

Abstract
Glioblastoma (GBM) is the most lethal primary brain tumor and is highly resistant to current treatments. GBM harbors glioma stem cells (GSCs) that not only initiate and maintain malignant growth but also promote therapeutic resistance including radioresistance. Thus, targeting GSCs is critical for overcoming the resistance to improve GBM treatment. Because the bone marrow and X-linked (BMX) nonreceptor tyrosine kinase is preferentially up-regulated in GSCs relative to nonstem tumor cells and the BMX-mediated activation of the signal transducer and activator of transcription 3 (STAT3) is required for maintaining GSC self-renewal and tumorigenic potential,pharmacological inhibition of BMX may suppress GBM growth and reduce therapeutic resistance.We demonstrate that BMX inhibition by ibrutinib potently disrupts GSCs, suppresses GBM malignant growth, and effectively combines with radiotherapy. Ibrutinib markedly disrupts the BMX-mediated STAT3 activation in GSCs but shows minimal effect on neural progenitor cells (NPCs) lacking BMX expression. Mechanistically, BMX bypasses the suppressor of cytokine signaling 3 (SOCS3)-mediated inhibition of Janus kinase 2 (JAK2), whereas NPCs dampen the JAK2-mediated STAT3 activation via the negative regulation by SOCS3, providing a molecular basis for targeting BMX by ibrutinib to specifically eliminate GSCs while preserving NPCs. Our preclinical data suggest that repurposing ibrutinib for targeting GSCs could effectively control GBM tumor growth both as monotherapy and as adjuvant with conventional therapies.
INTRODUCTION
Glioblastoma (GBM) is the most common malignant primary brain tumor, which universally shows extreme resistance to conventional therapies including radiation. Despite therapeutic advances in treating other solid tumors, there has been minimal improvement in the treatment and survival of GBM patients (1), highlighting an urgent need for therapeutic development. GBM displays remarkable cellular heterogeneity and hierarchy containing glioma stem cells (GSCs) with potent tumorigenic capacity (2, 3). GSCs are functionally defined by extensive self-renewal, multilineage differentiation, and potential to drive malignant growth in vivo (2, 4). GSCs promote therapeutic resistance, cancer invasion, tumor angiogenesis, immune evasion, and recruitment of tumor-supportive macrophages to facilitate malignant growth (2, 5, 6), indicating that targeting GSCs is an important strategy to improve GBM treatment. However, the similarity between GSCs and neural progenitor cells (NPCs) poses a major challenge for developing a GSC-specific treatment paradigm (3, 7). Because both GSCs and NPCs share features of stemness and use similar molecular pathways for their maintenance (7), targeting crucial regulators in common pathways may affect both GSCs and NPCs. Therefore, defining the differential regulatory mechanisms that distinguish GSCs from NPCs is critical for developing GSC-specific therapeutics.
In the search for GSC-specific molecular targets, we previously discovered that the bone marrow and X-linked (BMX) nonreceptor tyrosine kinase (also known as ETK) is preferentially expressed in GSCs but not in NPCs (8). BMX mediates the hyperactivation of the signal transducer and activator of transcription 3 (STAT3) in GSCs, which is required for GSC self-renewal and tumorigenic potential (8). As a member of the TEC family kinases (9), BMX can be activated by numerous signals (10,11) and is overexpressed in several cancers (8, 12,13). Functional inhibition of BMX impairs tumor cell proliferation and angiogenesis (8, 14). Moreover, BMX contributes to cancer cell resistance to radiation and chemotherapy (13, 15). In addition, BMX knockout mice display minimal phenotypes (16), suggesting that BMX is not required for embryo development and survival. Thus, pharmacologic inhibition of BMX kinase may have therapeutic potential with little toxicity to normal tissues.
BMX-mediated STAT3 activation is essential for maintaining the tumorigenic potential of GSCs (8). STAT3 activation is a critical signaling event required for both malignant and normal physiological functions (8, 17,18). The canonical STAT3 activation is stimulated by the interleukin-6 (IL-6) cytokine family, followed by the dimerization of the IL-6 receptor glycoprotein 130 (gp130) and the activating phosphorylation of Janus family kinases (JAKs) (18). The activated JAKs then phosphorylate STAT3, inducing its nuclear translocation to promote target gene transcription (17–19). To avoid overactivation in normal cells, STAT3 signaling is restrained by endogenous negative regulatory mechanisms mediated by the suppressor of cytokine signaling 3 (SOCS3). SOCS3 is a pivotal inhibitory factor that binds to the JAK2-gp130 receptor complex to inactivate JAK2 and therefore suppresses STAT3 activation (20, 21). Although STAT3 activity is tightly controlled in normal cells including NPCs, STAT3 hyperactivation is often detected in tumor cells, including GSCs in GBM (8, 19, 22). Defining the molecular mechanisms underlying the differential activation of STAT3 between NPCs and GSCs may facilitate the development of therapeutics selectively targeting GSCs.
Here, we defined a mechanism underlying the differential regulation of STAT3 activation that distinguishes GSCs from NPCs. In GSCs, BMX circumvents the SOCS3 inhibition of JAK2 to sustain STAT3 activation. By contrast, the JAK2-mediated STAT3 activation in NPCs is negatively regulated by SOCS3. These findings provide a molecular basis for targeting BMX to inhibit STAT3 activation specifically in GSCs but not in NPCs. Furthermore, we demonstrated the therapeutic potential of targeting BMX by a pharmacologic inhibitor, ibrutinib (originally identified as PCI-32765) (23), for GBM treatment. Ibrutinib is U.S. Food and Drug Administration (FDA)-approved for treating mantle cell lymphoma and chronic lymphocytic leukemia by inhibiting Bruton’s tyrosine kinase (BTK) (24,25), but ibrutinib can inhibit kinase activities of both BTK and BMX through irreversible binding to a conserved cysteine residue of BTK and BMX with similar affinity (23, 24). Here, we found that BTK expression was hardly detected in GSCs. Thus, ibrutinib should primarily inhibit BMX in GSCs. We demonstrated that ibrutinib suppressed the BMX-mediated STAT3 activation to disrupt GSC maintenance and impair GBM tumor growth and radioresistance. Notably, the effective dose of ibrutinib for inhibiting GSCs and GBM growth showed no detectable impact on NPCs, which lack expression of BMX and BTK. Our preclinical studies suggest that targeting GSCs through BMX inhibition by ibrutinib may effectively improve GBM treatment.
RESULTS
Targeting BMX by ibrutinib inhibits GBM growth to prolong animal survival
We previously discovered that BMX-mediated STAT3 activation was required for maintaining GSC tumorigenic potential (8). To translate this dependency into a clinical application, we examined whether pharmacologic inhibition of BMX by ibrutinib, a small-molecule inhibitor of BMX and BTK (23), could suppress tumor growth in GSC-derived orthotopic xenografts. Because BTK expression is hardly detected in human GSCs and NPCs and BMX is highly expressed in GSCs but not in NPCs (fig. S1A), ibrutinib should primarily target BMX kinase in GSCs to inhibit GBM growth. To test this possibility, we examined the effects of ibrutinib on GSC-driven tumor growth by in vivo bioluminescent imaging (Fig. 1A and fig. S1, B and C). The results showed that ibrutinib administration markedly inhibited GSC-driven tumor growth (Fig. 1,A and B , and fig. S1, C and D) and conferred a significant survival benefit relative to the vehicle control [Fig. 1C (P = 0.0017) and fig. S1E (P = 0.0018)]. These data demonstrate that ibrutinib treatment effectively suppresses GBM growth and increases animal survival.
Fig. 1. Ibrutinib is more effective than TMZ at inhibiting GSC-derived tumor growth to extend animal survival.
