umpolung operators) through the reduction of carbonyl derivatives has been a major focus.6 Since carbonyls are typically characterized by strongly negative redox potentials (E1/2 red = –1.93 V vs. SCE for benzaldehydes),7 the development of cooperative catalytic systems to effect said reduction potential have been highly explored to afford annulations, reductive couplings and radical–radical couplings (Fig. 1).8
Fig. 1. Selected annulation strategies.

Similarly, d3 umpolung operators in photoredox have been explored.9 Yoon et al. pioneered a cooperative catalytic approach to enone β-umpolung reactivity using a photoredox/Lewis acid approach to afford [2 + 2], and [3 + 2] cycloadditions.10 Following Yoon's seminal work on enone β-umpolung reactivity, recent reports have focused on using bifunctional and cooperative catalytic approaches in photoredox catalysis to access new chemical reactivity.11 However, new directions and opportunities remain unexplored in this area, primarily due to the inherent limitations of bifunctional catalytic manifolds, which restrict reactivity and generalizability as well.
As part of our program to generate new opportunities in β-umpolung chemistry, we recently reported the use of arylidene malonates as substrates in photoredox/Lewis acid cooperative catalysis to afford radical–radical cross coupling, radical dimerizations, and transfer hydrogenations.12 As a major goal of this study, we focused on designing a stabilized β-umpolung operator intermediate, with the hypothesis that a more stabile radical anion would enable underexplored chemical reactivity, namely intermolecular radical couplings, rather than the dimerization reactions often seen with enone-derived radical anions (e.g. cinnamates, E1/2 red = –2.3 V vs. SCE).13 In this regard, we demonstrated that arylidene malonates (E1/2 red = –1.57 V for phenyl arylidene malonate vs. SCE),14 demonstrated a drastic shift in reduction potential upon complexation with a Lewis acid (E1/2 red = –0.37 V for phenyl arylidene malonate vs. SCE). By utilizing arylidene malonates, this cooperative catalytic approach afforded a stabilized β-radical enolate intermediate exhibiting reactivity divergent from reductive species generated from conventional enones, presumably due to greater persistence of the resonance-stabilized radical anion. Herein we report a cooperative Lewis acid/photoredox reductive enolate annulation strategy to provide densely functionalized carbo and heterocycles.
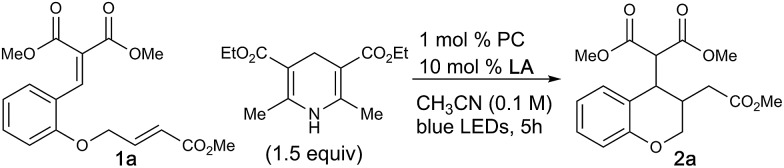
Chroman and related heterocycles are a diverse class of bioactive small molecules that our lab have previously prepared and investigated for their wide range of biological activities in anti-cancer models.15 We envisioned that the arylidene malonate-derived β-umpolung operator could grant access to previously unprepared derivatives. We initiated our studies with 1a, which was readily accessible in 2 steps from salicylaldehyde, using a variety of photocatalysts and blue LEDs, the results of which are summarized in Table 1. Gratifyingly, we found that the desired chromane product was formed with scandium triflate in acetonitrile using photocatalyst dF-Ir and Hantzsch ester (HEH) in 81% yield with a 1.2 : 1 dr. A variety of other bidentate Lewis acids were investigated, all of which were capable of affording the title reaction, albeit with decreased yield in comparison to scandium triflate (entries 1–4). A survey of transition metal photocatalysts identified dF-Ir as optimal. Interestingly, organocatalysts of the dicyanobenzene family performed as well as dF-Ir, with diphenyl aniline organocatalyst DPAIPN16 providing the desired product in comparable 85% yield and diastereoselectivity. Solvent evaluation confirmed acetonitrile to be the optimal solvent, whereas aprotic and protic (see ESI†) were shown to be less successful. Evaluations of alternative stoichiometric hydrogen atom donors (DIPEA, NEt3, NBu3, BT) were capable of delivering 2a in slightly decreased yields relative to the HEH; where a slight increase in diastereoselectivity was observed (2 : 1 dr observed with NEt3) at a precipitous loss of yield (entries 9–12). The rationale for this observed increase in diastereoselectivity could be due to coordination from the radical cation of NEt3 with the malonate, providing a facial selectivity to facilitate increased diastereoselectivity, or that the C–C bond formation is reversible and that the diastereoselectivity is determined, at least in part, by the relative rates of hydrogen atom transfer. As tertiary amines have been used previously for activation of carbonyls for reduction, we evaluated DIPEA, NEt3 and NBu3 in the absence of Sc(OTf)3, where drastically lower yields were observed (entries 13–15). In this instance, it is believed that upon initial single electron reduction, the oxidized nitrogen atom in DIPEA, NEt3 or NBu3 can form a 2-center/3-e– interaction,17 or after a [1,2]-H shift, can serve a hydrogen-bond donor,18 which results in the oxidized amine serving as both the terminal reductant and the Lewis acid necessary for activation of 1a.8d,e A series of control experiments demonstrated that the reaction did not take place in the absence of light, photocatalyst or Lewis acid (entries 16–18).
