

Abstract
Background
Epilepsy is one of the most common neurological disorders. It is estimated that up to 30% of patients with epilepsy continue to have epileptic seizures despite treatment with an antiepileptic drug. These patients are classified as drug‐resistant and require treatment with a combination of multiple antiepileptic drugs. Brivaracetam is a third‐generation antiepileptic drug that is a high‐affinity ligand for synaptic vesicle protein 2A. This review investigates the use of brivaracetam as add‐on therapy for epilepsy.
Objectives
To evaluate the efficacy and tolerability of brivaracetam when used as add‐on treatment for people with drug‐resistant epilepsy.
Search methods
We searched the following databases on 9 October 2018: the Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL); Medline (Ovid) 1946 to 8 October 2018; ClinicalTrials.gov; and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP). Originally we also searched SCOPUS as a substitute for Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL.
Selection criteria
We sought randomised controlled trials with parallel‐group design, recruiting people of any age with drug‐resistant epilepsy. We accepted studies with any level of blinding (double‐blind, single‐blind, or unblind).
Data collection and analysis
In accordance with standard methodological procedures expected by the Cochrane Collaboration, two review authors independently assessed trials for inclusion before evaluating trial quality and extracting relevant data. The primary outcome to be assessed was 50% or greater reduction in seizure frequency. Secondary outcomes were: seizure freedom, treatment withdrawal for any reason, treatment withdrawal due to adverse events, the proportion of participants who experienced any adverse events, and drug interactions. We used an intention‐to‐treat (ITT) population for all primary analyses, and we presented results as risk ratios (RRs) with 95% confidence intervals (CIs).
Main results
The review included six trials representing 2411 participants. Only one study included participants with both focal and generalised onset seizures; the other five trials included participants with focal onset seizures only. All six studies included adult participants between 16 and 80 years old, and treatment periods ranged from 7 to 16 weeks. We judged two studies to have low risk of bias and four to have unclear risk of bias. One study failed to provide details on the method used for allocation concealment, and one did not report all outcomes prespecified in the trial protocol. One study did not describe how blinding was maintained, and another noted discrepancies in reporting.
Participants receiving brivaracetam add‐on were significantly more likely to experience a 50% or greater reduction in seizure frequency than those receiving placebo (RR 1.81, 95% CI 1.53 to 2.14; 6 studies; moderate‐quality evidence). Participants receiving brivaracetam were also significantly more likely to attain seizure freedom (RR 5.89, 95% CI 2.30 to 15.13; 6 studies; moderate‐quality evidence). The incidence of treatment withdrawal for any reason (RR 1.27, 95% CI 0.94 to 1.74; 6 studies; low‐quality evidence), as well as the risk of participants experiencing one or more adverse events (RR 1.08, 95% CI 1.00 to 1.17; 5 studies; moderate‐quality evidence), was not significantly different following treatment with brivaracetam compared to placebo. However, participants receiving brivaracetam did appear to be significantly more likely to withdraw from treatment specifically because of adverse events compared with those receiving placebo (RR 1.54, 95% CI 1.02 to 2.33; 6 studies; low‐quality evidence).
Authors' conclusions
Brivaracetam, when used as add‐on therapy for patients with drug‐resistant epilepsy, is effective in reducing seizure frequency and can aid patients in achieving seizure freedom. However, add‐on brivaracetam is associated with a greater proportion of treatment withdrawals due to adverse events compared with placebo. It is important to note that only one of the eligible studies included participants with generalised epilepsy. None of the studies included participants under the age of 16, and all studies were of short duration. Consequently, these findings are mainly applicable to adult patients with drug‐resistant focal epilepsy. Future research should thus focus on investigating the tolerability and efficacy of brivaracetam during longer‐term follow‐up, and should also assess the efficacy and tolerability of add‐on brivaracetam in managing other types of seizures and its use in other age groups.
Plain language summary
Brivaracetam add‐on therapy for drug‐resistant epilepsy
Background
Epilepsy is a disorder characterised by multiple seizures. Most people can control their epilepsy with a single antiepileptic drug; however, some people require multiple antiepileptic drugs. These people are said to have drug‐resistant epilepsy. Brivaracetam is an antiepileptic drug that can be taken as add‐on treatment with another antiepileptic medication to try to manage drug‐resistant epilepsy.
Aim of the review
This review aimed to determine whether brivaracetam is effective and tolerable when used as add‐on treatment for people with drug‐resistant epilepsy.
Results
We were able to identify six studies that investigated brivaracetam as add‐on treatment for drug‐resistant epilepsy. These studies included a total of 2411 participants, aged 16 to 80. Most participants had focal epilepsy (i.e. epilepsy that originates in one area of the brain). People who received brivaracetam in addition to their normal antiepileptic medication were almost twice as likely to experience a 50% or greater reduction in the frequency of their seizures compared to people who were given placebo (i.e. a fake, inactive drug that should not affect epilepsy). People who received brivaracetam were also nearly six times more likely to achieve freedom from all seizures than those receiving placebo. People who received brivaracetam were more likely to withdraw from studies due to side effects, but they were not actually more likely to experience side effects when compared to people receiving placebo.
Quality of the evidence
Evidence taken from studies examining the effectiveness of brivaracetam was of moderate quality. This means that we can be fairly certain that study findings showing that brivaracetam is effective in reducing the frequency of seizures in drug‐resistant epilepsy are accurate. Evidence regarding the tolerability of brivaracetam, for example, the number of people who withdrew from these studies and the number of people who experienced side effects, however, was of low quality. This means that we cannot be sure that trial findings are completely accurate, and that more research is needed to fully investigate the tolerability of brivaracetam. All study participants were adults, and most had focal epilepsy. As a result, the review cannot inform us about how effective brivaracetam is in children or in individuals with other types of epilepsy, for example, generalised epilepsy, which is epilepsy that involves the whole brain.
Evidence is current to October 2018.
Summary of findings
Summary of findings for the main comparison. Brivaracetam compared to placebo for add‐on therapy for focal epilepsy.
| Brivaracetam compared to placebo for add‐on therapy for focal epilepsy | ||||||
| Patient or population: patients with drug‐resistant focal epilepsy Setting: outpatients Intervention: brivaracetam (all doses) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with brivaracetam | |||||
|
50% or greater reduction in seizure frequency (responder rate) Follow‐up (range): 7 to 16 weeks |
Study population | RR 1.81 (1.53 to 2.14) | 2411 (6 RCTs) | ⊕⊕⊕⊝ MODERATEa | Brivaracetam likely increases the 50% responder rate | |
| 189 per 1000 | 342 per 1000 (289 to 404) | |||||
|
Seizure freedom Follow‐up (range): 7 to 16 weeks |
Study population | RR 5.89 (2.30 to 15.13) | 2411 (6 RCTs) | ⊕⊕⊕⊝ MODERATEa,b,c | Brivaracetam likely results in a large increase in the number of patients achieving seizure freedom | |
| 4 per 1000 | 26 per 1000 (10 to 66) | |||||
|
Treatment withdrawal Follow‐up (range): 7 to 16 weeks |
Study population | RR 1.27 (0.94 to 1.74) | 2411 (6 RCTs) | ⊕⊕⊝⊝ LOWa,b | Brivaracetam might increase treatment withdrawal slightly | |
| 71 per 1000 | 90 per 1000 (67 to 124) | |||||
|
Proportion of participants who experienced adverse events leading to treatment withdrawal Follow‐up (range): 7 to 16 weeks |
Study population | RR 1.54 (1.02 to 2.33) | 2411 (6 RCTs) | ⊕⊕⊝⊝ LOWa,b | Brivaracetam may increase the proportion of participants who experience adverse events leading to treatment withdrawal | |
| 39 per 1000 | 60 per 1000 (40 to 91) | |||||
|
Proportion of participants who experienced any adverse events Follow‐up (range): 7 to 16 weeks |
Study population | RR 1.08 (1.00 to 1.17) | 2011 (5 RCTs) | ⊕⊕⊕⊝ MODERATEa | Brivaracetam probably slightly increases the proportion of participants who experience any adverse events | |
| 598 per 1000 | 646 per 1000 (598 to 700) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once for risk of bias: all studies were pharmaceutical sponsored and some included studies had incomplete methodological information.
bDowngraded once for imprecision: number of events does not suffice for optimal information size.
cUpgraded once for large effect: large effect size (RR > 5) observed for outcome.
Background
Description of the condition
Epilepsy is a chronic neurological disorder that is characterised by recurrent seizures. These seizures are caused by sudden, usually brief, excessive electrical discharges within a group of neurons. More than 50 million people in the world today have received a diagnosis of epilepsy, and approximately 2.4 million new cases occur each year, worldwide (WHO 2013). Antiepileptic drug monotherapy is generally accepted as the preferred initial management approach in epilepsy care. However, up to 30% of individuals with epilepsy do not respond adequately to conventional antiepileptic drug treatment, either due to recurrent seizures despite optimised antiepileptic drug therapy, or due to adverse effects (Van Paesschen 2013). Many of these people will use add‐on therapies. Therefore there is a clear need for antiepileptic drugs that can control the seizures of those who do not respond to conventional drug treatment. As a result, dozens of novel antiepileptic drugs have been marketed in the past two decades, and it is therefore very important that researchers assess the efficacy and tolerability of new antiepileptic drugs for all individuals.
Description of the intervention
Brivaracetam is a novel antiepileptic drug that has been investigated as add‐on therapy for epilepsy. Brivaracetam is a third‐generation antiepileptic agent that shares a similar chemical structure with levetiracetam and piracetam. Brivaracetam has been shown to have a wider antiepileptic spectrum and higher efficacy than levetiracetam in several animal models of structural and genetic epilepsy (Schulze‐Bonhage 2011). In 2005, brivaracetam was approved as an orphan drug for the treatment of progressive myoclonus epilepsies by the European Commission (Chu‐Shore 2010). In the same year, the US Food and Drug Administration (FDA) also approved brivaracetam as a treatment for symptomatic myoclonus (Johannessen Landmark 2008). Brivaracetam has been shown to suppress generalised photoparoxysmal electroencephalography (EEG) responses in a photosensitivity model as proof‐of‐principle of its efficacy in patients with epilepsy (Kasteleijn‐Nolst Trenité 2007). Brivaracetam was well tolerated as add‐on therapy in adults with drug‐resistant focal‐onset seizures, but it failed to show consistent efficacy in decreasing the frequency of seizures in phase IIb and phase III randomised controlled trials (French 2010; Van Paesschen 2013; Werhahn 2010).
Brivaracetam exhibits linear pharmacokinetics across a wide dose range (10 mg to 600 mg) when administrated as a single oral dose to healthy subjects. It is rapidly and completely absorbed and is weakly bound to plasma proteins (≤ 20%), with an elimination half‐life of seven to eight hours after oral administration (Schulze‐Bonhage 2011). Brivaracetam is metabolised primarily via hepatic hydrolysis of the acetamide group, and secondarily through hydroxylation mediated by cytochrome P450 (CYP) 2C19 (Nicolas 2012). It is extensively eliminated renally within 72 hours of ingestion (> 95%). In patients with hepatic impairment, total body clearance of brivaracetam is reduced and plasma half‐life is accordingly prolonged. However, the pharmacokinetic profile of brivaracetam in patients with renal impairment is similar to that in healthy participants (von Rosenstiel 2007). Researchers observed a slight decrease in plasma carbamazepine levels and a 2.5‐fold increase in plasma carbamazepine‐epoxide levels when brivaracetam was applied with other antiepileptic drugs at 400 mg per day. In addition, peak concentrations of a single dose of 600 mg phenytoin were decreased slightly when co‐administered with brivaracetam (Schulze‐Bonhage 2011). The manufacturers of brivaracetam have claimed that evidence from phase II/III trials has shown that no dose adjustment is required when brivaracetam is added to treatment with other antiepileptic drugs (Bialer 2010).
How the intervention might work
Brivaracetam is a high‐affinity synaptic vesicle protein SV2A ligand that is involved in presynaptic transmitter release. It shows inhibition of neuronal voltage‐dependent sodium (Na+) channels (French 2010; Schulze‐Bonhage 2011; Van Paesschen 2013).
Why it is important to do this review
To our knowledge, this is the first Cochrane systematic review that focuses on the use of brivaracetam as add‐on therapy for epilepsy. We summarise here available evidence on the efficacy and tolerability of brivaracetam as derived from randomised controlled trials.
Objectives
To evaluate the efficacy and tolerability of brivaracetam when used as add‐on treatment for people with drug‐resistant epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
Trials were required to meet all of the following criteria.
