

Abstract
Higher plants are well known for their value in affording clinically useful anticancer agents, with such compounds acting against cancer cells by a range of mechanisms of action. There remains a strong interest in the discovery and development of plant secondary metabolites as additional cancer chemotherapeutic lead compounds. In the present review, progress on the discovery of plant-derived compounds of the biflavonoid, lignan, sesquiterpene, steroid, and xanthone structural types is presented. Several potential anticancer leads of these types have been characterized from tropical plants collected in three countries as part of our ongoing collaborative multi-institutional project. Preliminary structure-activity relationships and work on in vivo testing and cellular mechanisms of action are also discussed. In addition, the relevant work reported by other groups on the same compound classes is included herein.
Graphical Abstract
INTRODUCTION
Cancer is continuing to occur among populations of countries all over the world. According to recent global cancer figures, there were over eight million cancer deaths in 2012, of which about 65% occur in the developing countries.1 In the United States, approximately 600,000 cancer deaths were expected to occur in 2018, even though there has been an overall drop of about 25% in cancer mortalities over the last 20 years.2
Natural products purified from organisms and their synthetic analogues have been used for many decades as cancer chemotherapeutic agents. Approximately 50% of the small molecules approved for cancer chemotherapy in Western medicine over the last 70 years are either structurally unmodified natural products or their synthetic derivatives.3 As described in detail by others previously, compounds that are either already approved or under clinical trials are of terrestrial (microbes or higher plants) or marine organism origin4,5 and are notable in acting via a wide variety of different cellular mechanisms.4,6 Recently, the cephalotaxine alkaloid, omacetaxine mepesuccinate (homoharringtonine; Synribo®), was given full approval by the U.S. FDA for the treatment of certain forms of chronic myeloid leukemia resistant to tyrosine kinase inhibitors, based on its mechanism as a protein translation inhibitor.3,7 Also, two new formulations of existing plant-derived natural product anticancer agents that offer enhanced solubility and/or pharmacokinetic parameters have been approved by the FDA, namely, a vincristine sulfate liposome injection (Marqibo®; for Philadelphia chromosome-negative adult acute lymphoblastic leukemia)8,9 and a nanoliposomal irinotecan formulation (Onivyde®; for advanced pancreatic cancer).9,10
Since 2007, our collaborative team has participated in a multi-institutional and multidisciplinary research program, funded by the U.S. National Cancer Institute through the program project (P01) mechanism. There are three main universities included, namely, The Ohio State University, the University of Illinois at Chicago, and the University of North Carolina at Greensboro, and the primary focus at these institutions is the search for new anticancer lead compounds from tropical plants, freshwater and terrestrial cyanobacteria, and filamentous fungi, respectively. The project has been supported by the active participation of a fungal biotechnology company (Mycosynthetix, Inc., Hillsborough, NC) and senior investigators from additional academic institutions and a private research institute, with the pharmaceutical company Bristol-Myers Squibb being a collaborator. The overall administrative framework of the project and a detailed description of the technical approaches taken by the research team, in addition to examples of promising lead bioactive compounds isolated and characterized structurally, have been published in a recent review article.11 Technical progress made by our group program project has appeared in the literature periodically.12–16
In the present contribution, we focus on recent progress of characterization of anticancer lead compounds from selected higher plants collected in the Dominican Republic, Indonesia, and Vietnam as part of this program project collaboration. The plant species from our work referred to below were collected sustainably by the University of Illinois at Chicago, in accordance with existing international treaty requirements, as explained in detail in a recent book chapter.17 This book chapter covers progress made on several of our previously isolated plant-derived lead compounds, including betulinic acid, pervilleine A, and silvestrol,17 which are therefore not included in the present review article. In addition to describing our own work, relevant investigations on the same compound classes reported by other groups are also discussed in each section of this review.