Table 1. Optimization of the reaction conditions.
| |||
| Entry | PC | Lewis acid | Yield a |
| 1 | 3 | Sc(OTf)3 | 81 |
| 2 | 3 | Mg(OTf)2 | 72 |
| 3 | 3 | La(OTf)3 | 55 |
| 4 | 3 | LiCl | 62 |
| 5 | 4 | Sc(OTf)3 | Trace |
| 6 | 5 | Sc(OTf)3 | 52 |
| 7 | 6 | Sc(OTf)3 | 75 |
| 8 | 7 | Sc(OTf)3 | 87 (85) b |
| 9 c | 7 | Sc(OTf)3 | 67 |
| 10 d | 7 | Sc(OTf)3 | 55 |
| 11 e | 7 | Sc(OTf)3 | 52 |
| 12 f | 7 | Sc(OTf)3 | 64 |
| 13 c | 7 | — | 41 |
| 14 d | 7 | — | 31 |
| 15 e | 7 | — | 35 |
| 16 | 7 | — | NR |
| 17 | — | Sc(OTf)3 | NR |
| 18 g | 7 | Sc(OTf)3 | NR |
| |||
aYield determined by GC with bibenzyl as internal standard. Product was observed in a 1.2 : 1 dr ratio.
bYield of isolated product.
cDIPEA used instead of HEH.
dNet3 used instead of HEH.
eNBu3 used instead of HEH.
fBT used instead of HEH.
gNo light. DIPEA = diisopropylethylamine, NEt3 = triethylamine, BT = 2-phenyl-dihydrobenzothiazoline.
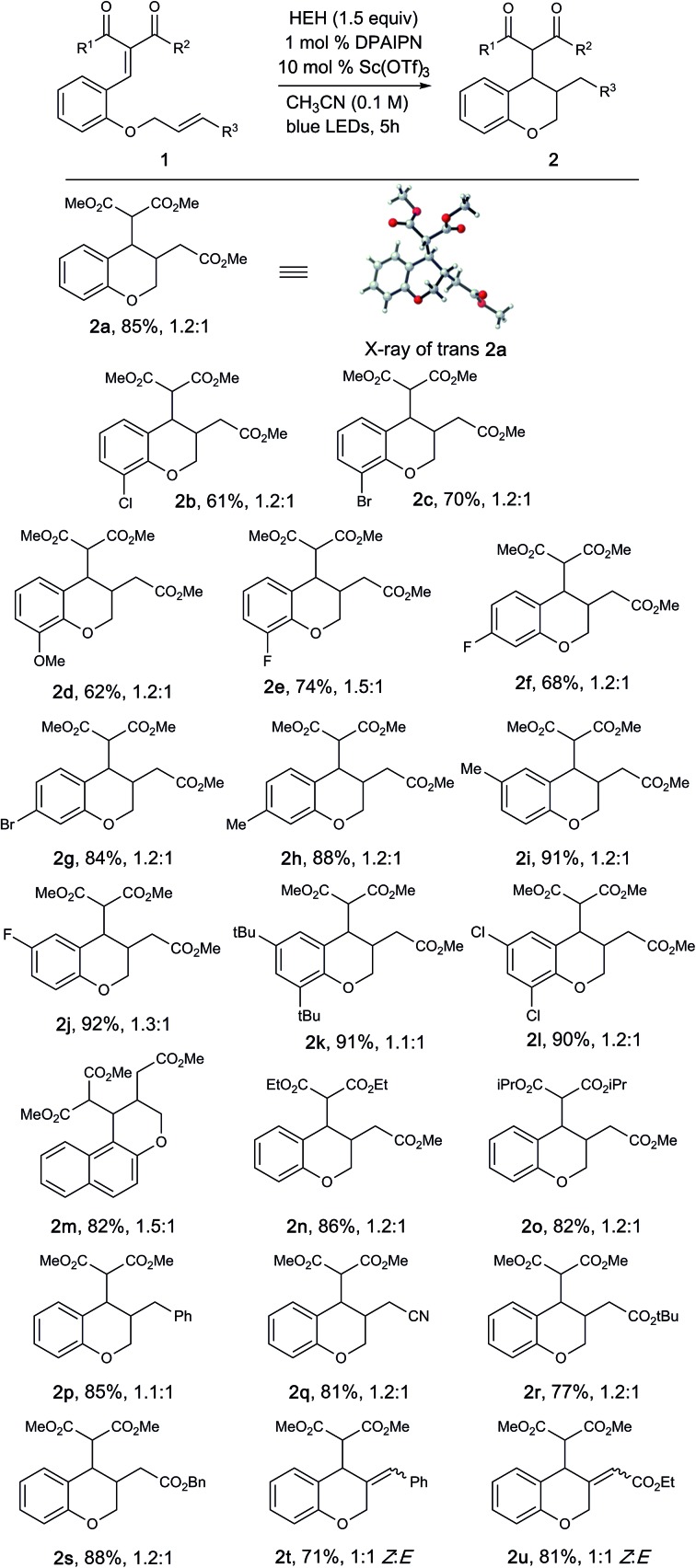
With these optimized conditions, we investigated a variety of substrates (Table 2). Generally, the desired products were obtained in good to excellent yields. Substrates bearing either electron-rich and electron-poor substituents were well tolerated; however, substrates with substitution at the 6-position did not provide the desired product, presumably due to reduced overlap between the enone and aryl π-systems. Diversity could be introduced into the dicarbonyl moiety to tolerate a variety of diesters (2n, 2o) in good to excellent yields, albeit as a complex mixture of diastereomers. The unsaturated