Randomised controlled trials using an adequate method of concealment of randomisation (e.g. allocation of sequentially numbered, sealed packages of medication; sealed, opaque envelopes; telephone randomisation). We excluded quasi‐randomised controlled trials in which treatment allocation was decided through methods such as alternate days of the week.
Double‐blind, single‐blind, or unblinded.
Placebo‐controlled or active‐controlled.
Parallel‐group design.
Types of participants
People of any age with drug‐resistant focal‐onset seizures (simple focal, complex focal, or secondary generalised tonic‐clonic seizures) or generalised‐onset seizures.
Types of interventions
The experimental group consisted of participants who received brivaracetam in addition to an existing antiepileptic drug regimen taken at the time of randomisation.
The control group consisted of participants who received a matched placebo or active comparator in addition to an existing antiepileptic drug regimen taken at the time of randomisation.
Types of outcome measures
Primary outcomes
50% or greater reduction in seizure frequency (responder rate)
The proportion of individuals with a 50% or greater reduction in seizure frequency during the treatment period compared with the pre‐randomisation baseline period was our primary outcome. This outcome was commonly reported in this type of study and could be calculated for studies that did not report it, provided that baseline seizure data were recorded.
Secondary outcomes
Seizure freedom: the proportion of participants with complete cessation of seizures at the end of the follow‐up period.
Treatment withdrawal: the proportion of participants having treatment withdrawn, for any reason, during the course of the treatment period. This provides a measure of global effectiveness. Treatment is likely to be withdrawn due to adverse effects, lack of efficacy, or a combination of both. This is an outcome to which the individual makes a direct contribution. In trials of short duration, it is likely that adverse effects will be the most common reason for withdrawal.
-
Adverse events:
The proportion of participants who experienced adverse events leading to treatment withdrawal.
The proportion of participants who experienced any adverse events.
Drug interactions: any drug interactions reported in the included studies.
Search methods for identification of studies
Electronic searches
Searches for this review were first run in April 2013. Subsequent searches were run in March 2015 and March 2017. The most recent searches were run on 9 October 2018, when we searched the following databases, with no language restrictions:
Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL), using the search strategy set out in Appendix 1.
Medline (Ovid), 1946 to 08 October 2018, using the search strategy set out in Appendix 2.
ClinicalTrials.gov, using the search strategy set out in Appendix 3.
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP), using the search strategy set out in Appendix 4.
Originally, we also searched SCOPUS as a substitute for Embase, but this is no longer necessary because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL.
Searching other resources
We reviewed the reference lists of retrieved studies to check for additional reports of relevant studies. We also contacted UCB, Inc. (manufacturers of brivaracetam) and epilepsy experts for ongoing studies and unpublished information.
Data collection and analysis
Selection of studies
The process of selecting studies for inclusion in the review involved merging search results using reference management software and removing duplicates of the same report. Two review authors (RB and MP) screened all titles, abstracts, and keywords of publications identified by the searches to assess trial eligibility. We excluded publications describing studies that clearly did not meet the inclusion criteria at this stage. We retrieved all potentially relevant papers, and two review authors (RB and MP) independently evaluated the full text of each paper, according to pre‐specified selection criteria. We resolved disagreements by discussion. If disagreements persisted, the third review author (AGM) arbitrated.
Data extraction and management
Two review authors (RB and MP) independently extracted the following information from included trials, if available. We resolved disagreements by discussion.
-
Methods
Study design
Method of randomisation
Allocation concealment
Blindness
Study duration
-
Participants
Age
Gender
Ethnicity
Type of seizure
Seizure frequency
Epilepsy duration
Inclusion criteria
Exclusion criteria
Total number of participants recruited
Total number of participants randomised
-
Interventions
Dosage
Administration method
Treatment duration
Number of background drugs
-
Outcomes
Primary outcome
Secondary outcomes
Adverse events
Drug interactions
-
Follow‐up data
Duration of follow‐up period
Total number of participants followed up
Number of losses to follow‐up
Reasons for treatment withdrawal
Assessment of risk of bias in included studies
Two review authors (RB and MP) independently assessed the risk of bias associated with included studies using the Cochrane 'Risk of bias' tool, as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane 'Risk of bias' tool comprises seven specific parameters: (1) random sequence generation, (2) allocation concealment, (3) blinding of participants and personnel, (4) blinding of outcome assessors, (5) incomplete outcome data, (6) selective outcome reporting, and (7) other bias. For each entry, review authors made the judgement ('low' risk of bias, 'high' risk of bias, or 'unclear' risk of bias) and provided support for the decision by an agreed review author comment or by a quote taken from the corresponding publication.
We then determined an overall judgement for risk of bias within each study, again in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Specifically, if we deemed a study to have an unclear risk of bias for one or more of the key domains, then we awarded that study an overall unclear risk of bias judgement. Accordingly, if we determined that a study had high risk of bias for one or more of the key domains, we awarded that study a high risk of bias judgement overall. Only if we judged a study to have low risk of bias across all seven domains did we award that study a low risk of bias judgement overall. We resolved any disagreements by discussion.
Measures of treatment effect
For dichotomous data, we used the risk ratio (RR) with 95% confidence interval (CI) for analysis.
For drug interactions, we described the outcome narratively.
Unit of analysis issues
According to guidance provided in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011), we did not encounter any unit of analysis issues.
Dealing with missing data
If data were insufficient or missing, we contacted the manufacturers and original investigators of relevant trials for additional information through personal communication. If we did not receive a response, we analysed available data according to the intention‐to‐treat (ITT) principle.
Assessment of heterogeneity
We evaluated clinical and methodological heterogeneity among trials by comparing the characteristics of participants (age, gender, seizure type, seizure frequency, duration of epilepsy), interventions (dosage, administration method and duration, co‐treatments), and study design (randomisation, allocation concealment, blinding methods) between studies.
We evaluated statistical heterogeneity among trials using the Chi² test with significance set at 0.1 along with the I² statistic.
A P value greater than 0.1 in the Chi² test (P > 0.1) indicated no significant statistical heterogeneity (Deeks 2011).
If a P value was less than or equal to 0.1 in the Chi² test, we interpreted heterogeneity according to percentage ranges of the I² statistic, as follows (Deeks 2011).
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity*.
50% to 90%: may represent substantial heterogeneity*.
75% to 100%: represents considerable heterogeneity*.
*The importance of the observed value of the I² statistic depends on (1) the magnitude and direction of effect and (2) the strength of evidence for heterogeneity (e.g. P value from the Chi² test or confidence interval for the I² statistic).
Assessment of reporting biases
We had originally planned to assess funnel plot asymmetry. Reasons for asymmetry include publication bias, outcome reporting bias, language bias, citation bias, poor methodological design, and heterogeneity. Unfortunately, however, our review included fewer than 10 studies; as a consequence, funnel plots would have been minimally informative. Therefore, we did not generate funnel plots as part of this review.
Data synthesis
We analysed the data using Review Manager version 5.3 (RevMan 2014). Heterogeneity determined the choice of a fixed‐effect or a random‐effects model. If clinically appropriate, and if we found no evidence of substantial statistical heterogeneity using the I² statistic (I² < 50%), we analysed data in a meta‐analysis using a fixed‐effect model. If we found substantial heterogeneity (I² ≥ 50%), we explored possible factors contributing to the heterogeneity and used a random‐effects model to perform meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We conducted subgroup analyses according to different dose groups of brivaracetam, such as 50 mg/d and 100 mg/d, for each outcome. In addition, we had planned to conduct subgroup analyses according to the different age groups of participants (children younger than 17 years versus adults); however, all of the studies exclusively comprised adult populations.
Sensitivity analysis
We had planned to conduct the following sensitivity analyses to test the robustness of the meta‐analysis, where possible.
Repeating the analysis with exclusion of unpublished studies.
Repeating the analysis with exclusion of studies published only as abstracts.
This sensitivity analysis was not required to be conducted in the current review, as all included studies were published journal articles.
Results
Description of studies
Results of the search
By conducting searches, we identified a total of 186 records for potential inclusion in the review (Figure 1). We removed 38 duplicate records, leaving 148 eligible records. We then discarded 17 of these records due to irrelevance. Of the 131 records remaining, we excluded a further 106 records at the stage of abstract and title screening, again due to irrelevance. We attempted to retrieve the full texts for the 25 records that remained after the initial screening stage. After accessing and assessing these full‐text articles, we determined that 23 records were eligible for inclusion in the review. All of the 23 records identified were linked to six individual studies (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013), which we subsequently included in the meta‐analyses.
1.
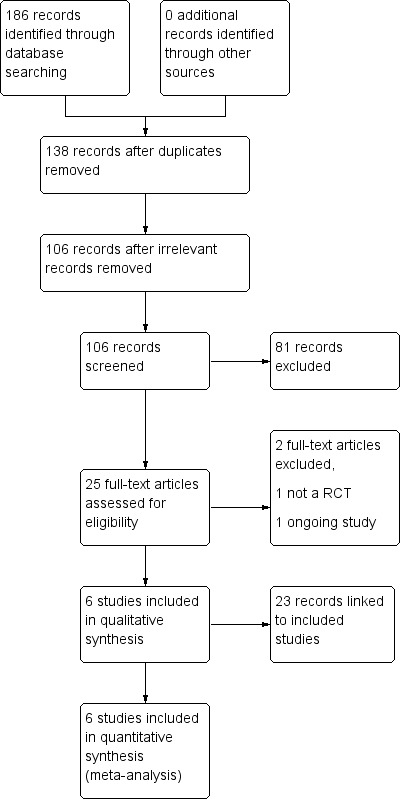
Study flow diagram.
Included studies
All six included studies were randomised, double‐blind, placebo‐controlled trials, with parallel‐group design. We have summarised the details of the included studies in the Characteristics of included studies tables.
Biton 2014 was a multi‐centre study, conducted across Australia, Brazil, Canada, Mexico, and the United States, and including a total of 400 participants. Participants were aged 16 to 70 and had drug‐resistant focal epilepsy. Most participants were receiving two concomitant antiepileptic drugs (AEDs) at baseline; however, some participants were receiving more than three AEDs. Participants were required to undergo an 8‐week baseline period before randomisation to one of four treatment groups. Participants next entered a 12‐week treatment period, during which they received 5, 20, or 50 mg/d brivaracetam treatment, or matching placebo, with no up‐titration. After completing the trial, participants were given the option to enter an open‐label extension.
French 2010 was, again, a multi‐centre study, with sites based in Brazil, India, Mexico, and the United States. The study included a total of 208 participants. All participants were between 16 and 65 years of age and had well‐characterised focal epilepsy. Participants were required to be taking one or two concomitant AEDs at baseline. Similar to Biton 2014, most were receiving two concomitant AEDs, and a small subset of participants were receiving more than three AEDs. Eligible participants were randomised to one of four treatment groups (5, 20, or 50 mg/d brivaracetam or matching placebo) after completion of the four‐week baseline period. The treatment period was seven weeks long and did not include an up‐titration period. Upon completion of the treatment period, participants were offered entry into a long‐term open‐label extension study.
Klein 2015, a multi‐centre study conducted at sites across North America, Western Europe, Eastern Europe, Latin America, and Asia, enrolled and randomised a total of 768 participants. Eligible participants were between 16 and 80 years of age and had well‐characterised drug‐resistant focal epilepsy. Most participants were receiving two concomitant AEDs at baseline. Only four participants (< 1%) were receiving three or more AEDs. Participants were required to complete an eight‐week baseline period before randomisation. After successful completion of the baseline period, participants were randomised into one of three treatment groups: 100 mg/d brivaracetam, 200 mg/d brivaracetam, or placebo. Participants then undertook a 12‐week treatment period, followed by a four‐week down‐titration period. Participants were then given the opportunity to enter an open‐label extension study.
Kwan 2014 was, likewise, a multi‐centre study that recruited a total of 480 participants from various sites, located in Austria, Belgium, Czech Republic, Germany, Hong Kong, India, Italy, Norway, Republic of South Africa, Russian Federation, Singapore, South Korea, Sweden, Taiwan, and Ukraine. Participants were aged 16 to 70, and 90% had drug‐resistant focal epilepsy. The remaining 10% had drug‐resistant generalised epilepsy. Participants were required to be taking one to three concomitant AEDs; most participants were receiving two or more AEDs (45.4%). It is notable that a much larger proportion of participants in this study (37.3%) were receiving three or more AEDs compared with the other studies. Participants completed a four‐week baseline period before they were randomised to one of two treatment arms: 20 to 150 mg/d brivaracetam or matching placebo, at a ratio of 3:1, respectively. As a consequence, a much larger number of participants were randomised to the experimental brivaracetam group than to the placebo control group. The study consisted of a 16‐week treatment period, which comprised an eight‐week dose‐finding phase and an eight‐week maintenance phase. During the dose‐finding phase, the dosage was up‐titrated in a stepwise manner on a two‐weekly basis, dependent on observed efficacy and participants' tolerability. The optimal dose achieved was then maintained over the final eight‐week period. After the treatment period, participants underwent a two‐week down‐titration period before they were offered entry into one of two open‐label follow‐up studies.