BENZOXANTHONE-TYPE PRENYLATED FLAVONOIDS
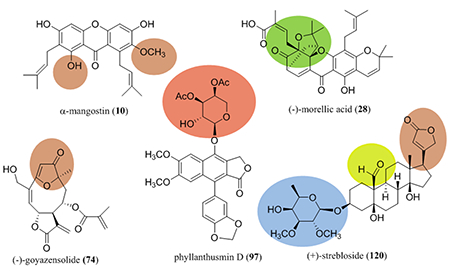
Benzoxanthone-type prenylated flavonoids are a group of 3-prenylated flavonoids that contain an additional D ring formed from a direct connection between C-12 of the isoprenyl group linked at the C-3 position and C-6’ of the flavonone unit. Representatives of this type of natural products have been isolated mainly from the genus Artocarpus (Moraceae) as potential anticancer agents.18 In our collaborative project, extracts of the twigs of Artocarpus rigida Blume (Moraceae), collected in Indonesia, were found to be cytotoxic toward HT-29 human colon cancer cells. Using column chromatography, several cytotoxic benzoxanthone-type prenylated flavonoids and their non-cytotoxic analogues, including (+)-artorigidin A (1), (+)-artorigidin B (2), (+)-cycloartobiloxanthone (3), (+)-artonin N (4), (+)-artonin G (5), (+)-artobiloxanthone (6), (+)-artonin O (7), and artorigidin C (8) were purified and characterized from this plant sample (Figure 1 and Table 1).18
Figure 1.
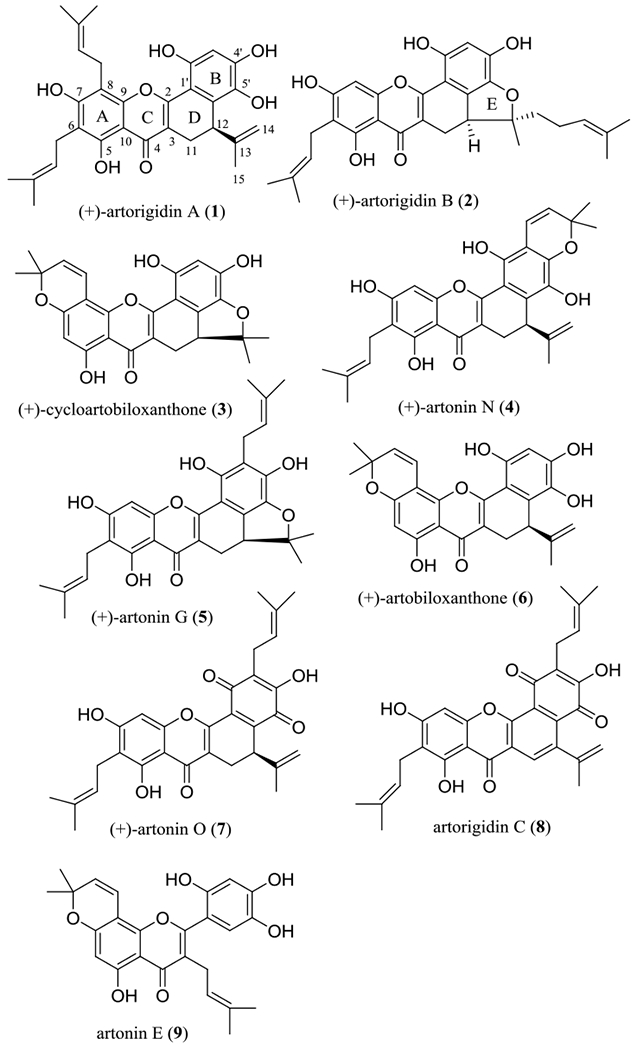
Structures of benzoxanthone-type prenylated flavonoids (1–8) isolated from the twigs of Artocarpus rigida and of artonin E (9).
Table 1.