electrophile could be varied to facilitate access to benzyl (2p) and nitrile (2q) substituted chromanes in excellent yields. Replacement of the olefin electrophile with alkyne electrophiles proceeded with excellent yields (2t, 2u), albeit as a mixture of Z/E isomers.
Table 2. Reactions of salicylaldehyde derived arylidene malonates a .
|
aReaction conditions: 1 (0.2 mmol), HEH (0.3 mmol), PC 7 (1 mol%), Sc(OTf)3 (10 mol%), degassed CH3CN (2.0 mL) was irradiated with a blue LED (456 nm) for 5 h. Reported yields are determined after isolation by column chromatography.
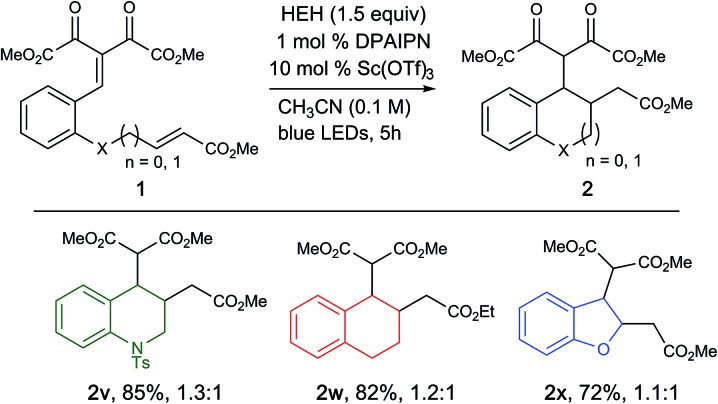
Furthermore, we were pleased to find that modification of the starting material to access tetrahydroquinolines (2v) and tetrahydronaphthalenes (2w) were also successful in high yields, illustrating the capability of this methodology to access a wide range of carbo and heterocyclic scaffolds in high efficiency. Attempts to access dihydrobenzofuran was successful, albeit with slightly diminished yields (2x, 72% yield), where the remaining mass balance is the saturated arylidene malonate species. This is presumably due to the decreased electrophilicity of the vinylogous carbonate starting material due to hyperconjugation (Table 3).19
Table 3. Reactions of arylidene malonates affording different core scaffolds a .
|
aReaction conditions: 1 (0.2 mmol), HEH (0.3 mmol), PC 7 (1 mol%), Sc(OTf)3 (10 mol%), degassed CH3CN (2.0 mL) was irradiated with a blue LED (456 nm) for 5 h. Reported yields are determined after isolation by column chromatography.