Ryvlin 2014 was also a multi‐centre study, with sites based across Poland, France, Germany, Spain, Italy, Switzerland, Hungary, Finland, The Netherlands, Belgium, the United Kingdom, and India. A total of 398 participants were enrolled into the study. All participants were aged 16 to 70 and had received a diagnosis of focal epilepsy. Participants were required to be receiving treatment with one or two AEDs at baseline, although a small proportion (4%) were receiving three or more AEDs. After completion of an eight‐week baseline period, participants were randomised to one of four treatment groups: 20 mg/d brivaracetam, 50 mg/d brivaracetam, 100 mg/d brivaracetam, or placebo. The study comprised a 12‐week treatment period (without up‐titration), followed by a two‐week down‐titration period, before participants were offered entry into an open‐label extension study.
Van Paesschen 2013 was, again, a multi‐centre study. This trial was conducted at multiple sites across Belgium, Czech Republic, Finland, France, Germany, The Netherlands, Poland, Spain, and the United Kingdom. A total of 157 participants were recruited into this study. Participants were aged 16 to 65 and had drug‐resistant focal epilepsy. They were required to be receiving one or two concomitant AEDs. Again, the largest proportion of participants were taking two concomitant AEDs at baseline, with only 6% taking three or more AEDs. Randomisation took place after completion of a four‐week baseline period. Participants were randomised to one of three treatment groups: 50 mg/d brivaracetam, 150 mg/d brivaracetam, or matching placebo. The treatment period consisted of a three‐week up‐titration followed by a seven‐week maintenance phase, and therefore lasted 10 weeks. After completion of the trial, participants were asked whether they wished to enter an open‐label extension study.
Excluded studies
We excluded one study at the full‐text screening stage because it was not an RCT but was instead a meta‐analysis of two studies that had already been included in the review (see Characteristics of excluded studies) (Lacroix 2007). We were unable to include another study because the study was ongoing and no results had so far been published (see Characteristics of ongoing studies) (NCT03083665). Additionally, it was not fully clear whether this study was eligible for inclusion because of the limited information provided regarding study design. If results of the NCT03083665 study have been published by the time of the next review update, we will reassess this study for inclusion.
Risk of bias in included studies
We judged that two studies had low risk of bias overall (French 2010; Klein 2015), whilst we judged that the other four studies had unclear risk of bias (Biton 2014; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Figure 2 and Figure 3 present summaries of the 'Risk of bias' associated with each of the included studies for each rating domain. We discuss below the individual rating domains for all included studies.
2.
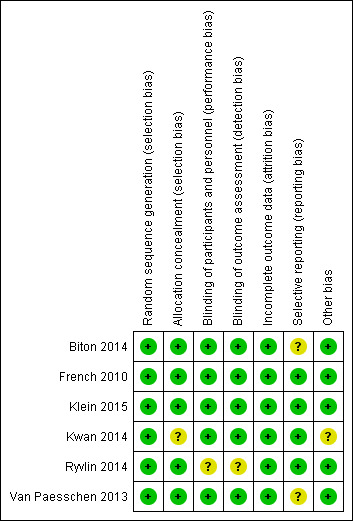
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
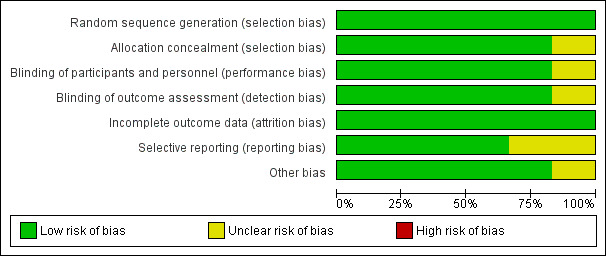
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
All six studies provided details regarding randomisation of participants. We therefore awarded a low risk of bias judgement for random sequence generation for all included studies. Five studies specified that randomisation was achieved using the random permuted blocks method with stratification (Biton 2014; French 2010; Klein 2015; Kwan 2014; Van Paesschen 2013). The remaining study instead used a central randomisation system, although, again, randomisation was stratified (Ryvlin 2014).
With regards to allocation concealment, three of the included studies described using an interactive voice response system (IVRS) to ensure allocation concealment (Biton 2014; Klein 2015; Ryvlin 2014). Meanwhile, two studies used a central randomisation system, which, again, enabled allocation to be effectively concealed (French 2010; Van Paesschen 2013). We judged that these five studies all had low risk of bias with regards to allocation concealment. In contrast, we assessed the remaining study to have unclear risk of bias after study authors failed to describe any methods used for allocation concealment (Kwan 2014).
Blinding
Five of the included studies were double‐blind and specified that they used matching placebo tablets to maintain blinding (Biton 2014; French 2010; Klein 2015; Kwan 2014; Van Paesschen 2013). One study further described that tablets of various strengths were used so that all participants took two tablets per dose, regardless of their actual randomised dosage of brivaracetam or allocation to placebo (Klein 2015). This further ensured that blinding was maintained. All patients and study personnel were adequately blinded by the matching placebo and, consequently, we assessed all five studies to be at low risk of performance bias.
Efficacy outcomes were self‐reported by patients in seizure diaries. Accordingly, patients were regarded as the outcome assessors. As described above, participants were effectively blinded by the matching placebo and, as a result, their reporting of outcomes was not affected or biased by treatment allocation. Likewise, because the studies were double‐blind, the investigators, including those responsible for data analysis, would also have been effectively blinded. We therefore assessed all five studies to have low risk of bias with regards to detection bias for outcome assessment (Biton 2014; French 2010; Klein 2015; Kwan 2014; Van Paesschen 2013).
The remaining study ‐ Ryvlin 2014 ‐ did not explain any methods used to maintain blinding. Consequently, we assessed this study as having unclear risk for both performance bias and detection bias.
Incomplete outcome data
We rated all of the included studies to be at low risk of attrition bias (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). All six studies reported the attrition rate and conducted an ITT analysis. In actuality, however, only two of these studies utilised a strict ITT population, whereby all participants who were randomised were analysed (Kwan 2014; Van Paesschen 2013). The other four studies instead used a modified ITT population, most commonly excluding participants who did not receive at least one dose of study drug (Biton 2014; French 2010; Klein 2015; Ryvlin 2014). For each study, no more than 1% of participants were excluded from the ITT population; therefore, we still assessed studies to be at low risk of attrition bias. All participants excluded from ITT analyses conducted within the studies were reinstated in the ITT analyses performed here, in this review.
Selective reporting
We assessed that four of the included studies were at low risk of reporting bias (French 2010; Klein 2015; Kwan 2014; Ryvlin 2014). Despite not supplying a trial protocol, each of the four studies reported results for all of the outcome measures prespecified in the methods section of their respective publications. Another study similarly reported the results of its prespecified outcomes (Biton 2014); however, study authors failed to provide results for the placebo group for one of the outcome measures ‐ the number of participants reporting one or more adverse events. This introduced reporting bias and precluded inclusion of this study in the meta‐analysis for that outcome. As a result, the study was deemed to have unclear risk of reporting bias. The remaining study ‐ Van Paesschen 2013 ‐ provided a trial protocol; however, not all intended outcomes identified in the trial protocol were reported in subsequent publications. Again, we assessed this study as having unclear risk of bias.
Other potential sources of bias
We identified another source of potential bias in the Kwan 2014 study, which randomised participants to the experimental brivaracetam group and the placebo control group at a ratio of 3:1, respectively. This produced an uneven distribution of participants between the two treatment groups. Unequal allocation ratios reduce the statistical power of a trial and negatively impact the ability of that trial to detect a therapeutic effect (Hey 2014). Kwan 2014 did, however, complete a power calculation and determined that a sample size of 376 participants would be required to detect a 16% reduction in baseline‐adjusted weekly focal seizure frequency compared to placebo. Kwan 2014 actually recruited 480 participants and, therefore, exceeded the estimated sample size. Thus, this trial should have retained adequate statistical power to be able to detect a therapeutic effect, despite the unequal allocation ratio.
Nevertheless, unequal allocation ratios are further associated with a greater placebo effect (Hey 2014). As a result, the unequal allocation ratio used could still distort the perceived therapeutic effect, despite the compensatory sample size calculation. For this reason, we awarded Kwan 2014 an unclear risk of bias with regards to other sources of bias.
Effects of interventions
See: Table 1
See Table 1 for the main comparison: brivaracetam compared to placebo for add‐on therapy for focal epilepsy.
Five of the included studies used well‐defined, escalated doses of brivaracetam for the experimental treatment groups (Biton 2014; French 2010; Klein 2015; Ryvlin 2014; Van Paesschen 2013). In contrast, the Kwan 2014 study utilised a flexible dosing regimen, whereby participants began on 20 mg/d brivaracetam or placebo, and then increased their dose up to 150 mg/d, depending on the efficacy that they experienced and their tolerability of the study drug. Although it was reported that most participants in both the brivaracetam and placebo treatment groups achieved the highest dosages of 100 mg/d and 150 mg/d, the dose was not standardised amongst participants. As a result, the data extracted from Kwan 2014 could not be included in the subgroup analysis for drug dose for any of the outcomes listed.
Notably, and also of importance to the analyses, two of the included studies each excluded eight participants from their ITT populations, despite having randomised these participants to a treatment group (Biton 2014; Klein 2015). Klein 2015 specified that participants must have received one or more doses of study drug and must have provided at least one post‐baseline diary entry, thus explaining the exclusion of some participants. Biton 2014 stated that participants must have received one or more doses of study drug, justifying the exclusion of four participants; however, researchers then excluded an additional four participants ‐ three due to serious non‐compliance and one as a clinical outlier. We reinstated the 16 excluded participants in the ITT analysis conducted in this review, to ensure that our ITT analysis fully adhered to the "once randomised, always analysed" principle. We repeated this for each of the outcomes analysed and reported on this below.
1. 50% or greater reduction in seizure frequency
All six included studies, involving a total of 2411 ITT participants, contributed to this outcome analysis (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Participants receiving brivaracetam were significantly more likely to achieve a 50% or greater reduction in seizure frequency, compared to those who received placebo (risk ratio (RR) 1.81, 95% confidence interval (CI) 1.53 to 2.14; Analysis 1.1). Subgroup analysis by dose of brivaracetam did not suggest any difference in 50% or greater reduction in seizure frequency dependent on dose. Doses of 20 mg/d (RR 1.64, 95% CI 1.18 to 2.27), 50 mg/d (RR 2.00, 95% CI 1.50 to 2.66), 100 mg/d (RR 1.81, 95% CI 1.42 to 2.30), and 200 mg/d (RR 1.76, 95% CI 1.33 to 2.33) brivaracetam were all associated with a significantly greater proportion of participants achieving a 50% or greater reduction in seizure frequency than placebo (Analysis 1.1).
1.1. Analysis.
Comparison 1 Brivaracetam vs placebo, Outcome 1 50% or greater reduction in seizure frequency (responder rate).
2. Seizure freedom
All six studies, consisting of 2411 ITT participants, were included in this outcome analysis (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Participants receiving brivaracetam were significantly more likely to experience seizure freedom, specifically, almost six times more likely, than those receiving placebo (RR 5.89, 95% CI 2.30 to 15.13; Analysis 1.2). We noted no significant heterogeneity within the data set (Chi² = 0.83, df = 5, P = 0.97, I² = 0%) for seizure freedom. Subgroup analysis stratified by dose did, however, show that only participants receiving the higher doses of 50 mg/d (RR 5.39, 95% CI 1.42 to 20.49), 100 mg/d (RR 7.19, 95% CI 1.93 to 26.85), and 200 mg/d (RR 5.24, 95% CI 1.16 to 23.68) were significantly more likely to achieve seizure freedom than those receiving placebo.
1.2. Analysis.
Comparison 1 Brivaracetam vs placebo, Outcome 2 Seizure freedom.