Cytotoxicity of Xanthone-type Compounds Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 1 | (+)-artorigidin A | Artocarpus rigida (twigs) | 9.3a | 18 |
| 2 | (+)-artorigidin B | A. rigida | 3.4a | 18 |
| 3 | (+)-cycloartobiloxanthone | A. rigida | 3.7a | 18 |
| 4 | (+)-artonin N | A. rigida | >10a | 18 |
| 5 | (+)-artonin G | A. rigida | >10a | 18 |
| 6 | (+)-artobiloxanthone | A. rigida | >10a | 18 |
| 7 | (+)-artonin O | A. rigida | 3.2a | 18 |
| 8 | artorigidin C | A. rigida | >10a | 18 |
| 9 | artonin E |
A. communis (bark) A. rotunda (root bark) |
0.06b | 20 |
| 10 | α-mangostin28 | Garcinia mangostana (stem bark) | 1.7a | 28 |
| 10 | α-mangostin29 | Cratoxylum cochinchinense (stems) | 4.1a | 29 |
| 11 | β-mangostin28 | G. mangostana | 1.7a | 28 |
| 12 | γ-mangostin29 | C. cochinchinense | 4.0a | 29 |
| 13 | 3-O-acetyl-α-mangostin | derived from α-mangostin | 8.8a | 29 |
| 14 | 3,6-di-O-acetyl-α-mangostin | derived from α-mangostin | 1.0a | 29 |
| 15 | 1,3,6-tri-O-acetyl-α-mangostin | derived from α-mangostin | 6.0a | 29 |
| 16 | 3,6-di-O-methyl-α-mangostin | derived from α-mangostin | >10a | 29 |
| 17 | 6-O-benzoyl-α-mangostin | derived from α-mangostin | 1.9a | 29 |
| 18 | 8-deoxygartanin | G. mangostana | >10a | 28 |
| 19 | gartanin | G. mangostana | >10a | 28 |
| 20 | 1,3,7-trihydroxy-2,4-diisoprenylxanthone | C. cochinchinense | >10a | 29 |
| 21 | garcinone D | G. mangostana | 2.3a | 28 |
| 22 | 9-hydroxycalabaxanthone | G. mangostana | 9.1a | 28 |
| 23 | 3-isomangostin | G. mangostana | 4.9a | 28 |
| 24 | 11α-mangostanin | G. mangostana | >10a | 28 |
| 25 | 3-isomangostin hydrate | derived from α-mangostin | 4.4a | 29 |
| 26 | 1-isomangostin hydrate | derived from α-mangostin | >10a | 29 |
| 27 | euxanthone | Polygala caudata (roots) | >10c
>10d |
33 |
HT-29 human colon cancer cell line (μM).
P-388 murine lymphocytic leukemia cell line (μg/mL).
SK-OV-3
OVCAR3 human ovarian cancer cell lines (μM).
A preliminary structure-cytotoxicity relationship study showed that the occurrence of a C-12-affixed β-oriented substituent and an additional E ring are required for a benzoxanthone-type prenylated flavonoid to show potent cytotoxicity toward HT-29 cells, as a result of comparing the activity of (+)-artonin O (7, IC50 3.2 μM) and (+)-cycloartobiloxanthone (3, IC50 3.7 μM) with those to their structurally similar analogues, artorigidin C (8, IC50 >10 μM) and (+)-artobiloxanthone (6, IC50 >10 μM), respectively. Comparison of the activities of (+)-artorigidin B (2, IC50 3.4 μM) and (+)-artonin G (5, IC50 >10 μM) indicates that a C-14 but not a C-3’ prenyl group is preferred for such activity (Figure 1 and Table 1).18
Interestingly, artonin E (9), a prenylated flavonoid isolated originally from the bark of Artocarpus communis Forst. & Forst. (syn. A. altilis Fosberg) (breadfruit) collected in Indonesia,19 containing a C-3 prenyl group rather than a benzoxanthone unit, is the most potent cytotoxic analogue of its type identified thus far from the genus Artocarpus. It has been reported that artonin E (9) exhibited cytotoxicity toward the P-388 murine lymphocytic leukemia cell line, with an IC50 value of 0.06 μg/mL (0.14 μM).20 It inhibited selectively SK-OV-3 human ovarian cancer cells when compared with effects on T1074 normal human ovarian cells.21 Also, artonin E (9) inhibited MDA-MB-231 triple negative and MCF-7 human breast cancer cell growth.22,23
Mechanistically, (+)-artorigidin A (1) has been found to inhibit NF-κB activity,18 and artonin E (9) has been proposed to dysregulate the mitochondrial pathways by induction of mitochondrial transmembrane potential (MTP) disruption, release of cytochrome c, and activation of the caspases-3, −8, and −9 proteins.21–23 These studies indicate that the benzoxanthone-type flavonoids mediate their cytotoxicity, at least in part, by induction of cancer cell apoptosis.
Several species of the genus Artocarpus are consumed as common tropical foods, including breadfruit [Artocarpus altilis Fosberg (syn. Artocarpus communis Forst. & Forst.)], chempedak [Artocarpus integer Merr. (syn. A. champeden Stokes)], and jackfruit (Artocarpus heterophyllus Lam.), and many Artocarpus species are also used in herbal medicines.21 The investigation of the cytotoxic benzoxanthones and other types of C-3 prenylated flavonoids identified from this genus could provide important information for the design of new anticancer agents with potentially reduced side effects.