Unfortunately, substrates designed to allow access to quaternary centers (either derived from 2′-hydroxyacetophenone or from senecioic acid) were not successful in this reaction, presumably due to decreased reactivity of the resulting β-radical enolate intermediate and decreasing electrophilicity of the tethered alkene respectively. The importance of the dicarbonyl moiety was validated as a means for Lewis acid coordination, as substrates derived from Meldrum's acid showed no conversion under the optimized conditions. An intermolecular reductive coupling between 8 and methyl acrylate was unsuccessful, where only transfer hydrogenation to afford 9 was observed. This likely indicates that reduction of the resulting radical anion proceeds more rapidly than radical conjugate addition (Fig. 2).
Fig. 2. Reductive annulation reaction extension.

Notably, we were able to demonstrate this reaction on multi-gram scale, where 2a was accessed in similar yields under identical reaction conditions, further highlighting the potential of this reaction. Additionally, the DPAIPN photocatalyst was recovered after column chromatography in 80% yield. The recovered DPAIPN was subsequently used in a multi-gram scale reaction without loss of yield, giving this methodology additional utility due to the ability to recover and reuse the catalyst (Fig. 3).
Fig. 3. Gram-scale reaction.

A practical advantage of this strategy is the ease of synthetically elaborating these products. A one-pot Krapcho/Dieckmann/Krapcho sequence with cis-2a proceeded in 85% yield, affording an interesting [6,6,5] fused ring system (Fig. 4).
Fig. 4. Krapcho/Dieckmann synthesis of fused ring system.

To probe the mechanism of this process, we investigated whether this photoredox process can propagate through a chain process rather than the presumed closed-catalytic photoredox cycle.20 We were pleased to find that upon using a “light/dark” experiment21 that product formation was only observed during periods of irradiation (Fig. 5). While supporting our mechanistic hypothesis, “light/dark” experiments are typically not sufficient to fully elucidate whether a process proceeds through a closed-catalytic cycle as opposed to a propagating chain process.22
Fig. 5. Light/dark experiments.

As a follow up to these studies, we demonstrated that this reaction has a quantum yield of 1.0, indicating that the reaction does not propagate through a radical chain process (see ESI† for details).
To further study the mechanism of the β-radical enolate formation, we employed fluorescence quenching techniques with 1a as a model substrate. A Stern–Volmer analysis revealed that 1a does not quench the excited state of DPAIPN (E1/2 red = –1.52 V vs. SCE) in acetonitrile at 25 °C (Fig. 6). However, inclusion of 100 mol% Sc(OTf)3 resulted in a large decrease in the measured fluorescence. Notably, control experiments demonstrated that Sc(OTf)3 itself does not quench the DPAIPN excited state, indicating that pre-complexation of Sc(OTf)3 with 1a is necessary for generation of the radical anion. Furthermore, variation of the stoichiometry of Sc(OTf)3 and 1a revealed that the quenching process exhibits a first-order dependence on each component (see ESI†). Notably, these results only provide evidence that a Sc(OTf)3/1a complex is necessary for arylidene malonate activation and is not indicative of oxidative quenching of DPAIPN by Sc(OTf)3/1a. To evaluate the possibility of a reductive quenching mechanism, we conducted Stern–Volmer analysis with the HEH, where quenching of DPAIPN fluorescence by the HEH is observed. Both 1a and a 1a/100 mol% Sc(OTf)3 complex were also added to the HEH for Stern–Volmer analysis, where minimal changes to the fluorescence quenching profile were observed. This trend was evident across 10, 25 and 50 mol% Sc(OTf)3 as well. This is likely indicative that the HEH is responsible for quenching the photo-excited DPAIPN, not the Sc(OTf)3/1a complex. Fluorescence quenching experiments with NBu3 was also evaluated, where the inclusion of 1a and a 1a/100 mol% Sc(OTf)3 complex resulted in no change to the quenching profile of DPAIPN by NBu3 (see ESI†).