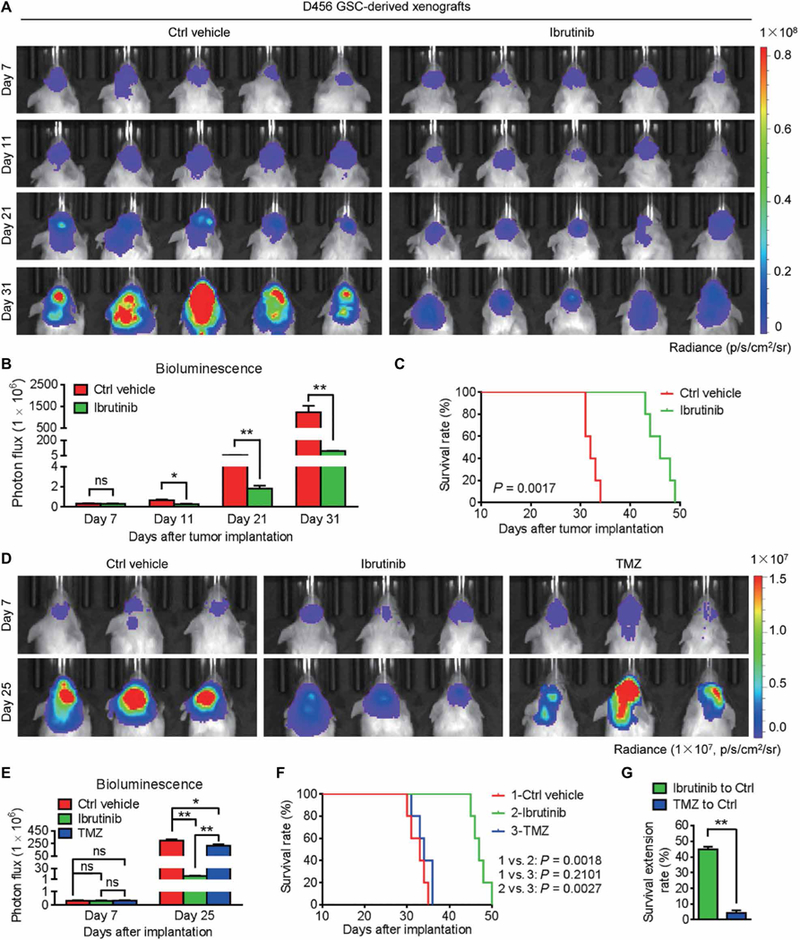
(A and B) In vivo bioluminescent images (A) and the quantification (B) of human GSC-derived xenografts in the brains of mice treated with ibrutinib or the vehicle control at the indicated time points. (C) Kaplan-Meier survival analysis of mice bearing GSC-derived xenografts treated with ibrutinib or the vehicle control. (D and E) In vivo bioluminescent images (D) and the quantification (E) of GSC-derived xenografts in mice treated with ibrutinib or TMZ at the indicated time points. (F) Kaplan-Meier survival analysis of mice bearing GSC-derived xenografts treated with the vehicle control, ibrutinib, or TMZ. (G) Survival extension of mice bearing GSC-derived GBM tumors treated with ibrutinib or TMZ relative to those treated with the vehicle control. Statistical analysis was performed using unpaired Student’s t test for two-group comparison, one-way analysis of variance (ANOVA) for multigroup comparison, or Kaplan-Meier method for survival analyses. Data are means ± SEM (B and E) or means ± SD (G). ns, not significant. *P < 0.05 and **P < 0.01. n = 5 for each group. p, photons; sr, steradian.
To further investigate the therapeutic value of ibrutinib for GBM treatment, we compared side by side the tumor-suppressive effects of ibrutinib and the most common anti-GBM chemotherapeutic agent temozolomide (TMZ). Ibrutinib was superior to TMZ treatment in impairing both in vitro viability of GSCs (fig. S1, F and G) and in vivo GSC-driven tumor growth (Fig. 1, D and E). Consequently, ibrutinib treatment significantly extended the average survival of the tumor-bearing mice (from 32.6 to 47.2 days; P = 0.0018) (Fig. 1F). By contrast, TMZ treatment showed little extension of animal survival (from 32.6 to 34 days; P = 0.2101) (Fig. 1F). The survival benefit from ibrutinib treatment was 10.4-fold more than that from TMZ treatment in the mice bearing GSC-derived tumors (Fig. 1G), indicating that ibrutinib is much more effective than TMZ. To address whether ibrutinib outperforming TMZ as an anti-GBM treatment is associated with the expression of O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme mediating TMZ resistance (26), we examined MGMT expression in GSC lines (D456 and T4121) used for the study. In contrast with high MGMT expression in T98G GBM cells used as a positive control (27), MGMT expression in D456 and T4121 GSCs was nearly undetectable (fig. S1H), suggesting that D456 and T4121 are not MGMT-driven TMZ-resistant cells. Collectively, these results indicate that ibrutinib is potentially more effective than TMZ for GBM treatment.
To further justify ibrutinib use in GBM treatment, we determined the bioavailability of ibrutinib in mouse brains bearing GSC-derived xenografts and found that the average maximum concentration (Cmax) of ibrutinib in mouse brains was 2.22 μM (fig. S2A), suggesting that ibrutinib could penetrate into mouse brains to exert tumoricidal effect. We also performed liver and lung autopsies from mice treated with ibrutinib or the vehicle control to histologically evaluate ibrutinib biosafety. The hematoxylin and eosin staining showed that the morphologies of mouse liver and lung were similar between the ibrutinib-treated group and the control group (fig. S2B). Immunohistochemical (IHC) staining ofcleaved caspase-3 as an apoptosis marker confirmed that ibrutinib treatment had no obvious influence on the viability of mouse hepatocytes or alveolar cells (fig. S2, C and D), suggesting that ibruti-nib is a well-tolerated agent with no severe toxicity to normal tissues.
Combination of ibrutinib treatment with radiation therapy improves survival of GBM-bearing mice
Because GSCs are highly resistant to conventional therapies and contribute to tumor relapse (4,28), we next examined whether disrupting GSCs through BMX inhibition by ibrutinib could improve the therapeutic efficacy of radiotherapy in mice bearing GSC-derived tumors. Whereas irradiation or ibrutinib treatment alone effectively suppressed tumor growth, the combined ibrutinib and irradiation treatment achieved the strongest tumor inhibition (Fig. 2, A and B) and conferred the longest survival extension among all the experimental groups (Fig. 2, C and D). The immunofluorescent staining of cleaved caspase-3 demonstrated that, whereas ibrutinib or radiation treatment alone induced apoptosis of tumor cells in GSC-derived xenografts (Fig. 2, E and F), the combination of ibrutinib treatment with radiation resulted in much more apoptosis in tumors (Fig. 2, E and F), confirming the combined suppressive effect of ibrutinib and radiation on GBM growth. Furthermore, immunofluorescent staining of Ki67 revealed a marked reduction of proliferating cells in xenografts treated with ibrutinib and radiation (Fig. 2, G and H). These data demonstrated the effectiveness of targeting GSCs by ibrutinib in combination with radiation therapy to enhance therapeutic efficacy.
Fig. 2. Ibrutinib treatment combines with radiation therapy to disrupt GBM tumor growth.
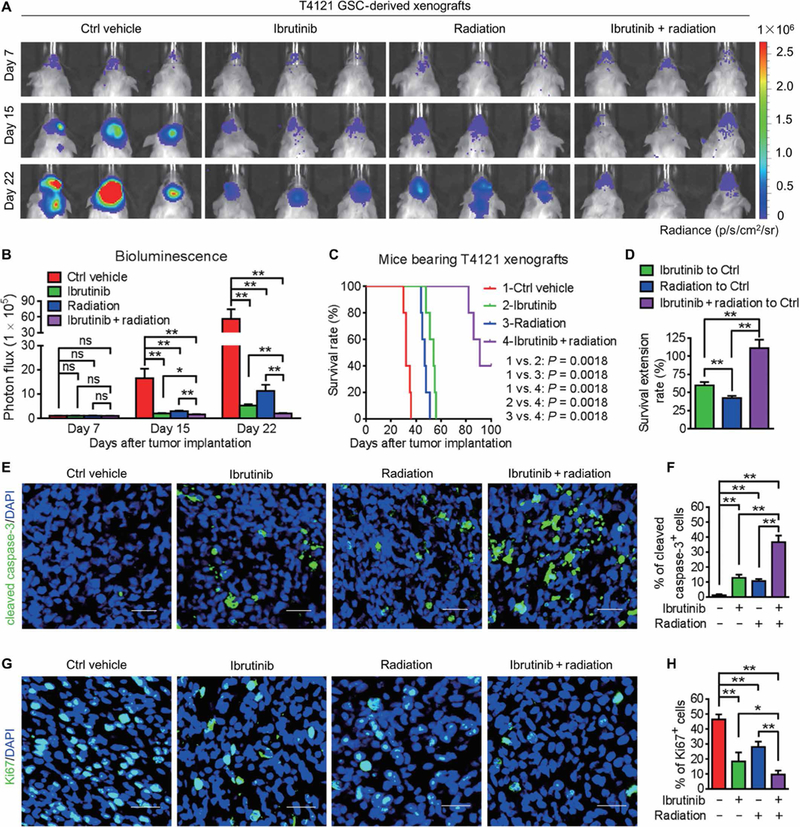
(A and B) In vivo bioluminescent images (A) and the quantifications (B) of GSC-derived xenografts in mice treated with the vehicle control, ibrutinib, and/or radiation at the indicated time points. (C) Kaplan-Meier survival analysis of mice bearing GSC-derived xenografts with indicated treatments. (D) Survival extension of mice bearing GSC-derived GBM tumors with indicated treatments. (E and F) Immunofluorescent staining (E) and the quantification (F) of cleaved caspase-3 (green) in GSC-derived xenografts from mice with indicated treatments. (G and H) Immunofluorescent staining (G) and the quantification (H) of Ki67 (green) in GSC-derived xenografts from mice with indicated treatments. Statistical analysis was performed using unpaired Student’s t test for two-group comparison, one-way ANOVA for multigroup comparison, or Kaplan-Meier method for survival analyses. Data are means ± SEM (B) or means ± SD (D, F, and H). *P < 0.05 and **P < 0.01. n = 5 for each group. Scale bars, 50 μm. DAPI, 4′,6-diamidino-2-phenylindole.