PRENYLATED XANTHONES
Naturally occurring xanthones are pyran-fused tricyclic diphenylene ketone oxides, an important group of natural products derived mainly from the plant families Bonnetiaceae and Clusiaceae.24 These secondary metabolites exhibit a variety of biological activities, of which prenylated xanthones, as represented by α-mangostin (10), the major active component of the botanical dietary supplement mangosteen [Garcinia x mangostana L. (Clusiaceae)], have been investigated extensively for their potential anticancer activity.24–27
Several prenylated xanthones have been characterized from our previous work on the stems of Cratoxylum cochinchinense (Lour.) Blume (Hypericaceae), collected in Vietnam, and the stem bark of Garcinia x mangostana, collected in Indonesia, including α-, β-, and γ-mangostins (10–12), 8-deoxygartanin (18), gartanin (19), 1,3,7-trihydroxy-2,4-diisoprenylxanthone (20), garcinone D (21), 9-hydroxycalabaxanthone (22), 3-isomangostin (23), and 11α-mangostanin (24). Some of these xanthones, including compounds 10–12 and 21–23, were characterized as the major cytotoxic components of C. cochinchinense and G. x mangostana, when evaluated against HT-29 human colon cancer cells (Figure 2 and Table 1).28,29
Figure 2.
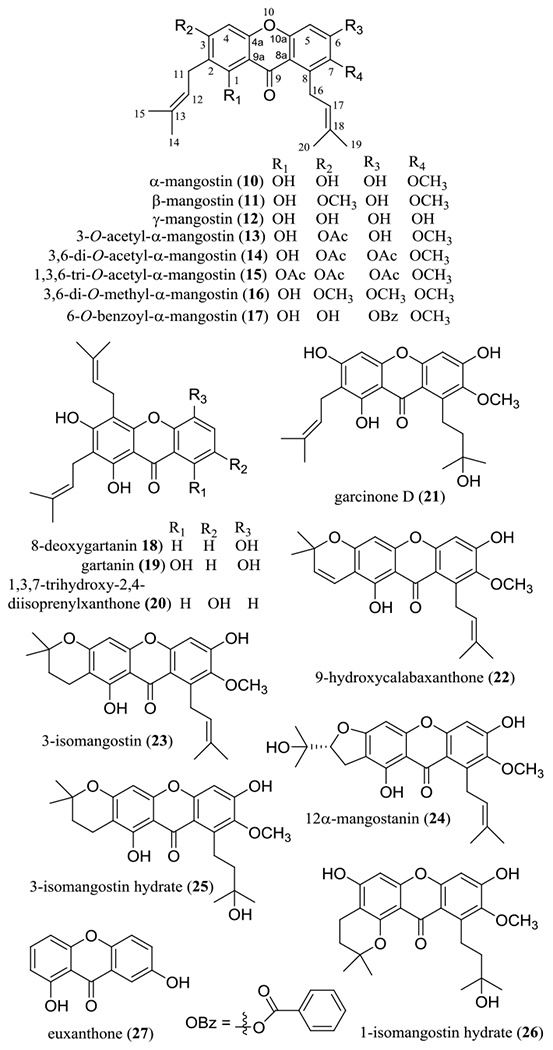
Structures of prenylated xanthones isolated from the stems of Cratoxylum cochinchinense (10, 12, and 20) and the stem bark of Garcinia mangostana (10, 11, 18, 19, and 21–24), and their semi-synthetic analogues (13–17, 25, and 26) and euxanthone (27).