Fig. 6. Stern–Volmer fluorescence quenching analysis.

While both processes are thermodynamically comparable, it is unlikely that an oxidative quenching mechanism predominates, primarily because of the decreased possibility of finding a Sc(OTf)3/1a complex due to catalytic Sc(OTf)3 relative to superstoichiometric HEH. We investigated the transformation of 1a to 2a using stoichiometric Sc(OTf)3 and found no significant difference in the reactivity profile or yield between using 10 and 100 mol% Sc(OTf)3. We found that using stoichiometric NBu3 was able to provide 2a without the presence of Sc(OTf)3 in 35% yield, indicating that reductive quenching of the photocatalyst is likely the initial step of this mechanism, where oxidative quenching is thermodynamically unfavourable (Table 1, entry 15). Moreover, as with transition metal photocatalysts, reductive quenching of DPAIPN is kinetically favourable relative to oxidative quenching, leading us to believe that a reductive quenching pathway is the primary pathway.23 Furthermore, Ir(ppy)3, which would only be viable in an oxidative quenching cycle, only yielded trace product (Table 1, entry 5).
To further understand the nature of the Sc(OTf)3/1a complex, UV-Vis spectroscopic characterization was carried out. Interestingly, the Sc(OTf)3/1a complex demonstrated a considerable difference in the UV-Vis spectrum relative to 1a alone, where there is an additional shoulder peak around 380–410 nm. While this demonstrates that the Sc(OTf)3/1a complex can absorb blue LED light, no reactivity is observed without the DPAIPN photocatalyst present, indicating that it is unlikely that the Sc(OTf)3/1a complex can generate any intermediates that result in consumption of 1a. To confirm that the Sc(OTf)3/1a complex does not undergo productive photoredox reaction pathways in the absence of DPAIPN, we irradiated 1a with 10 mol% Sc(OTf)3 and superstoichiometric DIPEA, NEt3, NBu3 and HEH. No conversion of 1a was observed in any case. Finally, the UV-Vis profile of 1a showed no change in the presence of either DIPEA, NEt3 or NBu3 (100 mol%), indicating that the amines themselves are not interacting with 1a, but instead need to undergo a single electron reduction to complex with 1a (see ESI† for details).
With this data in hand and in line with our previous work on photoredox/Lewis acid cooperative catalysis, we propose the following mechanism for the observed reactivity (Fig. 7): irradiation with visible light results in the formation of excited DPAIPN photocatalyst, a capable oxidant (E1/2 ox = 1.10 V vs. SCE). Reduction of the resulting DPAIPN excited state by HEH (E1/2 ox = 0.89 V vs. SCE) furnishes a strongly reducing DPAIPN catalyst (E1/2 red = –1.52 V vs. SCE). Subsequently, the reduced DPAIPN species transfers an electron to the Lewis acid–arylidene malonate complex, producing the nucleophilic radical anion and regenerating the ground state DPAIPN catalyst. The radical anion is able to add into the unsaturated bond, forming the chromane ring and a stabilized radical, which upon hydrogen atom transfer from the corresponding HEH radical cation, leads to the enolate complex. Subsequent proton transfer from the protonated HEH leads to the desired product.
Fig. 7. Proposed pathway.

Conclusions
A Lewis acid/photoredox cooperative catalytic manifold is capable of generating stabilized radical anion species from salicylaldehyde-derived arylidene malonates has been developed. This reactive intermediate undergoes intramolecular conjugate addition with pendent unsaturated electrophiles to afford structurally diverse chromanes. This platform sets the stage for further development of β-umpolung reactivity via photoredox catalysis, which is currently underway in our laboratory.
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences (R01GM073072, F31GM116532 to B. R. M. and T32GM105538 to R. C. B.) for financial support. R. C. B. was supported in part by the Chicago Cancer Baseball Charities at the Lurie Cancer Center of Northwestern University.