Ibrutinib treatment is effective for GSCs from a majority of GBMs tested
To determine the generalizability of ibrutinib treatment for varied GBM molecular subtypes (29), we examined ibrutinib impact on 10 GSC populations derived from primary GBMs with different molecular signatures (table S1). The results demonstrated that ibrutinib treatment caused a dose-dependent suppression of cell growth in all tested GSCs (fig. S3, A to C). Immunoblot analyses confirmed that all patient-derived GSC populations expressed BMX (fig. S3D). Moreover, GSCs with higher BMX expression were more sensitive to ibrutinib treatment (fig. S3, D and E). These data indicate that ibrutinib is effective at inhibiting the growth of most GSC subtypes. We further analyzed the expression of total BMX and the phosphorylated active BMX (pBMX-Y40) in human GBM specimens through IHC staining to predict the fractions of GBM patients that may benefit from ibrutinib therapy. Expression of BMX and pBMX-Y40 was detected in 38 of 42 (90.48%) cases of human GBM (fig. S3, F and G, and table S2), suggesting that most of the GBM patients may respond to the BMX-targeting agent ibrutinib. Collectively, these data underscore the broad potential of ibrutinib for treating most GBMs.
Ibrutinib treatment targets GSCs but not NPCs in vivo
To determine whether ibrutinib treatment specifically affects GSC populations in GBM xenografts, we examined GSCs by immunostaining for the stem cell marker SOX2 in the treated and control tumors. Ibrutinib-treated xenografts harbored significantly reduced percentages of SOX2+ tumor cells (P < 0.001; fig. S4, A and B), suggesting that ibrutinib inhibited GSC survival. Notably, ibrutinib treatment had no significant effect on the SOX2+ NPC populations in the subventricular zone (SVZ) of mouse lateral ventricles (fig. S4, C and D). Although proliferative Ki67+ tumor cells were markedly reduced in the ibrutinib-treated xenografts relative to the control xenografts (fig. S4, E and F), ibrutinib treatment showed no obvious impact on the proliferation of mouse NPCs residing in the SVZ (fig. S4, G and H). Moreover, ibrutinib treatment increased the proportion of apoptotic cells marked by cleaved caspase-3 in treated xenografts relative to the control tumors (fig. S4, I and J) while avoiding induction of cell apoptosis in mouse SVZ containing abundant NPCs (fig. S4, K and L). Consistent with ibrutinib functioning as a BMX inhibitor, pBMX-Y40 was reduced in ibrutinib-treated xenografts relative to control xeno grafts (fig. S4, M and N). However, expression of pBMX-Y40 was not detected in mouse NPCs in SVZs from the ibrutinib-treated or control mouse brains (fig. S4, O and P). Collectively, these data suggest that ibrutinib treatment in vivo selectively targets stemlike glioma cells expressing BMX in GBMs but shows little impact on NPCs lacking BMX expression.
Ibrutinib disrupts GSC maintenance but has no apparent effect on NPCs
To determine whether ibrutinib treatment affects GSC maintenance, we examined the impact of ibrutinib on GSC self-renewal in three human GSC populations (D456, T4121, and T387) isolated from patient-derived xenografts. In vitro limiting dilution assay and tumor sphere formation assay demonstrated that ibrutinib treatment suppressed GSC self-renewal potential (Fig. 3A) and tumor sphere formation (Fig. 3, B and C, and fig. S5, A and B). Furthermore, ibru tinib treatment effectively induced GSC apoptosis, as evaluated by the flow cytometry analyses of annexin V and propidium iodide (PI) as proapoptosis markers (Fig. 3D and fig. S5C). Consistent with these data, cleaved caspase-3 and cleaved poly(adenosine 5’-diphosphate-ribose) polymerase (PARP), two proapoptosis markers, were also increased in the GSCs after ibrutinib treatment in a dose-dependent manner (Fig. 3E and fig. S5D). To compare the effectiveness of ibrutinib on targeting GSCs and nonstem tumor cells (NSTCs), we examined the expression of BMX as the direct target of ibrutinib in these two populations and confirmed that BMX was preferentially expressed in GSCs relative to NSTCs (fig. S5E). In vitro cell viability analyses demonstrated that ibrutinib was more effective in targeting GSCs than NSTCs (fig. S5F). We then evaluated the sensitivity of GSCs and NPCs to ibrutinib and found that the mean effective concentration (EC50) of ibrutinib was much lower for human GSCs than for human NPCs (Fig. 3, F and G). As a result, the neurosphere formation ability of NPCs was not affected by ibrutinib treatment at the EC50 for eliminating GSCs (Fig. 3, H to J), confirming that ibrutinib has no detectable impact on the NPC maintenance at the effective dose of GSCs. Collectively, these data indicate that ibrutinib may display a high therapeutic index for GBM treatment.
Fig. 3. Ibrutinib disrupts the maintenance of GSCs but not of NPCs in vitro.
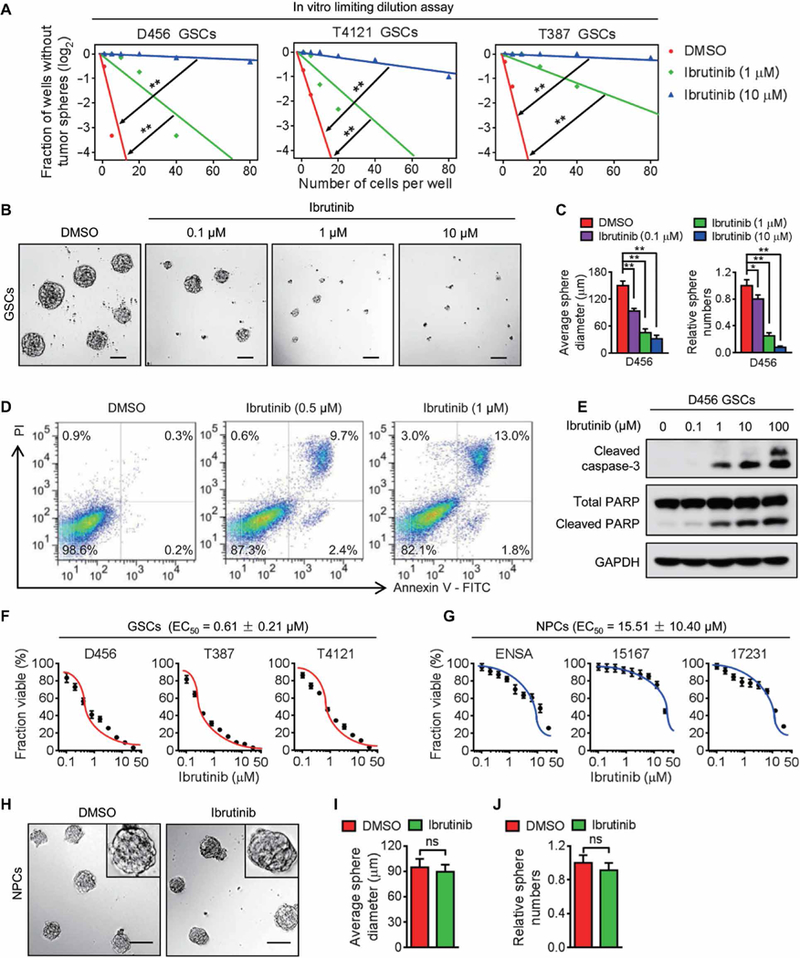
(A) In vitro limiting dilution assay of three individual human GSCs (D456, T4121, and T387) treated with indicated doses of ibrutinib or the vehicle control. (B and C) Representative images of GSC tumor spheres (B) and the quantification of sphere diameter [(C), left] and numbers [(C), right] of D456 GSCs treated with indicated doses of ibrutinib or the vehicle control. (D) Cell apoptosis assay of D456 GSCs treated with indicated doses of ibrutinib or the vehicle control. (E) Immunoblot analyses of cleaved caspase-3, cleaved PARP, PARP, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in D456 GSCs treated with indicated doses of ibrutinib or the vehicle control. (F and G) Dose-response curves of ibrutinib treatment in GSCs (F) and NPCs (G). Human GSCs and NPCs were exposed to increasing concentrations of ibrutinib (0 to 25.6 μM) for 72 hours, followed by in vitro cell viability analysis. EC50 of ibrutinib for GSCs and NPCs was measured using nonlinear regression analysis of the dose-response curves. (H to J) Representative images of neurospheres (H) and the quantification of sphere diameter (I) and numbers (J) of 15167 NPCs with indicated treatments. Statistical analysis was performed using unpaired Student’s t test for two-group comparison or oneway ANOVA for multigroup comparison. Data are means ± SD. *P < 0.05 and **P < 0.01. Scale bars, 100 μm. DMSO, dimethyl sulfoxide.