In addition, several derivatives of α-mangostin (10) have been partially synthesized and evaluated biologically, including 3-O-acetyl-α-mangostin (13), 3,6-di-O-acetyl-α-mangostin (14), 1,3,6-tri-O-acetyl-α-mangostin (15), 3,6-di-O-methyl-α-mangostin (16), 6-O-benzoyl-α-mangostin (17), 3-isomangostin hydrate (25), and 1-isomangostin hydrate (26) (Figure 2).29 A preliminary structure-activity relationship (SAR) study showed that the hydroxy group at the C-1 position and an isoprenyl group linked to the C-8 position are necessary for these xanthones to mediate their cytotoxicity toward HT-29 human colon cancer cells. Cyclization of the C-2 prenyl group with the C-3 hydroxy group resulted in the cytotoxic potency being retained or decreased slightly, but the activity was eliminated when the C-2 prenyl group is cyclized with the C-1 hydroxy group, as represented by compounds 25 and 26 (Table 1). Cytotoxic activity is retained when the C-3 hydroxy group is methylated (11) or the C-7 methoxy group is de-methylated (12), but this activity was not observed when methylation occurs at all of the C-3, C-6, and C-7 hydroxy groups (16). In addition, the resultant activity was improved by 3,6-di-acetylation (14) or 6-benzoylation (17), but it was decreased by either 3-mono-acetylation (13) or 1,3,6-tri-acetylation (15).28,29 These structure-cytotoxicity relationship conclusions have been supported by other studies, in which the mangostin xanthones showed selectivity toward a small panel of human cancer cells, with the diprenyl group and dipyrano and prenylated pyrano substituents contributing to the overall cytotoxic potency.30,31
Interestingly, γ-mangostin (12), a prenylated xanthone without methoxy groups, was found to induce apoptosis in HT-29 human colon cancer cells.32 Euxanthone (27), a simple xanthone without prenyl and methoxy groups that was isolated from the roots of Polygala caudata Rehder & E. H. Wilson (Polygalaceae), induced autophagy and apoptosis in SK-OV-3 and OVCAR3 human ovarian cancer cells by modulating pSTAT3/Bcl-2 signaling.33 These observations indicate that neither methoxy nor prenyl groups seem critical for a xanthone derivative to mediate its cytotoxicity toward human cancer cells.
The anticancer potential of α-mangostin (10) has been discussed in a recent review article.27 This compound has been found to inhibit matrix metalloproteinase (MMP)-2 and MMP-9 expression in U2OS human osteosarcoma cells and hence to show some potential for the treatment of osteosarcoma.34 It reduces the expression of both MMP-2 and MMP-9, increases E-cadherin expression, and suppresses the ERK signaling pathways in the MIAPaCa-2 and BxPC-3 pancreatic cancer cell lines to inhibit the invasion, migration, and growth of these cells.35
α-Mangostin (10) also exhibited a synergistic effect on the cytotoxicity of cisplatin [cis-diamminedichloroplatinum (II) (CDDP)] against HeLa human cervical cancer cells. Treatment of α-mangostin (10) plus CDDP showed significantly more potent cytotoxicity than the individual treatments toward HeLa cells in vitro, and this tendency was also observed in an in vivo cervical tumor model. Cervical tumor growth was inhibited significantly when 20–25 g female BALB/c nu/nu mice were inoculated with HeLa cells and treated with α-mangostin (10, orally, 12.5 mg/kg), CDDP (i.p., 3 mg/kg), or α-mangostin (10, orally, 12.5 mg/kg) plus CDDP (i.p., 3 mg/kg) once a week for four weeks. The antitumor potency of the combination treatment of α-mangostin plus CDDP was superior to the individual treatments conducted, indicating that α-mangostin (10) could be a promising lead for use as an adjuvant agent in cervical cancer chemotherapy. 36
Recently, α-mangostin (10) was found to effectively and selectively inhibit the ATP-binding cassette drug transporter ABCG2 to reverse multidrug resistance (MDR) in ABCG2-overexpressing MDR cancer cells at non-toxic concentrations.37 It also showed anti-inflammatory activity through attenuating TNF-α and IL-8 secretion in various cell lines, but it increased TNF-α output in both quiescent and lipopolysaccharide (LPS)-treated monocyte-derived macrophages (MDM).38
