
Fig. 5.
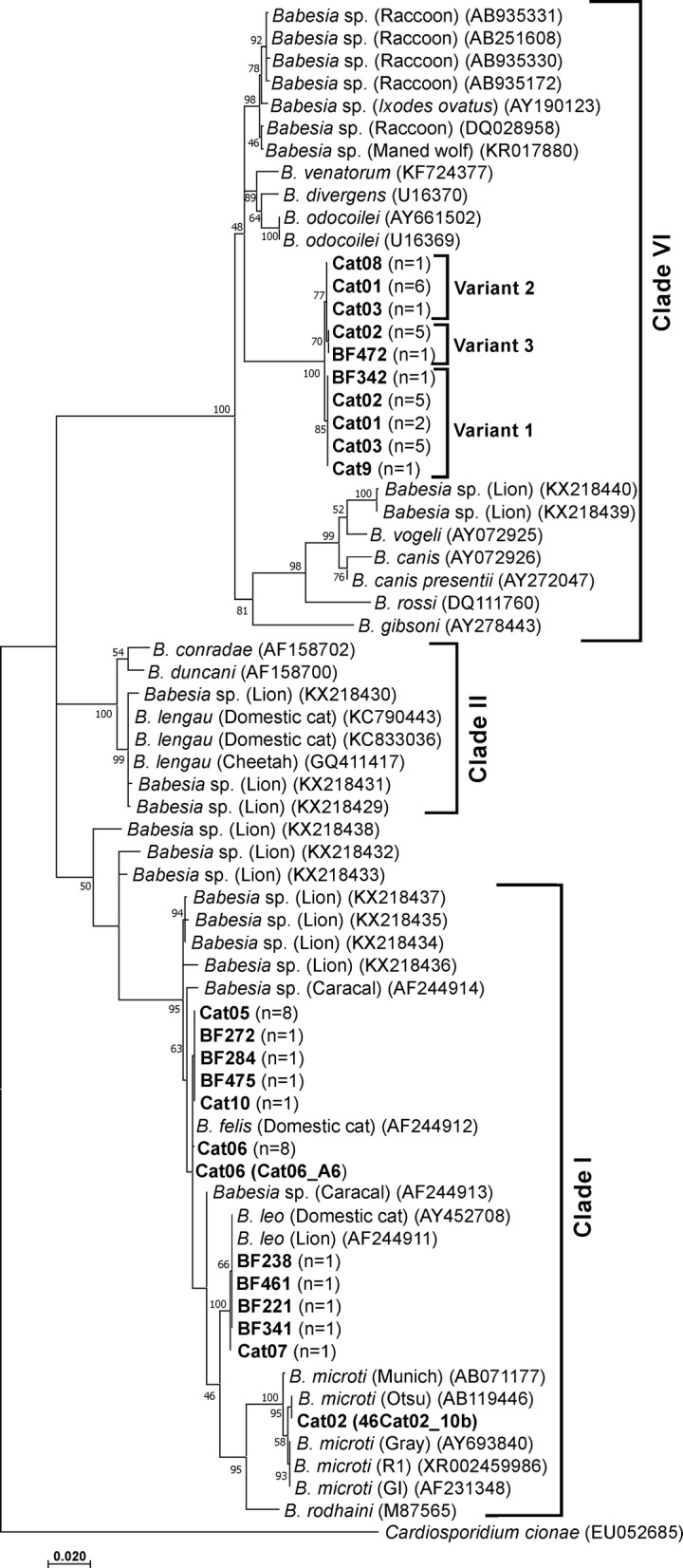
Maximum likelihood tree showing the evolutionary relationships of the Babesia 18S rDNA sequences obtained, with published sequences. The evolutionary history was inferred by using the Maximum Likelihood method based on the Hasegawa-Kishino-Yano model [38]. A discrete Gamma distribution was used to model evolutionary rate differences among sites [5 categories (+G, parameter = 0.4367)]. The rate variation model allowed for some sites to be evolutionarily invariable [(+I), 57.75% sites]. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 1208 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [34]