Ibrutinib disrupts BMX-mediated STAT3 activation in GSCs but shows negligible effect on STAT3 activation in NPCs
Because BMX kinase mediates STAT3 activation in GSCs (8), we investigated the effect of ibrutinib on the STAT3 activation in GSCs. Ibrutinib treatment markedly reduced pBMX-Y40 in a dose-dependent manner in three GSC lines (Fig. 4A and fig. S6A). Moreover, a time-course study showed that ibrutinib sustainably repressed pBMX-Y40 in GSCs for at least 16 hours after treatment (Fig. 4B and fig. S6B). We next investigated the effect of ibrutinib on the activating phosphorylation of STAT3 (pSTAT3-Y705). Ibrutinib treatment impaired STAT3 activation in a dose-and time-dependent manner (Fig. 4, A and B, and fig. S6, A and B) and inhibited the expression of NANOG and octamer-binding protein 4 as the downstream effectors of STAT3 signaling in GSCs (fig. S6C). Notably, in vitro cell viability analyses showed that ibrutinib was more effective in targeting GSCs than the STAT3 inhibitor stattic (fig. S6D). Because BMX is not expressed in NPCs (fig. S1A), we hypothesized that STAT3-activating phosphorylation in NPCs might not be affected by ibrutinib treatment at the dose effective in GSCs. Immunoblot analyses confirmed that ibrutinib treatment at 1 mM did not affect pSTAT3-Y705 in NPCs, whereas the same dose of ibrutinib markedly decreased pSTAT3-Y705 in GSCs (Fig. 4C). Similarly, in vivo treatment with ibrutinib reduced pSTAT3-Y705 in GSC-derived xenografts (Fig. 4D and fig. S6, E and F) but showed no apparent impact on the percentage of pSTAT3-Y705-positive cells in the SVZ regions containing abundant NPCs (Fig. 4, E and F). Together, these data demonstrate that ibrutinib specifically disrupts BMX-mediated STAT3 activation in GSCs but shows no detectable effects on STAT3 activation in NPCs.
Fig. 4. Ibrutinib inhibits the BMX-mediated STAT3 activation in GSCs.
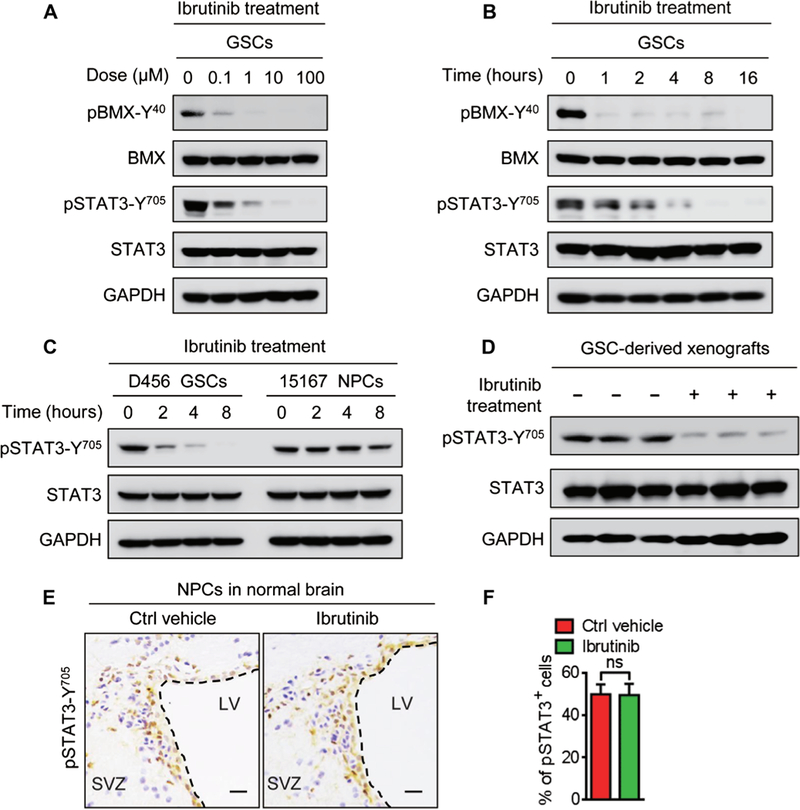
(A) Immunoblot analyses of pBMX-Y40, total BMX, pSTAT3-Y705, total STAT3, and GAPDH in D456 GSCs treated with indicated doses of ibrutinib or the vehicle control. (B) Immunoblot analyses of pBMX-Y40, BMX, pSTAT3-Y705, STAT3, and GAPDH in D456 GSCs treated with ibrutinib (1 μM) at the indicated time points. (C) Immunoblot analyses of pSTAT3-Y705, STAT3, and GAPDH showing the effects of ibrutinib (1 μM) on STAT3 phosphorylation in D456 GSCs and 15167 NPCs. (D) Immunoblot analyses of pSTAT3-Y705, STAT3, and GAPDH in D456 GSC-derived xenografts with indicated treatments. n = 3 for each group. (E and F) Representative IHC images (E) and the quantification (F) of pSTAT3-Y705 in the SVZ of mouse brains with indicated treatments. n = 5 for each group. Scale bars, 50 μm. Statistical analysis was performed using unpaired Student’s t test. Data are means ± SD. LV, lateral ventricle.
Constitutively active STAT3 reverses the suppressive effect of ibrutinib on GSCs
To determine whether the effect of ibrutinib on GSC maintenance is exerted through inhibition of BMX-mediated STAT3 activation, we introduced a constitutively active STAT3 (STAT3-C) (8, 30) and then examined whether STAT3-C could reverse the effect of ibrutinib treatment on GSCs. The expression of Flag-tagged STAT3-C (STAT3-C-Flag) in GSCs was confirmed by immunoblot analyses (Fig. 5A). Ectopic expression of STAT3-C largely rescued the proliferation and self-renewal of GSCs after ibrutinib treatment (Fig. 5, B and C, and fig. S7, A and B). Moreover, the ibrutinib-induced GSC apoptosis was diminished by the expression of STAT3-C in GSCs, as demonstrated by the reduced expressions of cleaved caspase-3 and cleaved PARP (Fig. 5D and fig. S7C) and the reduced proportion of apoptotic cells in STAT3-C-expressing GSCs treated with ibrutinib (Fig. 5E and fig. S7D). Together, these results demonstrate that restored STAT3 activity largely rescues GSC maintenance impaired by ibrutinib treatment.
Fig. 5. Constitutively active STAT3 largely reverses the suppressive effect of ibrutinib on GSCs.