The antitumor efficacy of α-mangostin (10) and 3,6-di-O-acetyl-α-mangostin (14) were evaluated at University of Illinois at Chicago, but both compounds were found to be inactive in the hollow fiber assay using immunodeficient NCr nu/nu mice implanted with HT-29 cells and dosed (i.p.) daily with 20.0 mg/kg of these two compounds for four days.28,29 In contrast, in our collaborative work performed at the Ohio State University, α-mangostin (10) was found to exhibit antitumor activity in a colon cancer xenograft model. After four-week-old BALB/c nu/nu mice were treated orally with α-mangostin (900 mg/kg, daily) for one week, mice were inoculated with HT-29 cells and treated continuously with α-mangostin (orally, 900 mg/kg, daily) for another two or four weeks. Colon tumor growth was inhibited significantly by the treatment with α-mangostin (900 mg/kg) when compared with the control treatment of a AIN-93G diet, and no obvious signs of toxicity were observed in mice after treatment.39
In other animal tumor models, α-mangostin (10) inhibited significantly tumor growth. For example, liver tumors were inhibited in five-week-old immunodeficient BALB/c male mice inoculated with SK-Hep-1 human hepatocarcinoma cells and administered (i.p.) with α-mangostin (10) (8 mg/kg, three times per week) for six weeks.40 Pancreatic tumor growth in a xenograft mouse model was found to be suppressed when athymic nude mice were inoculated with ASPC1 human pancreatic cancer cells and treated (i.p.) with α-mangostin (10) (6 mg/kg, five times per week) for eight weeks, and no toxicity from α-mangostin administration was observed in the normal pancreatic tissues of C57BL/6 wild-type mice.41 Prostate tumor growth was inhibited when seven- to eight-week-old athymic (nu/nu) male nude mice were inoculated with 22Rv1 human prostate cancer cells and given (oral gavage) α-mangostin (10) (100 mg/kg, five times per week) for five weeks.42 Also, tongue tumor growth was inhibited when male BALB/c nude mice were inoculated with YD-15 tongue mucoepidermoid carcinoma cells and treated (i.p.) with α-mangostin (10) (10 or 20 mg/kg, five times per week) for three weeks.43
In addition, α-mangostin (10) has been found to show some evidence of selective antitumor potential. For example, it exhibited low cytotoxicity against WRL-68 normal human liver cells, with an IC50 value of 65 μg/mL (158 μM)44 when compared with that toward SK-HEP-1 human hepatocarcinoma cells (IC50 19.6 μM).40 It did not show any adverse effects on body and organ weights, serum biochemical parameters, histopathology, and oxidative stress biomarkers of ICR female and male mice when mice were treated orally with this compound, using single doses of 100, 500 or 1000 mg/kg.44
The pharmacokinetic parameters of α-mangostin (10), γ-mangostin (12), and a methanol extract of the pericarps of G. x mangostana have been investigated in our collaborative work using male Sprague Dawley rats at the University of Florida. On oral administration (20 mg/kg, single dose), both 10 and 12 underwent intensive first-pass metabolism and were conjugated very rapidly. However, when administered in an extract, the conjugation of 10 or 12 was slower, and free compound exposure in the blood of the rats was increased.45
Mechanistic studies have shown that α-mangostin (10) induces mitochondrial-dependent apoptosis in SK-Hep-1 human hepatoma cells through inhibition of the p38 MAPK pathway and decreases the cell viability of ASPC1, BxPC3, PANC1, and PL-45 human pancreatic cancer cells, due, in part, to the inhibition of the NF-κB, STAT3, and MMP9 signaling pathways.40,41 It inhibited the proliferation of K562, KBM5, and KBM5-T315I human chronic myeloid leukemia (CML) cells targeting apoptosis and autophagy.46 Also, α-mangostin (10) was found to inhibit potently mitochondrial transmembrane potential (IC50 0.2 μM) in HT-29 human colon cancer cells29 and to decrease COLO 205 human colon cancer cell growth through increasing mitochondrial cytochrome c release, Bax, p53 and Bmf and reducing mitochondrial membrane potential.47
However, in an experimental colitis study, dietary α-mangostin (10) was found to exacerbate dextran sulfate sodium (DSS)-induced inflammation and injury in the colons of mice when ten-week-old female C57BL/6 mice were treated orally with α-mangostin (900 mg/kg, daily) for one week followed by DSS induction of colitis, and then the mice were treated continuously with α-mangostin (orally, 900 mg/kg, daily) for another one or two weeks. Thus, α-mangostin (10) has been suggested to be used with caution by patients with inflammatory bowel disorders and by healthy individuals.48
Based on the extensive investigations on α-mangostin (10) discussed, this xanthone compound could be modified to enhance its antitumor potential and to decrease its reverse effects for further development as a single chemotherapy agent or drug conjugate to contribute to the treatment of cancer, owing to its potent and multiple bioactivities, long-term history of human consumption, and widespread commercial availability.