3. Treatment withdrawal
All six studies, consisting of 2411 ITT participants, reported the number of treatment withdrawals and thus contributed to this outcome analysis (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). They reported no significant differences in the proportion of participants withdrawing from treatment when comparing those randomised to brivaracetam and those randomised to placebo (RR 1.27, 95% CI 0.94 to 1.74; Analysis 1.3). This was consistently found in each of the dose subgroups; no dose of brivaracetam was associated with a significantly higher rate of treatment withdrawal compared to placebo.
1.3. Analysis.
Comparison 1 Brivaracetam vs placebo, Outcome 3 Treatment withdrawal.
Notably, however, we detected more heterogeneity within the collective data set, consisting of all doses of brivaracetam (Chi² = 7.32, df = 5, P = 0.20, I² = 32%), as well as within the individual dose subgroups during subgroup analysis. This was particularly evident when compared to the complete absence of heterogeneity observed in the efficacy outcomes ‐ 50% or greater seizure reduction and seizure freedom. Heterogeneity was most prominent in the 5 mg/d (Chi² = 2.36, df = 1, P = 0.12, I² = 58%) and 100 mg/d (Chi² = 2.05, df = 1, P = 0.15, I² = 51%) brivaracetam subgroups, although it is important to note that the levels of heterogeneity remained statistically insignificant. Of greatest concern, the direction of effect varied between studies. French 2010 and Van Paesschen 2013 reported a greater incidence of treatment withdrawal amongst participants receiving placebo compared to those receiving brivaracetam, whereas Biton 2014 and Klein 2015 reported the opposite, with more participants randomised to brivaracetam withdrawing from treatment compared to those randomised to placebo.
4. Adverse events
All six studies, consisting of 2411 ITT participants, reported and stated the reasons for treatment withdrawal (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Data from all six studies were therefore included in the outcome analysis for the proportion of participants who experienced adverse events leading to treatment withdrawal. In contrast to treatment withdrawal for any reason, the analysis showed that receiving brivaracetam was associated with a significantly higher prevalence of participants withdrawing from treatment, specifically due to adverse events (RR 1.54, 95% CI 1.02 to 2.33; Analysis 1.4), compared to those receiving placebo. However, it is interesting to note that subgroup analysis revealed that none of the individual doses of brivaracetam were associated with a significantly higher proportion of treatment withdrawals due to adverse events than placebo. Furthermore, although not statistically significant, the data reported regarding 5 mg/d brivaracetam, compared to placebo, again displayed more heterogeneity (Chi² = 2.12, df = 1, P = 0.15, I² = 53%) than had been associated with the other outcomes. Most noticeably, French 2010 again observed the opposite treatment effect to that reported by the other studies included in this analysis.
1.4. Analysis.
Comparison 1 Brivaracetam vs placebo, Outcome 4 Proportion of participants who experienced adverse events leading to treatment withdrawal.
In contrast to the other outcome analyses, only five studies, comprising 2011 participants, fully reported the proportion of participants who experienced at least one adverse event, and thus contributed to the outcome analysis performed (French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Biton 2014 failed to report the incidence of participants in the placebo group reporting one or more adverse events and, therefore, was excluded from the analysis. Results show no significant difference in the proportion of participants experiencing one or more adverse events when receiving brivaracetam compared to those receiving placebo (RR 1.08, 95% CI 1.00 to 1.17; Analysis 1.5). Out of the six individual doses of brivaracetam tested during the subgroup analysis, only one dose ‐ 100 mg/d brivaracetam (RR 1.16, 95% CI 1.04 to 1.31) ‐ was associated with a significantly higher proportion of participants experiencing one or more adverse events compared to those receiving placebo. The effect size was fairly small, however, despite being significant. Specifically, there was a 16% increase in the number of participants reporting one or more adverse events when receiving 100 mg/d brivaracetam, compared to those receiving placebo.
1.5. Analysis.
Comparison 1 Brivaracetam vs placebo, Outcome 5 Proportion of participants who experienced any adverse events.
5. Drug interactions
Five of the included studies, including 1643 participants, described drug interactions in their publications (Biton 2014; French 2010; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Specifically, all five studies referenced the interaction of brivaracetam with concomitant levetiracetam use.
Biton 2014 noted that a smaller proportion of participants experienced a 50% or greater reduction in seizure frequency after receiving brivaracetam if they were using levetiracetam concomitantly. Furthermore, Biton 2014 recognised that the median per cent reduction from baseline in weekly partial onset seizure frequency was lower in participants using concomitant levetiracetam.
Equally, French 2010 demonstrated that a reduced proportion of participants achieved a 50% or greater reduction in seizure frequency, dependent on concomitant levetiracetam use. However, French 2010 was unable to comment on the significance of this result because of the small number of participants included in the observation.
Kwan 2014, similarly, reported that only 13% of participants receiving brivaracetam and taking concomitant levetiracetam experienced a 50% or greater reduction in seizure frequency compared to 34% of participants not using concomitant levetiracetam. Kwan 2014 also stated that participants using concomitant levetiracetam experienced a smaller baseline‐adjusted per cent reduction in weekly focal seizure frequency than levetiracetam naive participants,
Ryvlin 2014 agreed that, generally, a greater proportion of participants who were levetiracetam naive or had previously used levetiracetam but since discontinued its use achieved a 50% or greater reduction in seizure frequency. Likewise, participants concomitantly using levetiracetam in the Ryvlin 2014 study experienced a lesser reduction in seizure frequency.
Van Paesschen 2013 reported that 26% of participants receiving brivaracetam and using concomitant levetiracetam attained a 50% or greater reduction in seizure frequency, as opposed to 32% and 47% of participants with prior levetiracetam use and levetiracetam naive participants, respectively. Placebo responses showed the opposite trend but were also more consistent. Results show that 27% of participants receiving placebo and using concomitant levetiracetam were responders, achieving a 50% or greater reduction, whilst 22% of participants who received placebo with prior levetiracetam use or who were levetiracetam naive were responders.
All five studies consistently reported that a decreased proportion of participants randomised to brivaracetam achieved a 50% or greater reduction in seizure frequency when using levetiracetam concomitantly. These studies also implied that there was an overall decrease in the efficacy of brivaracetam with concomitant levetiracetam use, as demonstrated by the smaller reduction in seizure frequency observed.
Discussion
Summary of main results
This review evaluated the efficacy and tolerability of brivaracetam when used as an add‐on treatment for people with drug‐resistant epilepsy. Six studies, involving 2411 participants, contributed to the analyses performed in this review (Biton 2014; French 2010; Klein 2015; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). We assessed two of the included studies to have low risk of bias (French 2010; Klein 2015), and we deemed that four studies had unclear risk of bias (Biton 2014; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). Participants receiving brivaracetam were significantly more likely than those receiving placebo to experience a 50% or greater reduction in seizure frequency, and to achieve seizure freedom. Although participants receiving brivaracetam were significantly more likely to withdraw from treatment due to adverse events than those receiving placebo, the overall treatment withdrawal rate (withdrawal for any reason) was not significantly different between the two treatment groups. Moreover, the incidence of participants experiencing one or more adverse events was not significantly different between participants receiving brivaracetam and those given placebo. With regards to drug interactions, general consensus across all five included studies indicates that concomitant levetiracetam use diminishes the efficacy of brivaracetam with regards to both the responder rate and, more generally, the observed reduction in seizure frequency, despite no statistical analysis.
Subgroup analysis according to dosage suggested that no dose‐response relationship is associated with brivaracetam use. Notably, the effect size observed was fairly consistent across all doses. However, the subgroup analysis did provide some information regarding possible doses of brivaracetam for clinical use. Doses of 50 mg/d, 100 mg/d, and 200 mg/d brivaracetam were all associated with a significantly higher proportion of participants achieving a 50% or greater reduction in seizure frequency, as well as seizure freedom, compared to placebo. It is important to note that none of these doses was associated with a significantly higher treatment withdrawal rate for any reason or specifically due to adverse events experienced. A slightly higher incidence of participants reporting one or more adverse events was noted with use of 100 mg/d brivaracetam. It is interesting to note that 150 mg/d brivaracetam did not display a significant therapeutic effect compared to placebo. However, this subgroup yielded limited data, with only one study, involving only 104 participants, included in the subgroup analysis (Van Paesschen 2013). Consequently, this subgroup may have been underpowered; this could potentially explain the lack of efficacy noted.
The conclusions presented here should be applied cautiously due to the limited numbers of studies and participants included in each subgroup analysis.
Overall completeness and applicability of evidence
Although we did perform a subgroup analysis according to dose groups of brivaracetam, caution must be taken when interpreting and extrapolating the results. The number of participants included in each subgroup analysis ranged from 104 to 717 per subgroup analysis. This highlights that there could be possible inadequacies in statistical power for some of the subgroup analyses. As a consequence, this review can provide only limited information regarding the efficacy of specific brivaracetam doses.
We also intended to conduct a subgroup analysis involving the age of participants. Unfortunately, we were unable to perform this subgroup analysis as all six of the included studies utilised exclusively adult populations. We are therefore unable to comment on the efficacy of brivaracetam when used in children. Additionally, we are unable to adequately discuss the application of brivaracetam in drug‐resistant generalised epilepsy, although we included this population in the review. Notably, only the Kwan 2014 study included participants with drug‐resistant generalised epilepsy. Kwan 2014 did state that brivaracetam appeared to be more efficacious in participants with generalised epilepsy than in those with focal onset epilepsy. The small sample size of participants with generalised epilepsy, however, precluded any formal statistical analysis within the study and thus prevented any conclusions from being drawn. The finding does highlight the potential efficacy of brivaracetam in generalised epilepsy and emphasises the need for future research.
Quality of the evidence
We assessed two of the included studies to be at low risk of bias (French 2010; Klein 2015). Both studies described effective methods used for randomisation, allocation concealment, and blinding. We did not suspect attrition or reporting bias in either study. We assessed that the remaining four studies were at unclear risk of bias (Biton 2014; Kwan 2014; Ryvlin 2014; Van Paesschen 2013). We awarded each of the four studies an unclear risk of bias rating for one or two of the risk of bias domains. We assessed all other domains to be at low risk of bias. One study did not declare the method used for allocation concealment (Kwan 2014), whilst another study failed to adequately describe any method of blinding (Ryvlin 2014). We suspected two studies of selective reporting (Biton 2014; Van Paesschen 2013). Biton 2014 did not report data for the placebo group for one of the outcome measures, whilst Van Paesschen 2013 did not report all outcomes predefined in the trial protocol. We further assessed Kwan 2014 to be at unclear risk of other bias, namely, for using an unequal allocation ratio, which could lead to an exaggerated placebo effect.
As a result, during GRADE assessment, we downgraded the quality of evidence once for all outcomes due to concerns about unclear risk of bias across four of the included studies. We consequently rated the quality of evidence as moderate for the following outcomes: 50% responder rate and proportion of participants who experienced any adverse events. Notably, we also rated the quality of evidence for the outcome, seizure freedom, as moderate. In fact, we again downgraded the quality of evidence for seizure freedom due to very serious imprecision, resulting from the small number of events included within the outcome analysis. However, the downgrading was compensated for by the large effect size observed. This equated to an overall rating of moderate quality. For the remaining two outcomes ‐ treatment withdrawal for any reason and treatment withdrawal due to adverse events ‐ we rated the quality of evidence as low. We downgraded both outcomes once more for imprecision, again because of the small number of events constituting the analysis. For these two outcomes, the imprecision noted was not compensated for by a large effect size; consequently, the evidence remained assessed as of low quality.
We did consider downgrading the quality of evidence once again with regard to indirectness for all outcomes due to lack of data concerning the effect of add‐on brivaracetam in children and in patients with generalised epilepsy, specifically. However, we judged that the data provided by the included studies did sufficiently answer the original research question, that is, whether brivaracetam is efficacious and tolerable as an add‐on therapy for people with drug‐resistant epilepsy ‐ despite inclusion of no or limited data about these subgroups of participants. As a result, we did not think that indirectness was serious enough to permit downgrading the quality of evidence again. Instead, we emphasise that the findings reported are applicable only to adults, mainly to those with focal epilepsy. Findings might not necessarily be relevant or applicable to adults with generalised epilepsy.
As a result, we can be fairly confident that the conclusions made with regards to the outcomes ‐ 50% responder rate, seizure freedom, and proportion of participants likely to experience any adverse events ‐ are accurate. We are less certain about the accuracy of our observations concerning treatment withdrawal for any reason or specifically due to any adverse events experienced.
It is worth noting that all six studies were sponsored by UCB Pharma, the manufacturer of brivaracetam. Although it does not contribute to the risk of bias nor to GRADE assessment, this pharmaceutical sponsorship could potentially lead to funding bias. However, it is generally accepted that if a study is methodologically sound, and if the protocol is correctly adhered to, that study's conduct, and therefore findings, should not be affected by funding bias.