Footnotes
†Electronic supplementary information (ESI) available. CCDC 1835356. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00302a
References
- Jung M. E. Tetrahedron. 1976;32:3. [Google Scholar]
- (a) Curran D. P., van Elburg P. A. Tetrahedron Lett. 1989;30:2501. [Google Scholar]; (b) Curran D. P., Seong C. M. Tetrahedron. 1992;48:2175. [Google Scholar]; (c) Stork G., Mook R., Biller S. A., Rychnovsky S. D. J. Am. Chem. Soc. 1983;105:3741. [Google Scholar]; (d) Stork G., Suh H. S., Kim G. J. Am. Chem. Soc. 1991;113:7054. [Google Scholar]
- (a) Studer A., Curran D. P. Nat. Chem. 2014;6:765. doi: 10.1038/nchem.2031. [DOI] [PubMed] [Google Scholar]; (b) Studer A., Curran D. P. Angew. Chem., Int. Ed. 2016;55:58. doi: 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- (a) Prier C. K., Rankic D. A., MacMillan D. W. C. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaw M. H., Twilton J., MacMillan D. W. C. J. Org. Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Petronijević F. R., Nappi M., MacMillan D. W. C. J. Am. Chem. Soc. 2013;135:18323. doi: 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Terrett J. A., Clift M. D., MacMillan D. W. C. J. Am. Chem. Soc. 2014;136:6858. doi: 10.1021/ja502639e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jeffrey J. L., Petronijević F. R., MacMillan D. W. C. J. Am. Chem. Soc. 2015;137:8404. doi: 10.1021/jacs.5b05376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fava E., Millet A., Nakajima M., Loescher S., Rueping M. Angew. Chem., Int. Ed. 2016;55:6776. doi: 10.1002/anie.201511235. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fuentes de Arriba A. L., Urbitsch F., Dixon D. J. Chem. Commun. 2016;52:14434. doi: 10.1039/c6cc09172e. [DOI] [PubMed] [Google Scholar]; (f) Wang R., Ma M., Gong X., Panetti G. B., Fan X., Walsh P. J. Org. Lett. 2018;20:2433. doi: 10.1021/acs.orglett.8b00778. [DOI] [PubMed] [Google Scholar]; (g) Xu W., Ma J., Yuan X.-A., Dai J., Xie J., Zhu C. Angew. Chem., Int. Ed. 2018;57:10357. doi: 10.1002/anie.201805927. [DOI] [PubMed] [Google Scholar]
- (a) Qi L., Chen Y. Angew. Chem., Int. Ed. 2016;55:13312. doi: 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]; (b) Lee K. N., Lei Z., Ngai M.-Y. J. Am. Chem. Soc. 2017;139:5003. doi: 10.1021/jacs.7b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang H.-H., Yu S. J. Org. Chem. 2017;82:9995. doi: 10.1021/acs.joc.7b01425. [DOI] [PubMed] [Google Scholar]; (d) Chen M., Zhao X., Yang C., Xia W. Org. Lett. 2017;19:3807. doi: 10.1021/acs.orglett.7b01677. [DOI] [PubMed] [Google Scholar]; (e) Leitch J. A., Fuentes de Arriba A. L., Tan J., Hoff O., Martínez C. M., Dixon D. J. Chem. Sci. 2018;9:6653. doi: 10.1039/c8sc01704b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani O., Yanagida S., Takamuku S., Pac C. J. Org. Chem. 1987;52:2790. [Google Scholar]
- (a) Tarantino K. T., Liu P., Knowles R. R. J. Am. Chem. Soc. 2013;135:10022. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]; (b) Hopkinson M. N., Sahoo B., Li J. L., Glorius F. Chem.–Eur. J. 2014;20:3874. doi: 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]; (c) Yayla H. G., Knowles R. R. Synlett. 2014;25:2819. [Google Scholar]; (d) Nakajima M., Fava E., Loescher S., Jiang Z., Rueping M. Angew. Chem., Int. Ed. 2015;54:8828. doi: 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]; (e) Fava E., Nakajima M., Nguyen A. L. P., Rueping M. J. Org. Chem. 2016;81:6959. doi: 10.1021/acs.joc.6b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gentry E. C., Knowles R. R. Acc. Chem. Res. 2016;49:1546. doi: 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Miller D. C., Tarantino K. T., Knowles R. R. Top. Curr. Chem. 2016;374:30. doi: 10.1007/s41061-016-0030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Skubi K. L., Blum T. R., Yoon T. P. Chem. Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yoon T. P. Acc. Chem. Res. 2016;49:2307. doi: 10.1021/acs.accounts.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Neumann M., Zeitler K. Chem.–Eur. J. 2013;19:6950. doi: 10.1002/chem.201204573. [DOI] [PubMed] [Google Scholar]; (b) Pandey G., Hajra S., Ghorai M. K., Kumar K. R. J. Am. Chem. Soc. 1997;119:8777. [Google Scholar]; (c) Crimmins M. T. Chem. Rev. 1988;88:1453. [Google Scholar]; (d) Schuster D. I., Lem G., Kaprinidis N. A. Chem. Rev. 1993;93:3. [Google Scholar]; (e) Streuff J., Gansäuer A. Angew. Chem., Int. Ed. 2015;54:14232. doi: 10.1002/anie.201505231. [DOI] [PubMed] [Google Scholar]; (f) Larraufie M.-H., Pellet R., Fensterbank L., Goddard J.-P., Lacôte E., Malacria M., Ollivier C. Angew. Chem., Int. Ed. 2011;50:4463. doi: 10.1002/anie.201007571. [DOI] [PubMed] [Google Scholar]
- (a) Ischay M. A., Anzovino M. E., Du J., Yoon T. P. J. Am. Chem. Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Ischay M. A., Lu Z., Yoon T. P. J. Am. Chem. Soc. 2010;132:8572. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Du J., Espelt L. R., Guzei I. A., Yoon T. P. Chem. Sci. 2011;2:2115. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hurtley A. E., Cismesia M. A., Ischay M. A., Yoon T. P. Tetrahedron. 2011;67:4442. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu Z., Shen M., Yoon T. P. J. Am. Chem. Soc. 2011;133:1162. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yoon T. P. ACS Catal. 2013;3:895. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Du J., Yoon T. P. J. Am. Chem. Soc. 2009;131:14604. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tyson E. L., Farney E. P., Yoon T. P. Org. Lett. 2012;14:1110. doi: 10.1021/ol3000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Murphy J. J., Bastida D., Paria S., Fagnoni M., Melchiorre P. Nature. 2016;532:218. doi: 10.1038/nature17438. [DOI] [PubMed] [Google Scholar]; (b) Dell'Amico L., Fernández-Alvarez V. M., Maseras F., Melchiorre P. Angew. Chem., Int. Ed. 2017;56:3304. doi: 10.1002/anie.201612159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P. Nat. Chem. 2017;9:868. doi: 10.1038/nchem.2748. [DOI] [PubMed] [Google Scholar]; (d) Mazzarella D., Crisenza G. E. M., Melchiorre P. J. Am. Chem. Soc. 2018;140:8439. doi: 10.1021/jacs.8b05240. [DOI] [PubMed] [Google Scholar]; (e) Verrier C., Alandini N., Pezzetta C., Moliterno M., Buzzetti L., Hepburn H. B., Vega-Peñaloza A., Silvi M., Melchiorre P. ACS Catal. 2018;8:1062. [Google Scholar]; (f) de Assis F. F., Huang X., Akiyama M., Pilli R. A., Meggers E. J. Org. Chem. 2018;83:10922–10932. doi: 10.1021/acs.joc.8b01588. [DOI] [PubMed] [Google Scholar]; (g) Lin S.-X., Sun G.-J., Kang Q. Chem. Commun. 2017;53:7665. doi: 10.1039/c7cc03650g. [DOI] [PubMed] [Google Scholar]; (h) Bonilla P., Rey Y. P., Holden C. M., Melchiorre P. Angew. Chem., Int. Ed. 2018;57:12819. doi: 10.1002/anie.201808183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Goti G., Bieszczad B., Vega-Peñaloza A., Melchiorre P. Angew. Chem., Int. Ed. 2019;58:1213–1217. doi: 10.1002/anie.201810798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B. R., Scheidt K. A. Org. Lett. 2018;20:6877. doi: 10.1021/acs.orglett.8b02893. [DOI] [PubMed] [Google Scholar]