CAGED XANTHONES
The caged xanthones are a group of 5,7-diprenylated xanthones containing a saturated ring A. An increasing number of this type of xanthones has been identified mainly from the genus Garcinia (Clusiaceae), which comprises over 200 species with some having uses in systems of traditional medicine. Interest in these compounds is rising, especially in terms of evaluating their in vitro cancer cell cytotoxicity and in vivo antitumor efficacy.49,50
In our collaborative study, several cytotoxic caged xanthones were isolated from the stem bark of Garcinia lateriflora Blume (Clusiaceae), collected in Indonesia, using column chromatographic separation guided by cytotoxicity toward HT-29 human colon cancer cells. These included (–)-morellic acid (28), (–)-11,12-dihydro-12-hydroxymorellic acid (29), (–)-isomoreollic acid (30), (–)-isogaudichaudiic acid (31), and (–)-isogaudichaudiic acid E (32) (Figure 3).51 The structures of these compounds were elucidated by analysis of their spectroscopic data, and the absolute configuration at C-5, C-7, C-10a, and C-27 of (–)-morellic acid (28) was determined by analysis of its NMR parameters and the experimental and calculated electronic circular dichroism (ECD) spectra. The calculations were performed collaboratively at the University of Mississippi using the SYBYL 8.1 program, with MMFF94 molecular mechanic force-field calculations employed to search for the conformations possible. All ground-state geometries were optimized at the B3LYP/6–31G** level at 298 K, and harmonic frequency analysis was computed to confirm the minima. In turn, the absolute configuration of compounds 28–32 was determined by comparison of their NOESY NMR and ECD spectra with those of (–)-morellic acid (28).51
Figure 3.
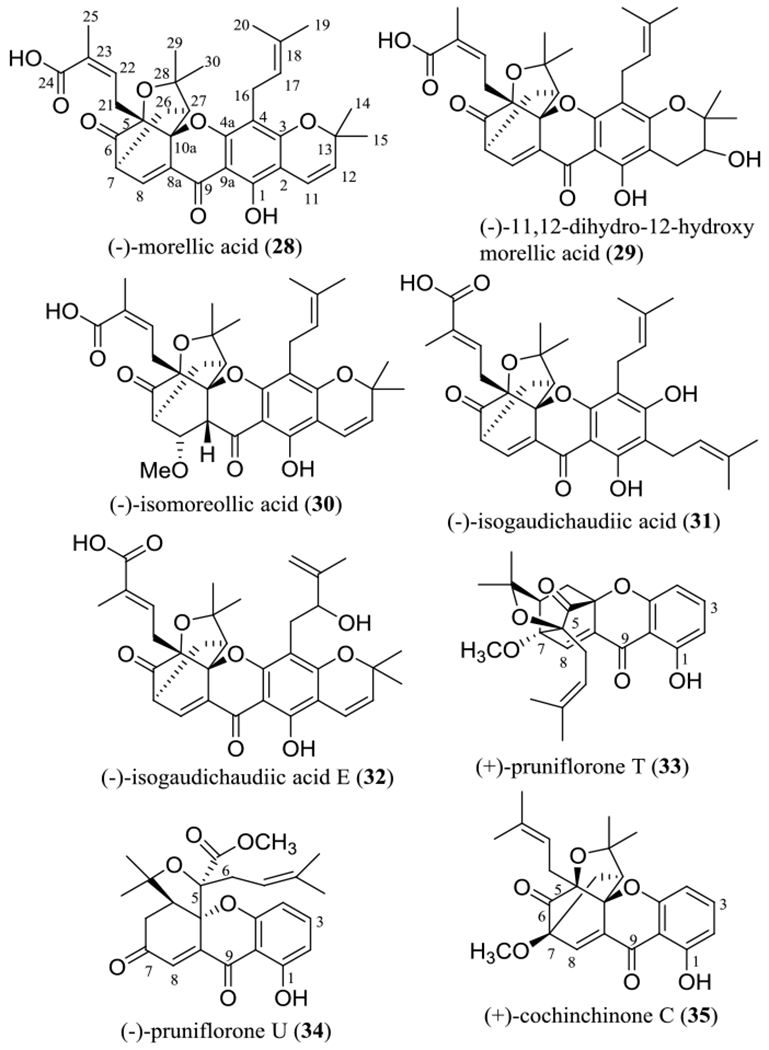
Structures of the cytotoxic caged xanthones 28–32 isolated from the stem bark Garcinia lateriflora and other related xanthones (33–35).
When evaluated toward HT-29 colon cancer cells, all the above-mentioned caged xanthones showed activity, with potency increasing in the sequence (–)-isogaudichaudiic acid (31, IC50 3.2 μM), (–)-11,12-dihydro-12-hydroxymorellic acid (29, IC50 2.9 μM), (–)-isogaudichaudiic acid E (32, IC50 2.6 μM), (–)-isomoreollic acid (30, IC50 1.9 μM), and (–)-morellic acid (28, IC50 0.36 μM) (Table 2).51 Investigations of the structure and cytotoxicity of these compounds and their analogues have indicated that the pyran ring connected at the C-2 and C-3 positions, the C-4 prenyl group, and the α,β-unsaturated carbonyl group including C-8, C-8a, C-9, C-9a, and C-4a, all contribute to the resultant activity toward HT-29 cells.