Potential biases in the review process
We are unaware of any sources of bias in our conduct of the review. As per the review protocol, we (two review authors) independently assessed the eligibility of studies identified by the search strategies for inclusion, independently extracted the relevant data, and independently completed both risk of bias and GRADE assessments. We requested all protocols as planned; however, we were provided with the trial protocol only for the Van Paesschen 2013 study. We also could not obtain missing data for the Biton 2014 study regarding the proportion of participants in the placebo group to experience one or more adverse events. Although both events could potentially bias the review, both instances were outside of our control.
Agreements and disagreements with other studies or reviews
The findings of our current review are consistent with the observations made in other systematic reviews, which similarly assessed the efficacy and tolerability of brivaracetam (Lattanzi 2016; Ma 2015; Tian 2015). These other systematic reviews likewise reported risk ratios for both the 50% responder rate and the seizure freedom rate. All review authors similarly concluded that brivaracetam is an efficacious add‐on therapy for drug‐resistant epilepsy. However, it is important to note that the systematic reviews identified specifically focused on the use of brivaracetam as an add‐on therapy for drug‐resistant focal epilepsy and, therefore, excluded participants with generalised epilepsy from their analyses. From this perspective, our review provides additional, novel information to that available in these other systematic reviews.
As observed here, in two of the reviews, the risk ratio for seizure freedom demonstrated an especially large effect for brivaracetam compared to placebo (Lattanzi 2016; Ma 2015). One review also completed a subgroup analysis according to dosage, and reported that any dose above 5 mg/d was associated with a significant therapeutic effect. In our review, we similarly observed that all doses of brivaracetam greater than 5 mg/d were associated with a significantly higher responder rate compared to placebo. However, we instead suggest that doses of 50 mg/d brivaracetam and greater are efficacious for managing drug‐resistant epilepsy. Doses of 50 mg/d and above of brivaracetam were consistently more effective than placebo across the two efficacy outcomes ‐ responder rate and seizure freedom. Neither 5 mg/d nor 20 mg/d brivaracetam was more efficacious than placebo with regard to seizure freedom.
With regards to drug interactions, Lattanzi 2016 further conducted a subgroup analysis to investigate the effect of levetiracetam status on responsiveness to brivaracetam. In accordance with our findings, Lattanzi 2016 emphasised that concomitant use of levetiracetam reversed the significant difference in the 50% responder rate normally observed with add‐on brivaracetam.
In addition to confirming the efficacy of brivaracetam, the other systematic reviews also assessed its tolerability. All three reviews emphasised that brivaracetam was well tolerated (Lattanzi 2016; Ma 2015; Tian 2015), and one review reported risk ratios for treatment withdrawal that were very similar to those reported here. Another review (Zhu 2017), which specifically investigated the safety and tolerability of brivaracetam, reported that brivaracetam was not significantly associated with serious adverse events nor treatment withdrawal for any reason or due to adverse events.
It is interesting to note that within our review, data taken from the French 2010 study appear to disagree with those from other included studies with regards to treatment withdrawal ‐ an outcome concerning tolerability. Specifically, French 2010 reported that treatment withdrawal for any reason and due to adverse events was greater amongst participants randomised to placebo than amongst those randomised to brivaracetam. Although the number of participants withdrawing from treatment during the study was low overall (placebo: 6 vs brivaracetam: 5), it is interesting to note that this study also reported the shortest treatment period (7 weeks vs 10 to 16 weeks in duration). Similarly, Van Paesschen 2013, which also reported a shorter treatment period compared to the other studies (10 weeks vs 12 to 16 weeks in duration), likewise reported a higher withdrawal rate for participants randomised to placebo compared to brivaracetam for treatment withdrawal for any reason. Length of the treatment period could thus potentially explain the heterogeneity observed.
It is apparent that the findings and conclusions of our current review regarding both efficacy and the safety profile of brivaracetam are consistent with those of currently available systematic reviews. This consequently generates further support for the argument that brivaracetam is effective in treating drug‐resistant epilepsy when used as an add‐on therapy.
Authors' conclusions
Implications for practice.
Moderate‐quality evidence shows that brivaracetam, when used as an add‐on for adults with drug‐resistant focal epilepsy, is effective in reducing seizure frequency and increasing the likelihood of people achieving seizure freedom. Limited information is available regarding the efficacy of brivaracetam in adults with drug‐resistant generalised epilepsy. However, a small sample trial suggested that brivaracetam could in fact display increased effectiveness in this population compared to when it is used in focal epilepsy. Additionally, our findings strongly suggest that brivaracetam should not be used in conjunction with concomitant levetiracetam due to the reduced efficacy reported.
Our current review suggests that a good tolerability profile is associated with brivaracetam. Evidence concerning treatment withdrawal ‐ an important outcome for determining drug safety ‐ was, however, of low quality and must therefore be interpreted cautiously. In contrast, evidence for the proportion of participants to experience any adverse events ‐ another outcome that contributes to drug safety ‐ was of moderate quality and demonstrated only a relatively slight increase in prevalence. We did not, however, investigate the prevalence of individual adverse events; this should be addressed in subsequent reviews.
We must again emphasise that the evidence for this review was derived from randomised controlled trials that exclusively studied adult populations, principally adult populations with drug‐resistant focal epilepsy ‐ not with generalised epilepsy. As a result, overall, this review shows that brivaracetam is a fairly tolerable and effective drug for use specifically in adults with drug‐resistant focal epilepsy.
Implications for research.
All current conclusions are based on relatively short‐term studies that have largely focused on populations with drug‐resistant focal epilepsy. More trials including participants with drug‐resistant generalised epilepsy are necessary for full assessment of whether brivaracetam also displays efficacy in this population, as suspected in this review. Additional trials should aim to incorporate multiple doses of brivaracetam to help ascertain a recommended specific dose for clinical use, and should be conducted over longer periods of time. Long‐term studies are required to assess the long‐term safety and tolerability of brivaracetam. After the safety profile of brivaracetam is ascertained, it would be recommended that additional studies should be conducted to determine the efficacy of brivaracetam in children. Together, these additional studies and subsequent meta‐analyses could more accurately inform clinical practice.
What's new
| Date | Event | Description |
|---|---|---|
| 1 August 2019 | Amended | Minor copyedits carried out |
Acknowledgements
We thank Graham Chan for his assistance in the search for relevant trials. We thank Qin Zhou, Cai‐you Hu, Wei Zhang, and Yong‐hong Huang for their contributions to the review protocol.
Appendices
Appendix 1. Cochrane Register of Studies (CRS Web) search strategy
1. (Brivaracetam):AB,KW,MC,MH,TI AND CENTRAL:TARGET
2. (monotherap* NOT (adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*)):TI AND CENTRAL:TARGET
3. #1 NOT #2
#3 AND >23/03/2017:CRSCREATED
Appendix 2. MEDLINE (Ovid) search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials (Lefebvre 2011).
1. Brivaracetam.tw.
2. exp Epilepsy/
3. exp Seizures/
4. (epilep$ or seizure$ or convuls$).tw.
5. 2 or 3 or 4
6. exp *Pre‐Eclampsia/ or exp *Eclampsia/
7. 5 not 6
8. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
9. clinical trials as topic.sh.
10. trial.ti.
11. 8 or 9 or 10
12. exp animals/ not humans.sh.
13. 11 not 12
14. 1 and 7 and 13
15. (monotherap$ not (adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$)).ti.
16. 14 not 15
17. remove duplicates from 16
18. limit 17 to ed=20170323‐20181009
19. 17 not (1$ or 2$).ed.
20. 19 and (2017$ or 2018$).dt.
21. 18 or 20
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsy | Brivaracetam | First posted from 03/23/2017 to 10/09/2018
Appendix 4. ICTRP search strategy
Condition: epilepsy
Intervention: Brivaracetam
Recruitment status: All
Date of registration between 23/03/2017 and 09/10/2018
Phases: All
Data and analyses
Comparison 1. Brivaracetam vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% or greater reduction in seizure frequency (responder rate) | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 5 mg/d | 2 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.97, 2.40] |
| 1.2 20 mg/d BRV | 3 | 504 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.18, 2.27] |
| 1.3 50 mg/d BRV | 4 | 611 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 [1.50, 2.66] |
| 1.4 100 mg/d BRV | 2 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.81 [1.42, 2.30] |
| 1.5 150 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [0.86, 3.65] |
| 1.6 200 mg/d BRV | 1 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.33, 2.33] |
| 1.7 All doses | 6 | 2411 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.81 [1.53, 2.14] |
| 2 Seizure freedom | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 5 mg/d BRV | 2 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.87 [0.65, 22.96] |
| 2.2 20 mg/d BRV | 3 | 551 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.98 [0.65, 13.61] |
| 2.3 50 mg/d BRV | 4 | 611 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.39 [1.42, 20.49] |
| 2.4 100 mg/d BRV | 2 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.19 [1.93, 26.85] |
| 2.5 150 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.32, 27.91] |
| 2.6 200 mg/d BRV | 1 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.24 [1.16, 23.68] |
| 2.7 All doses | 6 | 2411 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.89 [2.30, 15.13] |
| 3 Treatment withdrawal | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 5 mg/d BRV | 2 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [0.93, 4.09] |
| 3.2 20 mg/d BRV | 3 | 504 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.40, 1.55] |
| 3.3 50 mg/d BRV | 4 | 611 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.56, 1.77] |
| 3.4 100 mg/d BRV | 2 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.88, 2.35] |
| 3.5 150 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.18, 3.19] |
| 3.6 200 mg/d BRV | 1 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.89, 2.88] |
| 3.7 All doses | 6 | 2411 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.94, 1.74] |
| 4 Proportion of participants who experienced adverse events leading to treatment withdrawal | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 5 mg/d BRV | 2 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.06 [0.71, 5.96] |
| 4.2 20 mg/d BRV | 3 | 504 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.46, 2.72] |
| 4.3 50 mg/d BRV | 4 | 611 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.58, 2.76] |
| 4.4 100 mg/d BRV | 2 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.01, 3.59] |
| 4.5 150 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.26, 8.61] |
| 4.6 200 mg/d BRV | 1 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [0.83, 3.82] |
| 4.7 All doses | 6 | 2411 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [1.02, 2.33] |
| 5 Proportion of participants who experienced any adverse events | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 5 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.67, 1.39] |
| 5.2 20 mg/d BRV | 2 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.86, 1.30] |
| 5.3 50 mg/d BRV | 3 | 410 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.91, 1.25] |
| 5.4 100 mg/d BRV | 2 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [1.04, 1.31] |
| 5.5 150 mg/d BRV | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.73, 1.22] |
| 5.6 200 mg/d BRV | 1 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.99, 1.29] |
| 5.7 All doses | 5 | 2011 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [1.00, 1.17] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Biton 2014.