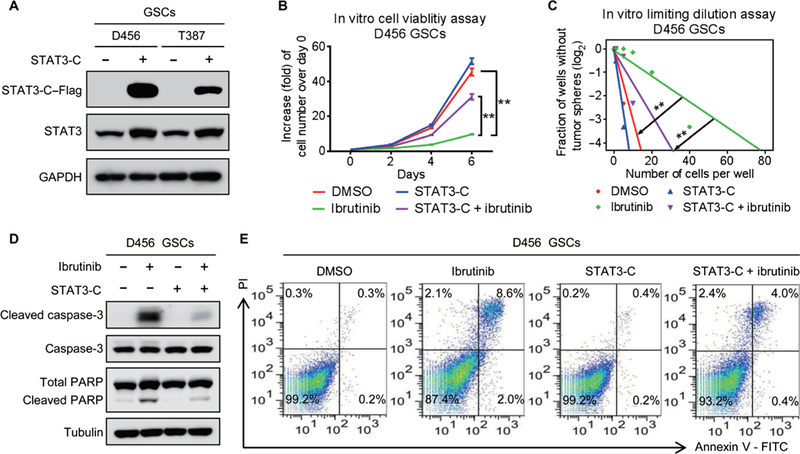
(A) Immunoblot analyses of STAT3-C–Flag, STAT3, and GAPDH in D456 and T387 GSCs transduced with STAT3-C–Flag or control vector. (B) In vitro cell viability assay of STAT3-C–expressing D456 GSCs treated with ibrutinib or the vehicle control. (C) In vitro limiting dilution assay of STAT3-C–expressing D456 GSCs treated with ibrutinib or the vehicle control. (D) Immunoblot analyses of cleaved caspase-3, caspase-3, cleaved PARP, PARP, and tubulin in STAT3-C–expressing D456 GSCs treated with ibrutinib or the vehicle control. (E) Fluorescence-activated cell sorting analyses of apoptosis of the STAT3-C–expressing D456 GSCs treated with ibrutinib or the vehicle control. Statistical analysis was performed using unpaired Student’s t test for two-group comparison or one-way ANOVA for multigroup comparison. Data are means ± SD. **P < 0.01
JAK2 kinase is inactivated in GSCs
Because ibrutinib has differential effects on STAT3 activation in GSCs and NPCs, it is critical to uncover the molecular mechanisms underlying the phenotype. It is well known that STAT3 activation in NPCs is mediated by JAK2 (31, 32). Although GSCs share some properties with NPCs, our previous study demonstrated that STAT3 activation in GSCs is mainly mediated by BMX kinase (8), indicating that the kinase activity of JAK2 and its effect on STAT3 activation may be differentially regulated in GSCs and NPCs. Immunoblot analyses showed that, whereas pSTAT3-Y705 was increased in both GSCs and NPCs upon IL-6 stimulation (Fig. 6A), JAK2-activating phosphorylation was only increased in NPCs but not in GSCs (Fig. 6A), suggesting that JAK2 is not the major upstream activator of STAT3 in GSCs. Coimmunoprecipitation analyses showed that JAK2 interacted with STAT3 and induced pSTAT3-Y705 in human NPCs upon IL-6 stimulation (Fig. 6B), whereas the interaction between JAK2 and STAT3 was almost undetectable in GSCs treated with IL-6 (Fig. 6B).To confirm the distinctive function of JAK2 in GSCs and NPCs, we used a selective JAK2 inhibitor ruxolitinib to block JAK2-mediated signaling and found that ruxolitinib treatment had no detectable impact on STAT3 activation in GSCs but abrogated JAK2-mediated STAT3 activation in NPCs upon IL-6 stimulation (Fig. 6C and fig. S8A). These results suggest that JAK2 kinase activity is inhibited in GSCs, and thus, JAK2 is not able to mediate STAT3 activation in GSCs.
Fig. 6. JAK2-mediated STAT3 activation is blocked in GSCs, and SOCS3 is responsible for JAK2 inactivation in GSCs.
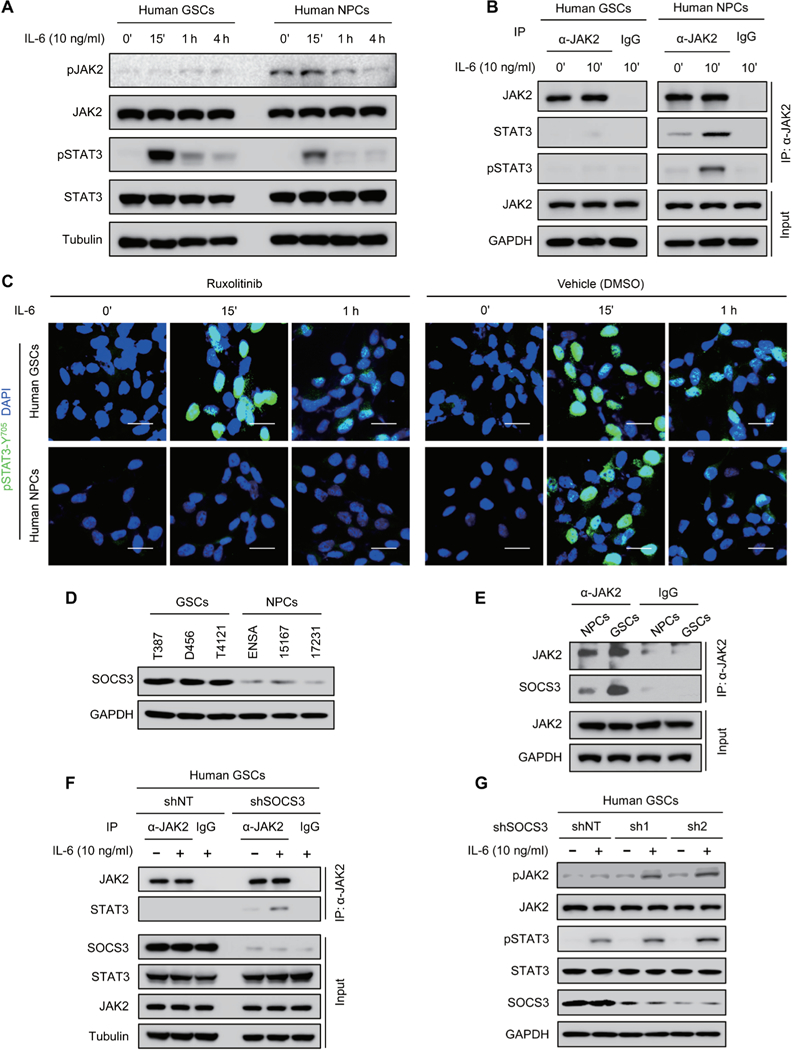
(A) Immunoblot analyses of pJAK2-Y1007/1008, JAK2, pSTAT3-Y705, STAT3, and tubulin in human GSCs (D456) and NPCs (15167) upon IL-6 stimulation. (B) Co-immunoprecipitation analyses of JAK2, STAT3, and pSTAT3-Y705 with anti-JAK2 antibody in IL-6— stimulated human GSCs (D456) and NPCs (15167). IgG, immunoglobulin G; IP, immunoprecipi-tation. (C) Immunofluorescent staining of pSTAT3-Y705 (green) in human GSCs (D456; top) and NPCs (15167; bottom) treated with JAK2 inhibitor ruxolitinib (100 nM) or the vehicle control. Scale bars, 25 μm. (D) Immunoblot analyses of SOCS3 and GAPDH in human GSCs and NPCs. (E) Coimmunoprecipitation analysis of SOCS3 with anti-JAK2 antibody in human GSCs (D456) and NPCs (ENSA). (F)Coimmuno precipitation analyses of STAT3 with anti-JAK2 antibody in D456 GSCs expressing short hairpin RNA (shRNA) against SOCS3 (shSOCS3) or nontargeting shRNA (shNT). (G) Immunoblot analyses of pJAK2-Y1007/1008, JAK2, pSTAT3-Y705, STAT3, SOSC3, and GAPDH in D456 GSCs expressing shSOCS3 (sh1 or sh2) or shNT with or without IL-6 stimulation. The symbol (‘) means minutes.