- (a) Shono T., Nishiguchi I., Ohmizu H. J. Am. Chem. Soc. 1977;99:7396. [Google Scholar]; (b) Zhao G., Yang C., Guo L., Sun H., Lin R., Xia W. J. Org. Chem. 2012;77:6302. doi: 10.1021/jo300796j. [DOI] [PubMed] [Google Scholar]
- Gang D., Jun Y., Xiao-ping Y., Hui-jun X. Tetrahedron. 1990;46:5967. [Google Scholar]
- (a) Farmer R. L., Scheidt K. A. Chem. Sci. 2013;4:3304. doi: 10.1039/C3SC50424G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Farmer R. L., Biddle M. M., Nibbs A. E., Huang X., Bergan R. C., Scheidt K. A. ACS Med. Chem. Lett. 2010;1:400. doi: 10.1021/ml100110x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Biddle M. M., Lin M., Scheidt K. A. J. Am. Chem. Soc. 2007;129:3830. doi: 10.1021/ja070394v. [DOI] [PubMed] [Google Scholar]; (d) Nibbs A. E., Baize A.-L., Herter R. M., Scheidt K. A. Org. Lett. 2009;11:4010. doi: 10.1021/ol901676f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McDonald B. R., Nibbs A. E., Scheidt K. A. Org. Lett. 2015;17:98. doi: 10.1021/ol503303w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xu L., Gordon R., Farmer R., Pattanayak A., Binkowski A., Huang X., Avram M., Krishna S., Voll E., Pavese J., Chavez J., Bruce J., Mazar A., Nibbs A., Anderson W., Li L., Jovanovic B., Pruell S., Valsecchi M., Francia G., Betori R., Scheidt K., Bergan R. Nat. Commun. 2018;9:2454. doi: 10.1038/s41467-018-04465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nibbs A. E., Scheidt K. A. Eur. J. Org. Chem. 2012;2012:449. doi: 10.1002/ejoc.201101228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Luo J., Zhang J. ACS Catal. 2016;6:873. [Google Scholar]; (b) Romero N. A., Nicewicz D. A. Chem. Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Humbel S., Côte I., Hoffmann N., Bouquant J. J. Am. Chem. Soc. 1999;121:5507. [Google Scholar]
- Akalay D., Dürner G., Bats J. W., Bolte M., Göbel M. W. J. Org. Chem. 2007;72:5618. doi: 10.1021/jo070534j. [DOI] [PubMed] [Google Scholar]
- Foy N. J., Forbes K. C., Crooke A. M., Gruber M. D., Cannon J. S. Org. Lett. 2018;20:5727–5731. doi: 10.1021/acs.orglett.8b02442. [DOI] [PubMed] [Google Scholar]
- Cismesia M. A., Yoon T. P. Chem. Sci. 2015;6:5426. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Miyake Y., Nakajima K., Nishibayashi Y. J. Am. Chem. Soc. 2012;134:3338. doi: 10.1021/ja211770y. [DOI] [PubMed] [Google Scholar]; (b) Wallentin C.-J., Nguyen J. D., Finkbeiner P., Stephenson C. R. J. J. Am. Chem. Soc. 2012;134:8875. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]; (c) Sahoo B., Hopkinson M. N., Glorius F. J. Am. Chem. Soc. 2013;135:5505. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; (d) Mizuta S., Engle K. M., Verhoog S., Galicia-López O., O'Duill M., Médebielle M., Wheelhouse K., Rassias G., Thompson A. L., Gouverneur V. Org. Lett. 2013;15:1250. doi: 10.1021/ol400184t. [DOI] [PubMed] [Google Scholar]; (e) Oh S. H., Malpani Y. R., Ha N., Jung Y.-S., Han S. B. Org. Lett. 2014;16:1310. doi: 10.1021/ol403716t. [DOI] [PubMed] [Google Scholar]
- (a) Burnett G. M., Melville H. W. Chem. Rev. 1954;54:225. [Google Scholar]; (b) McIntosh R. G., Eager R. L., Spinks J. W. T. Science. 1960;131:992. doi: 10.1126/science.131.3405.992. [DOI] [PubMed] [Google Scholar]; (c) McIntosh R. G., Eager R. L., Spinks J. W. T. Can. J. Chem. 1965;43:3490. [Google Scholar]
- (a) Pitre S. P., McTiernan C. D., Scaiano J. C. Acc. Chem. Res. 2016;49:1320. doi: 10.1021/acs.accounts.6b00012. [DOI] [PubMed] [Google Scholar]; (b) Arias-Rotondo D. M., McCusker J. K. Chem. Soc. Rev. 2016;45:5803. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.