| Methods |
Study design: phase 3, randomised, double‐blind, PBO‐controlled, parallel‐group, multi‐centre Countries: Australia, Brazil, Canada, Mexico, and the USA Duration: 1. Prospective baseline period (8 weeks) 2. Treatment period (12 weeks) 3. Down‐titration period (1 week) or entry into long‐term open‐label follow‐up study |
|
| Participants |
Randomised population BRV 50 mg/d = 102 BRV 20 mg/d = 100 BRV 5 mg/d = 99 PBO = 99 ITT populationa: BRV 50 mg/d = 101 BRV 20 mg/d = 100 BRV 5 mg/d = 97 PBO = 98 mITT populationb: BRV 50 mg/d = 101 BRV 20 mg/d = 99 BRV 5 mg/d = 96 PBO = 96 Safety populationc: BRV 50 mg/d = 101 BRV 20 mg/d = 100 BRV 5 mg/d = 97 PBO = 98 Age (mean and SD)d: ≥ 16 to 70 years BRV 50 mg/d = 38.9 (12.3) BRV 20 mg/d = 37.3 (13.3) BRV 5 mg/d = 38.9 (11.6) PBO = 37.5 (12.6) Gender, male, n (%)d: BRV 50 mg/d = 51 (50.5%) BRV 20 mg/d = 52 (52.0%) BRV 5 mg/d = 49 (50.5%) PBO = 43 (43.9%) Ethnicity white, n (%)d: BRV 50 mg/d = 77 (76.2%) BRV 20 mg/d = 70 (70.0%) BRV 5 mg/d = 73 (75.3%) PBO = 66 (67.3%) Types of seizure: drug‐resistant focal onset seizures |
|
| Interventions | All treatment groups received their respective treatment in 2 equally divided doses per day: BRV 50 mg/d (BID) BRV 20 mg/d (BID) BRV 5 mg/d (BID) PBO (BID) |
|
| Outcomes |
Primary outcomes: 1. Per cent reduction over PBO in adjusted FOS frequency per week during the treatment period 2. Per cent reduction over PBO in 28‐day adjusted FOS frequency during the treatment period Secondary outcomes: 1. ≥ 50% responder rate based on per cent reduction in seizure frequency/week from baseline to the treatment period 2. Seizure freedom rate Safety and tolerability outcomes: 1. Adverse events (AEs) and severity 2. Laboratory tests 3. Physical and neurological examination findings 4. Vital signs 5. Electrocardiography (ECG) recordings |
|
| Notes | Trial registry number: N01253, NCT00464269 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a central randomization method (random permuted blocks) that stratified for concomitant LEV use at study entry ("yes" or "no")" |
| Allocation concealment (selection bias) | Low risk | Quote: "treatment was assigned via an Interactive Voice Response System using a central randomization method" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "matching placebo" was used to maintain blinding Quote: "patients and investigators were blinded to treatment" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: patients acted as outcome assessors; patients self‐reported seizure frequency by completion of "seizure daily record card" and were effectively blinded by matching placebo. Investigators, including data analysts/statisticians, were also effectively blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported; modified intent‐to‐treat analysis was conducted, resulting in the exclusion of 3 patients for non‐compliance and 1 patient as a clinical outlier. Due to the small number of patients excluded and the valid reasoning provided, low risk of bias was still awarded |
| Selective reporting (reporting bias) | Unclear risk |
Comment: protocol was not provided; all outcomes defined in the methods were reported in the results; however, no data were reported for the number of patients taking placebo who reported at least 1 adverse event Quote: "the incidence of treatment‐emergent adverse events (TEAEs) was similar in all four treatment groups. At least one TEAE was reported during the treatment period of 69 (71.1%) of 97 patients on BRV 5 mg/day, 79 (79.0%) of 100 on BRV 20 mg/day, and 76 (75.2%) of 101 on BRV 50 mg/day" |
| Other bias | Low risk | Comment: none detected |
French 2010.
| Methods |
Study design: phase 2b, randomised, double‐blind, PBO‐controlled, parallel‐group, multi‐centre Countries: Brazil, India, Mexico, and the USA Duration: 1. Prospective baseline period (4 weeks) 2. Treatment period w/o up‐titration (7 weeks) 3. 2‐week drug‐free period or entry into long‐term open‐label follow‐up study |
|
| Participants |
Randomised population: BRV 50 mg/d = 52 BRV 20 mg/d = 52 BRV 5 mg/d = 50 PBO = 54 ITT populationa: BRV 50 mg/d = 52 BRV 20 mg/d = 52 BRV 5 mg/d = 50 PBO = 54 Safety populationc: BRV 50 mg/d = 52 BRV 20 mg/d = 52 BRV 5 mg/d = 50 PBO = 54 Age (mean and SD)d: ≥ 16 to 65 years BRV 50 mg/d = 30.9 (11.6) BRV 20 mg/d = 35.3 (13.7) BRV 5 mg/d = 32.7 (12.2) PBO = 33.6 (11.3) Gender, male, n (%)d: BRV 50 mg/d = 28 (53.8) BRV 20 mg/d = 28 (53.8) BRV 5 mg/d = 30 (60.0) PBO = 24 (44.4) Ethnicity white, n (%)d: BRV 50 mg/d = 12 (23.1) BRV 20 mg/d = 22 (42.3) BRV 5 mg/d = 16 (32.0) PBO = 23 (42.6) Types of seizure: drug‐resistant focal onset seizures |
|
| Interventions | All treatment groups received tablets, administered in 2 equally divided doses per day, without up‐titration BRV 50 mg/d (BID) BRV 20 mg/d (BID) BRV 5 mg/d (BID) PBO (BID) |
|
| Outcomes |
Primary outcome: 1. Per cent reduction over PBO in 28‐day adjusted FOS frequency during the treatment period Secondary outcomes: 1. Absolute and percentage reduction from baseline in weekly FOS frequency during the treatment period 2. ≥ 50% responder rate for FOS frequency/week from baseline during the treatment period 3. Seizure freedom rate Safety and tolerability outcomes: 1. Adverse events (AEs) 2. Laboratory tests 3. Physical and neurological examination findings 4. Vital signs 5. Electrocardiography (ECG) recordings |
|
| Notes | Trial registry number: N01193, NCT00175825 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "central randomization (random permuted blocks)... stratified for the intake of LEV... and of CBZ" |
| Allocation concealment (selection bias) | Low risk | Quote: "once a patient was eligible to be randomized, the investigator called the Central Randomization Center to receive a kit number to assign to the patient" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the study was blinded, by use of matching placebo tablets which were identical in shape, size, and color to BRV tablets" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "efficacy assessments were made from information recorded by the patients on daily record cards" Comment: patients were the outcome assessors and were adequately blinded throughout the study; moreover, the study was double‐blind, meaning that investigators, including those responsible for data analysis, would also have been effectively blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported and intention‐to‐treat analysis was conducted; however, a modified population was actually used. Two participants were excluded as they did not take at least 1 dose of study drug. Due to the small number of patients excluded and the valid reasoning, a small risk of bias was still awarded |
| Selective reporting (reporting bias) | Low risk | Comment: protocol was not provided; however, all outcomes defined in methods were reported in results |
| Other bias | Low risk | Comment: none detected |
Klein 2015.
| Methods |
Study design: phase 3, randomised, double‐blind, PBO‐controlled, parallel‐group, multi‐centre Countries: North America, Western Europe, Eastern Europe, Latin America, and Asia Duration: 1. Prospective baseline period (8 weeks) 2. Treatment period (12 weeks) 3. Down‐titration period (4 weeks) 4. Drug‐free period (2 weeks) or entry into a long‐term follow‐up study |
|
| Participants |
Randomised population: BRV 200 mg/d = 251 BRV 100 mg/d = 254 PBO = 263 ITT populationa: BRV 200 mg/d = 249 BRV 100 mg/d = 252 PBO = 259 Safety populationc: BRV 200 mg/d = 250 BRV 100 mg/d = 253 PBO = 261 Age (mean and SD)d: ≥ 16 to 80 years BRV 200 mg/d = 39.8 (12.8) BRV 100 mg/d = 39.1 (13.4) PBO = 39.8 (12.5) Gender, female, n (%)d: BRV 200 mg/d = 117 (46.8%) BRV 100 mg/d = 151 (59.7%) PBO = 128 (49.0%) Ethnicity white, n (%)d: BRV 200 mg/d = 182 (72.8%) BRV 100 mg/d = 182 (71.9%) PBO = 189 (72.4%) Types of seizure: drug‐resistant focal onset seizures |
|
| Interventions | All treatment groups received oral film‐coated tablets, administered in 2 equally divided doses per day, without up‐titration BRV 200 mg/d (BID) BRV 100 mg/d (BID) PBO (BID) |
|
| Outcomes |
Primary outcomes: 1. Per cent reduction over PBO in 28‐day adjusted FOS frequency during the treatment period 2. ≥ 50% responder rate based on per cent reduction in seizure frequency from baseline to the treatment period Secondary outcomes: 1. Per cent reduction in seizure frequency from baseline to the treatment period 2. Categorised per cent reduction from baseline in seizure frequency over the treatment period 3. Seizure freedom rate Safety and tolerability outcomes: 1. Adverse events (AEs) 2. Laboratory tests 3. Vital signs 4. Electrocardiography (ECG) recordings |
|
| Notes | Trial registry number: N01358, NCT01261325 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a 1:1:1 central randomization (random permuted blocks with a block size of three) stratified by country, LEV status (never used vs. prior use), and number of AEDs previously used or discontinued prior to study entry" |
| Allocation concealment (selection bias) | Low risk | Quote: "patients were assigned to a treatment group at enrollment by an interactive voice/computer response system (IVRS), which was accessed by the investigator" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "all personnel who were involved with the study were blinded to the patients’ treatment... Oral film‐coated tablets of BRV 10, 25, and 50 mg and matching PBO tablets were used; these tablet strengths were used both to help maintain the blinding" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: patients reported seizure frequency using seizure diaries and were therefore the outcome assessors; patients were sufficiently blinded by the matching placebo. Investigators, including data analysts, were also effectively blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported and intention‐to‐treat analysis conducted; 8 participants were excluded from the ITT population for the reasons specified: discontinuation for unspecified reasons before study drug administration (n = 4), loss to follow‐up (n = 1), discontinuation due to a TEAE (n = 2), and withdrawal of consent (n = 1). Due to the small number of participants excluded, low risk of bias was still awarded |
| Selective reporting (reporting bias) | Low risk | Comment: protocol was not provided; however, all outcomes defined in methods were reported in results |
| Other bias | Low risk | Comment: none detected |
Kwan 2014.
| Methods |
Study design: phase 3, randomised, double‐blind, PBO‐controlled, parallel‐group, flexible‐dose, multi‐centre Countries: Austria, Belgium, Czech Republic, Germany, Hong Kong, India, Italy, Norway, Republic of South Africa, Russian Federation, Singapore, South Korea, Sweden, Taiwan, and Ukraine Duration: 1. Prospective baseline period (4 weeks) 2. Treatment period (16 weeks) including 8‐week dose‐finding and 8‐week maintenance 3. Down‐titration period (2 weeks) and drug‐free period (2 weeks) or entry into a long‐term follow‐up study |
|
| Participants |
Randomised population: BRV = 359 PBO = 121 ITT populationa: BRV = 359 PBO = 121 Safety populationc: BRV = 359 PBO = 121 Age (mean and SD)d: ≥ 16 to 70 years BRV = 35.6 (11.5) PBO = 36.5 (11.5) Gender, male, n (%)d: BRV = 181 (50.4) PBO = 69 (57.0) Ethnicity white, n (%)d: BRV = 209 (58.2) PBO = 69 (57.0) Types of seizure: drug‐resistant focal onset or generalised epilepsy |
|
| Interventions | All treatment groups received tablets administered in 2 equally divided doses per day: BRV 20, 50, 100, 150 mg/d (BID) PBO (BID) For participants randomised to BRV, BRV was initiated at 20 mg/d. Participants were then up‐titrated in a stepwise manner to 50, 100, or 150 mg/d at 2‐week intervals based on the investigator’s assessment of efficacy and tolerability |
|
| Outcomes |
Safety and tolerability outcomes: 1. Adverse events (AEs) 2. Discontinuations due to AEs 3. Vital signs 4. Physical and neurological examination findings 5. Laboratory tests 6. Electrocardiography (ECG) recordings Primary efficacy outcome: 1. Per cent reduction in baseline‐adjusted FOS frequency/week during the treatment period over PBO Secondary outcomes: 1. Median per cent reduction from baseline in FOS frequency/week 2. ≥ 50% responder rate in FOS frequency/week 3. Seizure freedom rate 4. Time to first, fifth, and 10th focal seizure |
|
| Notes | Trial registry number N01254, NCT00504881 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized 3:1 in random permuted blocks to BRV or PBO at the end of the baseline period. Randomization was stratified by epilepsy type (focal or generalized) (International League Against Epilepsy, 1989), concomitant levetiracetam (LEV) use (yes or no), and geographic region" |
| Allocation concealment (selection bias) | Unclear risk | Comment: details regarding allocation concealment were not provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "matching PBO tablets" Comment: appropriate measures were taken to maintain blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
Quote: "the date and number of seizures were recorded using a daily record card" Comment: outcomes were self‐reported by the participants who remained appropriately blinded throughout the study; moreover, the study was double‐blind, meaning that investigators, including those responsible for data analysis, would also have been effectively blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported and intent‐to‐treat analysis conducted, which correctly included all randomised participants |
| Selective reporting (reporting bias) | Low risk | Comment: protocol was not provided; however, all outcomes defined in methods were reported in results |
| Other bias | Unclear risk |
Quote: "patients were randomized 3:1 in random permuted blocks to BRV or PBO" Comment: 3:1 randomisation ratio produces uneven treatment group sizes, which reduces the statistical power and can augment the placebo effect (Hey 2014) |
Ryvlin 2014.