The negative regulator SOCS3 is responsible for JAK2 inactivation in GSCs
To understand how the JAK2-STAT3 signaling cascade is disrupted in GSCs, we interrogated a potential negative regulator of JAK2 in GSCs. Because SOCS3 has been reported to suppress the JAK2-mediated STAT3 activation by binding to JAK2 in many cells (20,21), we sought to determine whether SOCS3 could affect the function of JAK2 in GSCs. Immunoblot analyses showed that SOCS3 protein was increased in GSCs relative to NPCs (Fig. 6D). Moreover, a greater proportion of SOCS3 was found in the complex with JAK2 in GSCs relative to that in NPCs by coimmunoprecipitation analysis (Fig. 6E), suggesting that JAK2 activity in GSCs may be suppressed by SOCS3. To address this possibility, we examined whether disruption of SOCS3 could restore JAK2 activity in GSCs. Coimmuno-precipitation results demonstrated that silencing SOCS3 expression restored the interaction of JAK2 with STAT3 in GSCs after IL-6 stimulation (Fig. 6F). Moreover, active phosphorylation of JAK2 and STAT3 was increased in GSCs expressing shSOCS3 relative to those expressing shNT after IL-6 stimulation (Fig. 6G), confirming that increased SOCS3 impairs JAK2 activity in GSCs.
BMX interacts with gp130 to mediate STAT3 activation in GSCs
Having observed that JAK2 activity is inhibited by SOCS3 in GSCs, we investigated how BMX maintains sustainable activation of STAT3 in GSCs. Because the upstream activators of BMX in GSCs in response to IL-6 stimulation remain largely unknown and gp130 has been identified as a crucial receptor of IL-6 in GSCs (18, 22), we examined whether BMX could respond to IL-6 stimulation by coupling with gp130. Reciprocal coimmunoprecipitation analyses showed that BMX bound to gp130 upon IL-6 stimulation and that BMX was actively phosphorylated (Fig. 7A and fig. S8B). We then investigated whether BMX could interact with STAT3 to mediate STAT3 activation in GSCs. Reciprocal coimmunoprecipitation analyses demonstrated that BMX bound to STAT3 upon IL-6 stimulation and induced STAT3-activating phosphorylation (Fig. 7B and fig. S8C). Immunoblot analyses confirmed that silencing BMX by shRNA abolished STAT3 activation upon IL-6 stimulation in GSCs (Fig. 7C and fig. S8D). Furthermore, exogenous expression of wild-type BMX (BMX-WT) or constitutively active BMX (BMX-C) enhanced STAT3 activation upon IL-6 stimulation (Fig. 7D and fig. S8E), whereas introduction of the dominant-negative BMX (BMX-DN) in GSCs competitively suppressed the effect of endogenous BMX and reduced STAT3-activating phosphorylation (Fig. 7D and fig. S8E). Together, these results indicate that BMX couples with the upstream IL-6 receptor gp130 and the downstream STAT3 to activate STAT3 signaling in GSCs.
Fig. 7. BMX interacts with STAT3 and gp130 to mediate STAT3 activation in GSCs.
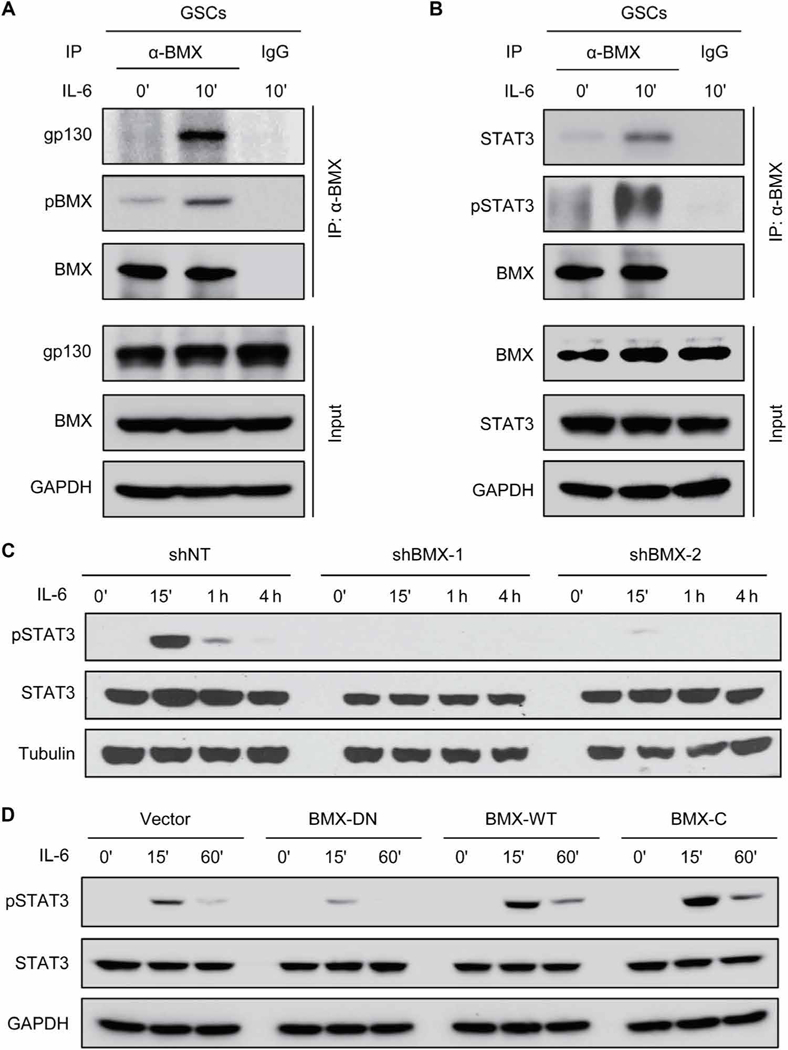
(A) Coimmunoprecipitation of gp130 and pBMX with anti-BMX antibody in D456 GSCs upon IL-6 stimulation. (B) Coimmunoprecipitation of STAT3 and pSTAT3-Y705 with anti-BMX antibody in D456 GSCs upon IL-6 stimulation. (C) Immunoblot analyses of pSTAT3-Y705, STAT3, and tubulin in D456 GSCs expressing shBMX-1, shBMX-2, or shNT upon IL-6 stimulation. (D) Immunoblot analyses of pSTAT3-Y705, STAT3, and GAPDH in GSCs expressing BMX-DN, BMX-WT, or BMX-C upon IL-6 stimulation (10 ng/ml). The symbol (‘) means minutes.
BMX bypasses the SOCS3-mediated negative regulation of JAK2 to sustainably activate STAT3 in GSCs
Because SOCS3 is a typical negative regulator of JAK2-STAT3 signaling and STAT3 hyperactivation in GSCs is primarily mediated by BMX, we investigated whether BMX could bypass SOCS3’s negative regulation of JAK-STAT3 signaling in GSCs. Immunoblot analyses demonstrated that forced expression of SOCS3 did not affect pBMX-Y40 or pSTAT3-Y705 in GSCs in response to IL-6 stimulation (Fig. 8A). By contrast, forced expression of SOCS3 suppressed the binding of JAK2 with STAT3 in human NPCs (fig. S9A), followed by the reduction of pSTAT3-Y705 (fig. S9B). Furthermore, coimmunoprecipitation analysis showed that SOCS3 did not interact with BMX in GSCs (Fig. 8B), indicating that BMX is not negatively regulated by SOCS3 in GSCs. To further determine whether BMX bypasses the JAK2 inhibition by SOCS3 in GSCs, we examined the impact of SOCS3 overexpression on the maintenance of GSCs or NPCs and found that forced SOCS3 expression did not affect GSC maintenance but potently inhibited survival and growth of human NPCs (Fig. 8C). Collectively, these data demonstrate that BMX bypasses the SOCS3-mediated negative regulation to constitutively activate STAT3 in GSCs. The differential activation and regulation of STAT3 in GSCs and NPCs enable their differential sensitivity to BMX inhibition by ibrutinib (Fig. 8D), resulting in a high therapeutic index and selective targeting of GSCs to inhibit GBM growth.
Fig. 8. BMX bypasses SOCS3 negative regulation on JAK2 to sustainably activate STAT3 in GSCs.
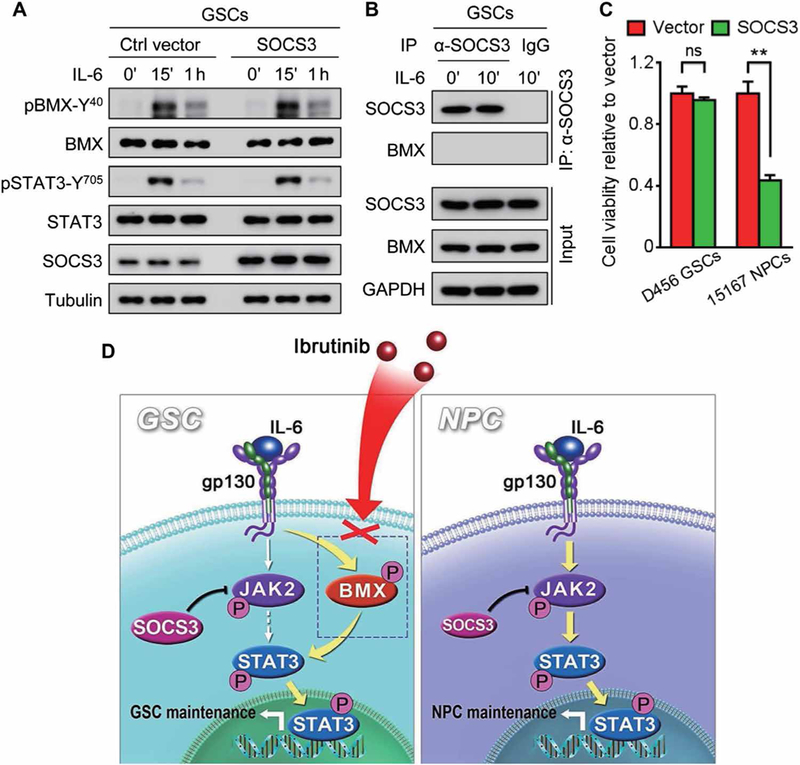
(A) Immunoblot analyses of pBMX-Y40, BMX, pSTAT3-Y705, STAT3, SOCS3, and tubulin in D456 GSCs transduced with SOCS3 or the control vector upon IL-6 stimulation. (B) Coimmunoprecipitation of BMX with anti-SOCS3 antibody in D456 GSCs upon IL-6 stimulation. Precipitation with normal mouse IgG was used as a negative control. Total cell lysates (input) were immunoblotted with antibodies against SOCS3, BMX, and GAPDH. (C) In vitro cell viability analyses of human GSCs (D456) and NPCs (15167) expressing SOCS3 or the control vector. (D) Schematic diagram of targeting GSCs through BMX inhibition by ibrutinib and the underlying molecular mechanisms. BMX bypasses the SOCS3-mediated inhibition of JAK2 to sustain activation of STAT3 in GSCs (left), whereas JAK2-mediated STAT3 activation in NPCs (right) is negatively regulated by SOCS3, providing a molecular basis for targeting BMX by ibrutinib to specifically inhibit STAT3 activation in GSCs but not in NPCs. The symbol (‘) means minutes. Statistical analysis was performed using unpaired Student’s t test. Data are means ± SD. **P < 0.01.
DISCUSSION
GBM is the most aggressive and deadly brain tumor with dismal prognosis despite multimodal therapies (1). GBM tumors exhibit remarkable cellular heterogeneity, with a population of GSCs at the apex of the differentiation hierarchy (2). GSCs not only display potent tumor-initiating or tumor-propagating potential but also promote malignant behaviors associated with disease progression and relapse (2, 4, 8), suggesting that elimination of GSCs is crucial for improving GBM treatment and overcoming therapeutic resistance. However, a GSC-targeting drug is still unavailable in clinical practice. Thus, the development of anti-GSC therapeutics based on GSC-specific targets is urgently needed.
In the search for GSC-specific functional targets, we have identified the nonreceptor tyrosine kinase BMX as a molecular target. There are several advantages for using BMX as a therapeutic candidate: (i)BMX-mediated STAT3 activation is a critical dependency for maintaining GSC self-renewal and tumorigenic potential, whose pharmacological disruption can impair GSCs and inhibit GBM growth;(ii)the preferential BMX up-regulation in GSCs compared with NPCs or other brain parenchyma (8) suggests a favorable therapeutic index for BMX inhibition; (iii) GSC-specific evasion of SOCS3-mediated JAK2 inhibition by BMX preserves homeostatic activity of JAK2-STAT3 axis important in normal cells; (iv) BMX is a nonreceptor tyrosine kinase with a cysteine residue in the kinase domain that is amenable for development of irreversible and specific small-molecule inhibitors; and (v) BMX contributes to the resistance of cancer cells to radiation and chemotherapy (13, 15), suggesting a synergistic potential of BMX inhibition with conventional radiation or chemotherapy. Our current study demonstrates that disrupting GSCs through BMX inhibition by ibrutinib effectively suppresses GBM growth and increases survival of animals with orthotopic xenografts. Concordant with BMX dispensability in normal development and survival (16), targeting BMX in vivo had no detectable effect on NPCs and other normal cells in the brain. Our in vivo experiments confirmed that the combination of ibrutinib and radiation markedly inhibited GBM growth and extended animal survival. Because of preferential BMX expression in infrequent GSC populations of the bulk tumor, BMX expression may not be an effective prognostic biomarker. Nevertheless, BMX is a promising druggable target to specifically eliminate GSCs.
Ibrutinib is an FDA-approved oral drug for the treatment of hematological malignancies (24, 33), with notable response rates and an acceptable side-effect profile (24, 34). The therapeutic effect of ibrutinib on blood malignancies was thought to mainly result from its inhibition of BTK kinase activity (23, 33), which suppresses signal transduction of the B cell receptor pathway responsible for hematological malignancies (23, 24, 35). Here, we expand the potential application of ibrutinib to treat human GBMs primarily through inhibiting the BMX-STAT3 pathway. Notably, because STAT3-C could not completely reverse the effect of ibrutinib on GSCs, it is possible that other signaling pathways may also be affected by ibrutinib. Multiple kinases homologous to BTK, including EGFR (epidermal growth factor receptor), have been predicted as potential ibrutinib targets, although their sensitivity to ibrutinib is much less than that of BMX and BTK (23). Further investigations are warranted to determine whether ibrutinib could inhibit the activity of these BTK homologous kinases in GBMs.
As an FDA-approved oral drug, ibru-tinib has been well evaluated regarding its pharmacokinetics and efficacy in previous clinical trials. The half-life of ibru-tinib is about 4 to 18 hours through oral administration in human patients with hematological malignancies (33, 35), suggesting that the pharmacokinetics of ibrutinib may not limit its application in clinic. In addition, ibrutinib is an irreversible inhibitor that binds to the cysteine residue at the kinase domain, which persistently suppresses BMX kinase activity to achieve anticancer effects, thus further reducing cellular toxicity. It has been reported that ibrutinib is quite safe, is well tolerated, and has a promising response rate in several long-term clinical trials in hematological malignancies (24, 33). Thus, repurposing ibrutinib for GBM treatment should pose few contraindications.
Beyond its suppressive effect on GSCs, ibrutinib treatment could also target GSC-derived pericytes that highly express BMX to specifically disrupt the blood-tumor barrier (BTB) and thus increase drug penetration into tumors (36). Therefore, ibrutinib application in GBM treatment should disrupt GSCs and BTB, which should enhance delivery of antitumor agents that poorly penetrate BTB and effectively combine with chemotherapy or other targeted therapies to benefit GBM patients. In addition, other studies have demonstrated that ibrutinib is able to penetrate through the blood-brain barrier (BBB) to treat primary and recurrent brain lymphoma (35, 37). Because BBB prevents most antitumor drugs from penetrating into the brain, BBB penetration by ibrutinib should expand the scope of this drug to treat primary or metastatic brain tumors with high expression of BMX or BTK.
In consideration of the promising preclinical data on ibrutinib in GSC-derived GBM xenograft models, clinical trials using ibrutinib to treat primary and recurrent GBMs may demonstrate a longterm benefit and therapeutic value for GBM patients. A potential limitation is whether long-term treatment with ibrutinib may result in drug resistance, which needs to be further investigated. Nevertheless, the effective tumoricidal activity of ibrutinib on GSC-derived xenografts from our preclinical studies provides strong evidence for its clinical application to combat malignant brain tumors and overcome resistance to conventional therapies.
MATERIALS AND METHODS
Study design
The aim of this study was to investigate the effect of ibrutinib on GSCs and its therapeutic relevance for GBM treatment. To this end, we performed controlled laboratory experiments using patient-derived GSCs, mice bearing GSC-derived xenografts, and human GBM tissues. For in vitro studies, multiple patient-derived GSCs were used, and experiments included three or more biological replicates and at least three technical replicates. For in vivo studies, mice were randomized into control or treatment groups at the seventh day after tumor implantation based on the bioluminescent signal of initial tumor burden. Sample size was five mice per group, and all mice were included to determine statistical significance of differences in tumor growth and animal survival between groups. For histology of mouse tissues (mainly brains), three to five mice per treatment group were randomly selected, and three to five sections per mouse were analyzed. For histology of human tissues, tumor sections from all 42 primary GBMs were used, and experiments were performed twice. The studies were not performed blindly.