| Methods |
Study design: phase 3, randomised, double‐blind, PBO‐controlled, multi‐centre Countries: Poland, India, France, Germany, Spain, Italy, Switzerland, Hungary, Finland, The Netherlands, Belgium, and the United Kingdom Duration: 1. Prospective baseline period (8 weeks) 2. Treatment period (12 weeks) 3. Down‐titration period (2 weeks) and drug‐free period (2 weeks) or entry into a long‐term follow‐up study |
|
| Participants |
Randomised population: BRV 100 mg/d = 100 BRV 50 mg/d = 99 BRV 20 mg/d = 99 PBO = 100 ITT populationa: BRV 100 mg/d = 100 BRV 50 mg/d = 99 BRV 20 mg/d = 99 PBO = 100 Safety populationc: BRV 100 mg/d = 100 BRV 50 mg/d = 99 BRV 20 mg/d = 99 PBO = 100 Age (mean and SD)d: ≥ 16 to 70 years BRV 100 mg/d = 38.0 (13.1) BRV 50 mg/d = 38.9 (13.6) BRV 20 mg/d = 35.7 (12.5) PBO = 36.4 (13.0) Gender, male, n (%)d: BRV 100 mg/d = 58 (58.0) BRV 50 mg/d = 54 (54.5) BRV 20 mg/d = 61 (61.6) PBO = 54 (54.0) Ethnicity white, n (%)d: BRV 100 mg/d = 76 (76.0) BRV 50 mg/d = 76 (76.8) BRV 20 mg/d = 76 (76.8) PBO = 77 (77.0) Type of seizure: drug‐resistant focal onset seizures |
|
| Interventions | All treatment groups received their respective treatment in 2 equally divided doses per day: BRV 100 mg/d (BID) BRV 50 mg/d (BID) BRV 20 mg/d (BID) PBO (BID) |
|
| Outcomes |
Primary outcome: 1. Per cent reduction over PBO in baseline‐adjusted FOS frequency/week over the treatment period Secondary outcomes: 1. Median per cent reduction in seizure frequency/week from baseline to the treatment period 2. ≥ 50% responder rate based on per cent reduction in seizure frequency/week from baseline to the treatment period 3. Seizure freedom rate Safety and tolerability outcomes: 1. Adverse events (AEs) 2. Laboratory tests 3. Physical and neurological examination findings 4. Vital signs 5. Electrocardiography (ECG) recordings |
|
| Notes | Trial registry number: N01252, NCT00490035 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "central randomisation... stratified by geographic region and concomitant use of LEV" |
| Allocation concealment (selection bias) | Low risk | Quote: "treatment was assigned using central randomization via an interactive voice response system (IVRS)" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind" Comment: no evidence or explanation of blinding provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "patients recorded the occurrence of seizures on daily record cards" Comment: participants were responsible for the self‐reporting of outcome measures; however, no information is provided on blinding of participants or study personnel |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported and intent‐to‐treat analysis conducted; 1 participant was excluded from any analysis; however, that participant died from a subdural haematoma before taking any study drug |
| Selective reporting (reporting bias) | Low risk | Comment: protocol was not provided; however, all outcomes defined in methods were reported in results |
| Other bias | Low risk | Comment: none detected |
Van Paesschen 2013.
| Methods |
Study design: phase 2b, randomised, double‐blind, PBO‐controlled, parallel‐group, multi‐centre Countries: Belgium, Czech Republic, Finland, France, Germany, The Netherlands, Poland, Spain, and the United Kingdom Duration: 1. Prospective baseline period (4 weeks) 2. Treatment period (10 weeks: 3 weeks up‐titration and 7 weeks maintenance) 3. Conversion period (2 weeks): entry into a long‐term open‐label follow‐up study or down‐titration (2 weeks) |
|
| Participants |
Randomised population: BRV 150 mg/d = 52 BRV 50 mg/d = 53 PBO = 52 ITT populationa: BRV 150 mg/d = 52 BRV 50 mg/d = 53 PBO = 52 Safety populationc: BRV 150 mg/d = 52 BRV 50 mg/d = 53 PBO = 52 Age (mean and SD)d: ≥ 16 to 65 years BRV 150 mg/d = 34.4 (10.1) BRV 50 mg/d = 38.2 (12.1) PBO = 40.0 (11.7) Gender, male, n (%)d: BRV 150 mg/d = 21 (40.4) BRV 50 mg/d = 24 (45.3) PBO = 25 (48.1) Ethnicity white, n (%)d: BRV 150 mg/d = 52 (100.0) BRV 50 mg/d = 53 (100.0) PBO = 51 (98.1) Type of seizure: drug‐resistant focal onset seizures |
|
| Interventions | All treatment groups received their respective treatment via oral capsules in 2 equally divided doses per day: BRV 150 mg/d (BID) BRV 50 mg/d (BID) PBO (BID) Participants randomised to BRV 150 mg/d began the up‐titration period on a dose of 50 mg/d. After 1 week, the dosage was increased to 100 mg/d and was then increased again to 150 mg/d after 2 weeks. Patients were permitted 1 fallback during the maintenance period to 100 mg/d Participants randomised to BRV 50 mg/d started at a dose of 25 mg/d and were up‐titrated to 50 mg/d after 1 week. They were again permitted 1 fallback to 25 mg/d during the maintenance period. Participants randomised to placebo continued to receive placebo during the up‐titration and maintenance periods |
|
| Outcomes |
Primary outcome: 1. Per cent reduction in baseline‐adjusted FOS frequency/week over PBO during the maintenance period Secondary outcomes: 1. Reduction in FOS frequency/week over PBO during the treatment period 2. Per cent reduction from baseline in FOS frequency/week (maintenance and treatment periods) 3. ≥ 50% responder rate in FOS seizure frequency from baseline during maintenance and treatment periods 4. Seizure freedom rate Safety and tolerability outcomes: 1. Treatment‐emergent adverse events (TEAEs) 2. Physical and neurological examinations 3. Vital signs 4. Clinical laboratory tests 5. Electrocardiography (ECG) recordings |
|
| Notes | Trial registry number: N01114, NCT00175929 Sponsored by the manufacturer of BRV (UCB Pharma) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "central randomization method (random permuted blocks) stratified for concomitant use of LEV... and carbamazepine (CBZ)" |
| Allocation concealment (selection bias) | Low risk | Quote (from protocol): "each investigator will receive numbered subjects’ kits. When a subject is determined to be eligible for randomization (at visit 2), the Investigator or designee will call the Central Randomization Center (CRC) and will be assigned a subject’s kit number, according to the operating manual given by CRC" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "matching placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "efficacy assessments were made using data recorded by the patients on daily record cards and assessed by the investigator at each study visit" Comment: participants were adequately blinded by matching placebo; the study was double‐blind, meaning that investigators, including those responsible for data analysis, would also have been blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition was reported and intent‐to‐treat analysis conducted; no randomised participants were excluded from the ITT population |
| Selective reporting (reporting bias) | Unclear risk |
Comment: protocol was provided; not all outcomes defined in the protocol are reported in the journal article Quote: "secondary efficacy outcomes included..." |
| Other bias | Low risk | Comment: none detected |
AE: adverse event; AED: antiepileptic drug; BID: twice a day; BRV: brivaracetam; CBZ: carbamazepine; ECG: electrocardiogram; FOS: focal onset seizure; ITT: intention‐to‐treat; IVRS: interactive voice response system; LEV: levetiracetam; mITT: modified intention‐to‐treat; PBO: placebo; TEAEs: treatment‐emergent adverse effects.
aITT population was defined as all randomised patients who received at least one dose (≥ 1) of study drug, with the exception of Klein 2015, who defined ITT as all randomised patients who received at least one dose (≥ 1) of study drug and had at least one (≥ 1) post‐baseline seizure diary entry.
bBiton 2014 used a modified intent‐to‐population, excluding four participants (three for extreme non‐compliance and one as a clinical outlier).
cSafety population was defined as all randomised patients who received at least one dose (≥ 1) of study drug in Klein 2015. For all other studies, the safety population was identical to the intent‐to‐treat population.
dCalculated using the safety population.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Lacroix 2007 | Study was a meta‐analysis of 2 trials already included in the review |
Characteristics of ongoing studies [ordered by study ID]
NCT03083665.
| Trial name or title | A Randomized, Double‐blind, Placebo‐controlled, Multicenter, Parallel‐group Study to Evaluate the Efficacy and Safety of Adjunctive Brivaracetam in Asian Subjects (≥ 16 to 80 Years of Age) With Partial Seizures With or Without Secondary Generalization |
| Methods | Randomised, double‐blind, placebo‐controlled, multi‐centre study with parallel‐group design Countries: Japan, Malaysia, Philippines, Singapore, Taiwan, and Thailand |
| Participants |
Age: 16 to 80 years Type of seizure: uncontrolled focal onset seizures with or without secondary generalisation |
| Interventions | All treatment groups received tablets, administered in 2 equally divided doses per day, without up‐titration Film‐coated tablets BRV 50 mg/d PBO |
| Outcomes |
Primary outcome: 1. Per cent change in FOS frequency during the 12‐week treatment period Secondary outcomes: 1. ≥ 50% responder rate based on FOS frequency per 28 days from baseline to the treatment period 2. Per cent change in FOS frequency per 28 days from baseline to treatment period 3. Categorised per cent change in FOS frequency per 28 days from baseline to treatment period 4. All seizure frequency (focal, generalised, and unclassified epileptic seizures) per 28 days during the 12‐week treatment period 5. Percentage of participants who are seizure free (focal, all epileptic seizures) during the 12‐week treatment period 6. Time to nth (first, fifth, tenth) focal seizure during the 12‐week treatment period Safety and tolerability outcomes: 1. Brivaracetam plasma concentration 2. Adverse events (AEs) and severity 2. Laboratory tests 3. Electrocardiogram (ECG) 4. Vital signs 5. Physical and neurological examination findings 6. Mental and psychiatric status |
| Starting date | 22 August 2017 |
| Contact information | UCBCares@ucb.com |
| Notes | Sponsored by UCB Pharma |
AE: adverse event; BRV: brivaracetam; ECG: electrocardiography; FOS: focal‐onset seizures; PBO: placebo.
Differences between protocol and review
Review authorship has changed since publication of the review protocol. Rebecca Bresnahan and Mariangela Panebianco have since been instated as two review authors, with Rebecca Bresnahan primarily responsible for the conduct and reporting of the review. Qin Zhou, Cai‐you Hu, Wei Zhang, and Yong‐hong Huang remain acknowledged for their writing of the original protocol and for their contribution to the background section and methods section of the current review, which we adapted from the original review protocol.
We had stated in the protocol that we would assess funnel plot asymmetry as an indication of publication bias. However, our review included fewer than 10 studies, so we did not produce any funnel plots for defined outcomes.
We had further specified that we would conduct subgroup analyses according to the different dose groups of brivaracetam, as well as the different age groups of participants. However, we were able to conduct subgroup analysis only according to dose groups. All of the included studies comprised purely adult patient populations; consequently, subgroup analysis according to age group was not possible.
Finally, we had planned to conduct sensitivity analyses. Specifically, we had intended to repeat the meta‐analyses whilst excluding unpublished studies and then whilst excluding studies that had been published only as abstracts. All of the included studies were published as full‐length journal articles; therefore, neither sensitivity analysis was necessary.
Contributions of authors
Rebecca Bresnahan: assessed study eligibility and performed data extraction and risk of bias assessment. Responsible for the primary conduct and writing of this current review update. Mariangela Panebianco: assessed study eligibility and performed data extraction and risk of bias assessment for this current review update. Anthony Marson: arbitrated discussions when necessary.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research (NIHR), UK.
This review was supported by the National Institute for Health Research, via Cochrane Programme Grant funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Declarations of interest
Rebecca Bresnahan: nothing to declare. Mariangela Panebianco: nothing to declare. Anthony Marson: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
Edited (no change to conclusions)
References
References to studies included in this review
Biton 2014 {published data only}
- Biton V, Berkovic SF, Abou‐Khalil B, Sperling MR, Johnson ME, Lu S. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double‐blind, placebo‐controlled trial. Epilepsia 2014;55(1):57‐66. [DOI] [PubMed] [Google Scholar]
- NCT00464269. Double‐blind, Randomized Study Evaluating the Efficacy and Safety of Brivaracetam in Adults With Partial Onset Seizures [An International, Double‐blind, Parallel‐group, Placebo‐controlled, Randomized Study: Evaluation of the Efficacy and Safety of Brivaracetam in Subjects (>= 16 to 70 Years Old) With Partial Onset Seizures]. https://clinicaltrials.gov/ct2/show/NCT00464269 (first received 23 April 2007).
- Pack AM. Brivaracetam, a novel antiepileptic drug: is it effective and safe? Results from one phase III randomized trial. Epilepsy Currents 2014;14(4):196‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
French 2010 {published data only}
- French J, Rosenstiel P. Efficacy and tolerability of brivaracetam as adjunctive treatment for adults with refractory partial‐onset seizures. Epilepsia 2007;48 Suppl 7:78. [Google Scholar]
- French JA, Brodsky A, Rosenstiel P, Brivaracetam N01193 Study Group. Efficacy and tolerability of 5, 20 and 50 mg/day brivaracetam (UCB 34714) as adjunctive treatment in adults with refractory partial‐onset seizures. Epilepsia 2007;48 Suppl 6:400. [Google Scholar]
- French JA, Costantini C, Brodsky A, Rosenstiel P. Adjunctive brivaracetam for refractory partial‐onset seizures: a randomized, controlled trial. Neurology 2010;75(6):519‐25. [DOI] [PubMed] [Google Scholar]
- NCT00175825. A Dose‐ranging Study With Brivaracetam in Patients From 16 to 65 Years With Refractory Partial Onset Seizures. [A Multicenter, Double‐blind, Randomized, Placebo‐controlled, 4 Parallel Groups, Dose‐ranging Trial Evaluating the Efficacy and Safety of Brivaracetam Used as Adjunctive Treatment at Doses of 5, 20 and 50 mg/Day in b.i.d. Administration (Oral Tablets of 2.5 or 10 mg) for a Maximum of 7 Weeks in Subjects From 16 to 65 Years With Refractory Epilepsy Suffering From Partial Onset Seizures Whether or Not Secondarily Generalized]. https://clinicaltrials.gov/ct2/show/NCT00175825 (first received 15 September 2005).
Klein 2015 {published data only}
- Klein P, Doty P, Elmoufti S, Whitesides J, Gold M. Predictors of response in patients with epilepsy in a double‐blind, placebo‐controlled study of brivaracetam. Epilepsia 2015;56 Suppl 1:111. [Google Scholar]
- Klein P, Schiemann J, Sperling M, Whitesides J, Liang W, Stalvey T. A randomized, double‐blind, placebo‐controlled, multicenter, parallel‐group study to evaluate the efficacy and safety of brivaracetam in adult patients with partial onset seizures. Epilepsy Currents 2015;15 Suppl 1:379. [DOI] [PubMed] [Google Scholar]
- Klein P, Schiemann J, Sperling MR, Whitesides J, Liang W, Stalvey T, et al. A randomized, double‐blind, placebo‐controlled, multicenter, parallel‐group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial‐onset seizures. Epilepsia 2015;56(12):1890‐8. [DOI] [PubMed] [Google Scholar]
- NCT01261325. Brivaracetam Efficacy and Safety Study in Subjects With Partial Onset Seizures (BRITE) [A Randomized, Double‐blind, Placebo‐controlled, Multicenter, Parallel‐group Study to Evaluate the Efficacy and Safety of Brivaracetam in Subjects (≥16 to 80 Years Old) With Partial Onset Seizures]. https://clinicaltrials.gov/ct2/show/NCT01261325 (first received 16 December 2010).
Kwan 2014 {published data only}
- EUCTR2006‐006346‐34‐BE. An international, randomized, double‐blind, parallel‐group, placebo‐controlled, flexible dose study: evaluation of the safety and efficacy of brivaracetam in subjects (>=16 to 70 years old) suffering from localization‐related or generalized epilepsy. [An international, randomized, double‐blind, parallel‐group, placebo‐controlled, flexible dose study: evaluation of the safety and efficacy of brivaracetam in subjects (>=16 to 70 years old) suffering from localization‐related or generalized epilepsy.]. https://www.clinicaltrialsregister.eu/ctr‐search/trial/2006‐006346‐34/BE (first received 28 March 2007).
- Kwan P, Johnson M, Merschhemke M, Lu S. Adujunctive brivaracetam in adults with uncontrolled generalized seizures: sub‐population analysis of the results of a randomized, double‐blind, placebo‐controlled trial [abstract no: 1.267]. Proceedings of the 64th Annual Meeting of the American Epilepsy Society; 2010 December 3‐7; San Antonio (TX) [accessed 14 February 2011]. San Antonio, Texas, 2010.
- Kwan P, Johnson ME, Falter U, Pietteur VB, Brodsky AC, Rosenstiel P. Safety and tolerability of brivaracetam as adjunctive treatment in adults with refractory epilepsy: randomized, double‐blind, placebo‐controlled trial [abstract no: 1.219]. Epilepsia 2009;50 Suppl 11:107‐8. [Google Scholar]
- Kwan P, Johnson ME, Merschhemke M, Lu S. Safety and tolerability of adjunctive brivaracetam in adults with uncontrolled epilepsy: randomized, double‐blind, placebo‐controlled trial [abstract no: p514]. Epilepsia 2010;51 Suppl 4:152. [Google Scholar]
- Kwan P, Trinka E, Paesschen W, Rektor I, Johnson ME, Lu S. Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: results of a phase III, double‐blind, randomized, placebo‐controlled, flexible‐dose trial. Epilepsia 2014;55(1):38‐46. [DOI] [PubMed] [Google Scholar]
- NCT00504881. Brivaracetam as Add‐on Treatment in Adolescents and Adults Suffering From Epilepsy [An International, Randomized, Double‐blind, Parallel‐group, Placebo‐controlled, Flexible Dose Study: Evaluation of the Safety and Efficacy of Brivaracetam in Subjects (≥ 16 to 70 Years Old) Suffering From Localization‐related or Generalized Epilepsy.]. https://clinicaltrials.gov/ct2/show/NCT00504881 (first received 20 July 2007).
Ryvlin 2014 {published data only}
- NCT00490035. Double‐blind, Randomized Study Evaluating the Efficacy and Safety of Brivaracetam in Adults With Partial Onset Seizures [A Multi‐center, Double‐blind, Parallel‐group, Placebo Controlled, Randomized Study: Evaluation of the Efficacy and Safety of Brivaracetam in Subjects (>= 16 to 70 Years Old) With Partial Onset Seizures.]. https://clinicaltrials.gov/ct2/show/NCT00490035 (first received 22 June 2007).
- Ryvlin P, Werhahn KJ, Blaszczyk B, Johnson ME, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double‐blind, randomized, placebo‐controlled trial. Epilepsia 2014;55(1):47‐56. [DOI] [PubMed] [Google Scholar]
Van Paesschen 2013 {published data only}
- NCT00175929. A Study of Brivaracetam in Subjects With Partial Onset Seizures [A Multicenter, Double‐blind, Randomized, Placebo‐controlled, 3 Parallel Groups, Dose‐ranging Trial Evaluating the Efficacy and Safety of Ucb 34714 Used as Adjunctive Treatment at Doses of 50 and 150 mg/Day in b.i.d. Administration (Oral Capsules of 25 mg) for a Maximum of 12 Weeks in Subjects From 16 to 65 Years With Refractory Epilepsy Suffering From Partial Onset Seizures Whether or Not Secondarily Generalized]. https://clinicaltrials.gov/ct2/show/NCT00175929 (first received 15 September 2005).
- Paesschen W, Brodsky A, Brivaracetam N01114 Study Group. Efficacy and tolerability of 50 and 150 mg/day brivaracetam (UCB 34714) as adjunctive treatment in adults with refractory partial‐onset epilepsy. Epilepsia 2007;48 Suppl 6:329. [Google Scholar]
- Paesschen W, Hirsch E, Johnson M, Falter U, Rosenstiel P. Efficacy and tolerability of adjunctive brivaracetam in adults with uncontrolled partial‐onset seizures: a phase IIb, randomized, controlled trial. Epilepsia 2013;54(1):89‐97. [DOI] [PubMed] [Google Scholar]
- Paesschen W, Rosenstiel P. Efficacy and tolerability of brivaracetam as adjunctive treatment for adults with refractory partial‐onset epilepsy. Epilepsia 2007;48 Suppl 7:56‐7. [Google Scholar]
References to studies excluded from this review
Lacroix 2007 {published data only}
- Lacroix B, Rosenstiel P, Sargentini‐Maier ML. Population pharmacokinetics of brivaracetam in patients with partial epilepsy. Epilepsia 2007;48 Suppl 6:333‐4. [Google Scholar]
References to ongoing studies
NCT03083665 {published data only}
- NCT03083665. A Study to Evaluate the Efficacy and Safety of Adjunctive Brivaracetam in Asian Subjects (>=16 to 80 Years of Age) With Epilepsy [A Randomized, Double‐blind, Placebo‐controlled, Multicenter, Parallel‐group Study to Evaluate the Efficacy and Safety of Adjunctive Brivaracetam in Asian Subjects (>=16 to 80 Years of Age) With Partial Seizures With or Without Secondary Generalization]. https://clinicaltrials.gov/ct2/show/NCT03083665 (first received 20 March 2017).
Additional references
Bialer 2010
- Bialer M, Johannessen S, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X). Epilepsy Research 2010;92(2‐3):89‐124. [DOI] [PubMed] [Google Scholar]
Chu‐Shore 2010
- Chu‐Shore CJ, Thiele EA. New Drugs for Pediatric Epilepsy. Pediatric Neuropharmacotherapeutics 2010;17(4):214‐23. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG. Chapter 9. Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from http://handbook.cochrane.org/.
Hey 2014
- Hey SP, Kimmelman J. The questionable use of unequal allocation in confirmatory trials. Neurology 2014;82(1):77‐9. [PUBMED: 24306005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG. Chapter 8. Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from http://handbook.cochrane.org/.
Johannessen Landmark 2008
- Johannessen Landmark C, Johannessen SI. Pharmacological management of epilepsy: recent advances and future prospects. Drugs 2008;68(14):1925‐39. [DOI] [PubMed] [Google Scholar]
Kasteleijn‐Nolst Trenité 2007
- Kasteleijn‐Nolst Trenité DG, Genton P, Parain D, Masnou P, Steinhoff BJ, Jacobs T, et al. Evaluation of brivaracetam, a novel SV2A ligand, in the photosensitivity model. Neurology 2007;69(10):1027‐34. [DOI] [PubMed] [Google Scholar]
Lattanzi 2016
- Lattanzi S, Cagnetti C, Foschi N, Provinciali L, Silvestrini M. Brivaracetam add‐on for refractory focal epilepsy. Neurology 2016;86(14):1344‐52. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6. Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from http://handbook.cochrane.org/.
Ma 2015
- Ma J, Huang S, You C. Adjunctive brivaracetam for patients with refractory partial seizures: a meta‐analysis of randomized placebo‐controlled trials. Epilepsy Research 2015;114:59‐65. [DOI] [PubMed] [Google Scholar]
Nicolas 2012
- Nicolas JM, Chanteux H, Rosa M, Watanabe S, Stockis A. Effect of gemfibrozil on the metabolism of brivaracetam in vitro and in human subjects. Drug Metabolism and Disposition 2012;40(8):1466‐72. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schulze‐Bonhage 2011
- Schulze‐Bonhage A. Brivaracetam for the treatment of epilepsy. Expert Opinion on Pharmacotherapy 2011;12(12):1959‐66. [DOI] [PubMed] [Google Scholar]
Tian 2015
- Tian X, Yuan M, Zhou Q, Wang X. The efficacy and safety of brivaracetam at different doses for partial‐onset epilepsy: a meta‐analysis of placebo‐controlled studies. Expert Opinion on Pharmacotherapy 2015;16(12):1755‐67. [DOI] [PubMed] [Google Scholar]
von Rosenstiel 2007
- Rosenstiel P. Brivaracetam (UCB 34714). Neurotherapeutics 2007;4(1):84‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Werhahn 2010
- Werhahn KJ, Biton V, Johnson ME, Merschhemke M, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from two randomized, double‐blind, placebo‐controlled trials [abstract no: p507]. Epilepsia 2010;51 Suppl 4:150. [DOI] [PubMed] [Google Scholar]
WHO 2013
- World Health Organization. Epilepsy. Available from www.who.int/mental_health/neurology/epilepsy/en/index.html. Accessed 5 June 2014.
Zhu 2017
- Zhu L‐N, Chen D, Chen T, Xu D, Chen S‐H, Liu L. The adverse event profile of brivaracetam: a meta‐analysis of randomized controlled trials. Seizure 2017;45:7‐16. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Zhou 2015
- Zhou Q, Hu CY, Zhang W, Huang YH. Brivaracetam add‐on therapy for epilepsy. Cochrane Database of Systematic Reviews 2015, Issue 2. [DOI: 10.1002/14651858.CD011501] [DOI] [Google Scholar]