Statistical analysis
PASW Statistics 18 and GraphPad Prism 6 were used for all statistical analyses in this study. Kolmogorov-Smirnov test was used to assess the normal distribution of data. The significance was determined by a two-tailed unpaired Student’s t test or one-way ANOVA, with P < 0.05 being considered statistically significant. Survival analyses were performed using the Kaplan-Meier method, with the log-rank test for comparison. The survival extension rate was calculated according to the following formula: survival extension rate (%) = [(A - B)/B] × 100%, where A was the average survival of mice treated with ibrutinib, radiation, or ibrutinib plus radiation, and B was the average survival of mice treated with the vehicle control. EC50 of ibrutinib was calculated using nonlinear regression analyses based on doseresponse curves. The investigators were not blinded to allocation during experiments and outcome assessment. All quantitative data are means ± SD or means ± SEM, as indicated. See the detailed Supplementary Materials and Methods.
Supplementary Material
Acknowledgments:
We thank the Neuro-Oncology Department of Southwest Hospital and the Brain Tumor and Neuro-Oncology Center of Cleveland Clinic for providing the GBM surgical specimens. We thank B. C Prager from the Division of Regenerative Medicine, University of California, and Q. Xiong of the School of Computer and Information Science, Southwest University for the assistance of analyzing the GBM subtype signatures.
Funding: This research work was supported by grants from National Key Research and Development Program of China (2016YFA0101200 to X.-W.B. and SQ2017ZY040203 to Y.-F.P.), NIH (United States) R01 grants (CA184090, NS091080, and NS099175) to S.B., and NIH grants (CA197718, CA154130, CA169117, CA171652, NS087913, and NS089272) to J.N.R.
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/10/443/eaah6816/DCl Materials and Methods
REFERENCES AND NOTES
- 1.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ, Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 60, 166–193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN, Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB, Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN, Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, Bao S, Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN, Bao S, Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park H-J, Kim J-K, Jeon H-M, Oh S-Y, Kim S-H, Nam D-H, Kim H, The neural stem cell fate determinant TLX promotes tumorigenesis and genesis of cells resembling glioma stem cells. Mol. Cells 30, 403–408 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, MacSwords J, Eyler CE, McLendon RE, Heddleston JM, Shou W, Hambardzumyan D, Lee J, Hjelmeland AB, Sloan AE, Bredel M, Stark GR, Rich JN, Bao S, Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 19, 498–511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamagnone L, Lahtinen I, Mustonen T, Virtaneva K, Francis F, Muscatelli F, Alitalo R, Smith CI, Larsson C, Alitalo K, BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene 9, 3683–3688 (1994). [PubMed] [Google Scholar]
- 10.Ekman N, Arighi E, Rajantie I, Saharinen P, Ristimaki A, Silvennoinen O, Alitalo K, The Bmx tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 19, 4151–4158 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Semaan N, Alsaleh G, Gottenberg J-E, Wachsmann D, Sibilia J, Etk/BMX, a Btk family tyrosine kinase, and Mal contribute to the cross-talk between MyD88 and FAK pathways. J. Immunol. 180, 3485–3491 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Dai B, Kim O, Xie Y, Guo Z, Xu K, Wang B, Kong X, Melamed J, Chen H, Bieberich CJ, Borowsky AD, Kung H-J, Wei G, Ostrowski MC, Brodie A, Qiu Y, Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 66, 8058–8064 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Zhu W, Zhang J, Guo L, Tyrosine kinase Etk/BMX protects nasopharyngeal carcinoma cells from apoptosis induced by radiation. Cancer Biol. Ther. 11, 690–698 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Holopainen T, Lopez-Alpuche V, Zheng W, Heljasvaara R, Jones D, He Y, Tvorogov D, D’Amico G, Wiener Z, Andersson LC, Pihlajaniemi T, Min W, Alitalo K, Deletion of the endothelial Bmx tyrosine kinase decreases tumor angiogenesis and growth. Cancer Res. 72, 3512–3521 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Fox JL, Storey A, BMX negatively regulates BAK function, thereby increasing apoptotic resistance to chemotherapeutic drugs. Cancer Res. 75, 1345–1355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajantie I, Ekman N, Iljin K, Arighi E, Gunji Y, Kaukonen J, Palotie A, Dewerchin M, Carmeliet P, Alitalo K, Bmx tyrosine kinase has a redundant function downstream of angiopoietin and vascular endothelial growth factor receptors in arterial endothelium. Mol. Cell Biol. 21, 4647–4655 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Aksoy I, Gonnot F, Osteil P, Aubry M, Hamela C, Rognard C, Hochard A, Voisin S, Fontaine E, Mure M, Afanassieff M, Cleroux E, Guibert S, Chen J, Vallot C, Acloque H, Genthon C, Donnadieu C, De Vos J, Sanlaville D, Guérin J-F, Weber M, Stanton LW, Rougeulle C, Pain B, Bourillot P-Y, Savatier P, Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat. Commun. 6, 7095 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H, Pardoll D, Jove R, STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, Matthews V, Schmid RM, Kirchner T, Arkan MC, Ernst M, Greten FR, gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA, Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 36, 239–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ, SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20, 469–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, Priebe W, Villarreal D, Kang SG, Joyce C, Sulman E, Wang Q, Marini FC, Andreeff M, Colman H, Lang FF, Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells 33, 2400–2415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ, The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U.S.A. 107, 13075–13080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA, Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J.Med. 369, 507–516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S, Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 369, 32–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JEC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R, MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Wickström M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjörnsson B, Sandén E, Darabi A, Siesjo P, Kool M, Kogner P, Baryawno N, Johnsen JI, Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 6, 8904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF, A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin YH, Satani N, Martinez-Ledesma E, Zheng S, Chang E, Sauvé CG, Olar A, Lan ZD, Finocchiaro G, Phillips JJ, Berger MS, Gabrusiewicz KR, Wang G, Eskilsson E, Hu J, Mikkelsen T, DePinho RA, Muller F, Heimberger AB,Sulman EP, Nam DH, Verhaak RGW, Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr., Stat3 as an oncogene. Cell 98, 295–303 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Kim YH, Chung JI, Woo HG, Jung YS, Lee SH, Moon CH, Suh-Kim H, Baik EJ, Differential regulation of proliferation and differentiation in neural precursor cells by the Jak pathway. Stem Cells 28, 1816–1828 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Yoshimatsu T, Kawaguchi D, Oishi K, Takeda K, Akira S, Masuyama N, Gotoh Y, Non-cell-autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development 133, 2553–2563 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, Sukbuntherng J, Izumi R, Hamdy A, Hedrick E, Fowler NH, Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grisafi D, Maestro A, Grumi C, Piazzoni L, Tirone G, Fiore W, Tessari R, Gianardi V, Gatti M, Tasca F, Generali D, Ravelli A, Lanza F, Scaglione F, Ibrutinib: From bench side to clinical implications. Med. Oncol. 32, 225 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, Yang Y, Cole DE, Melani C, Higham CS, Desai JV, Ceribelli M, Chen L, Thomas CJ, Little RF, Gea-Banacloche J, Bhaumik S, Stetler-Stevenson M, Pittaluga S, Jaffe ES, Heiss J, Lucas N, Steinberg SM, Staudt LM, Wilson WH, Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell 31, 833–843.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X, Huang Z, Zhai K, Ke SQ, Ping YF, Feng H, Rich JN, Yu JS, Bao S, Bian XW, Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell 21, 591–603.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernard S, Goldwirt L, Amorim S, Brice P, Brière J, de Kerviler E, Mourah S, Sauvageon H, Thieblemont C, Activity of ibrutinib in mantle cell lymphoma patients with central nervous system relapse. Blood 126, 1695–1698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung J, Kim LJY, Wang X, Wu Q, Sanvoranart T, Hubert CG, Prager BC, Wallace LC, Jin X, Mack SC, Rich JN, Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2, 90019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Zhou W, Cheng L, Chen C, Huang Z, Fang X, Wu Q, He Z, Xu S, Lathia JD, Ping Y, Rich JN, Bian XW, Bao S, Tetraspanin CD9 stabilizes gp130 by preventing its ubiquitin-dependent lysosomal degradation to promote STAT3 activation in glioma stem cells. Cell Death Differ. 24, 167–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y, Smyth GK, ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.