Table 2.
Cytotoxicity and Hsp90 ATPase and PTP1B Inhibition of Caged Xanthones Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 28 | (–)-morellic acid | Garcinia lateriflora (stem bark) | 0.36a | 51 |
| 29 | (–)-11,12-dihydro-12-hydroxymorellic acid | G. lateriflora | 2.9a | 51 |
| 30 | (–)-isomoreollic acid | G. lateriflora | 1.9a | 51 |
| 31 | (–)-isogaudichaudiic acid | G. lateriflora | 3.2a | 51 |
| 32 | (–)-isogaudichaudiic acid E | G. lateriflora | 2.6a | 51 |
| 33 | (+)-pruniflorone T | Cratoxylum formosum ssp. pruniflorum (roots) | >5b | 52 |
| 34 | (–)-pruniflorone U | C. formosum | >5b | 52 |
| 33 + | (+)-pruniflorone T plus | C. formosum | 0.11b | 52 |
| 34 | (–)-pruniflorone U | |||
| 35 | (+)-cochinchinone C | C. formosum | 0.36b | 52 |
| 36 | epicochinchinoxanthone | synthetic caged xanthone | 4.1c 5.9d |
53 |
| 37 | epicochinchinoxanthone-3-ether | synthetic caged xanthone | 3.3c 3.2d |
53 |
| 38 | 17,18-epoxy-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 2.7c 0.31d |
53 |
| 39 | 17,18-dihydroxy-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 1.7c 0.86d |
53 |
| 40 | 19-hydroxy-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 2.5c 3.8d |
53 |
| 41 | 19-carbonyl-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 1.0c 5.0d |
53 |
| 42 | 19-bromo-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 0.96c 1.6d |
53 |
| 43 | 2-bromo-19-hydroxy-epicochinchinoxanthone-3-ether | synthetic caged xanthone | 0.56c 2.2d |
53 |
| 44 | epiisobractatin | Garcinia bracteata (leaves) | 1.2e 2.1f |
54 |
| 45 | 13-hydroxyisobractatin | G. bracteata | 1.1e 2.1f |
54 |
| 46 | bractatin | G. bracteata | 0.40e 1.0f |
54 |
| 47 | 3-O-methyobractatin | G. bracteata | 0.20e 0.80f |
54 |
| 48 | DDO-6101 | synthetic caged xanthone | 14.3g 0.81h |
55 59 |
| 49 | 19-hydroxy-DDO-6101 | synthetic caged xanthone | 3.7g |
55 |
| 50 | isomorellin (50) | Garcinia hanburyi (resin) | 3.3i | 56 |
| 51 | forbesione (51) | G. hanburyi | 3.5i | 56 |
| 52 | DDO-6306 (52) | synthetic caged xanthone | 0.52h |
59 |
| 53 | (+)-cochinchinoxanthone |
Cratoxylum cochinchinense (stems) Cratoxylum cochinchinense (roots) |
5.8a 6.6j | 29 61 |
HT-29 human colon cancer cell line (μM).
MCF-7 human breast cancer cell line (μg/mL).
A549 human lung cancer cell line (μM).
BGC-823 human gastric cancer cell line (μM).
HL-60 human leukemia cell line (μM).
K562 human leukemia cell line (μM).
Hsp90 ATPase inhibition (μM).
HepG2 human hepatoma cell line (μM).
KKU-100 human cholangiocarcinoma cell line (μM).
Protein tyrosine phosphatase 1B (PTP1B) inhibition (μM).
In another research report from other groups, a neo-caged xanthone, (+)-pruniflorone T (33), a rearranged caged xanthone, (+)-pruniflorone U (34), and a normal caged xanthone, (–)-cochinchinone C (35) (Figure 3), were isolated from the roots of Cratoxylum formosum Benth. & Hook. f. ex Dyer ssp. pruniflorum (Kurz) Gogelein (Hypericaceae) collected in Thailand. When evaluated against MCF-7 human breast cancer cells, only the normal caged xanthone, cochinchinone C (35), showed activity, with an IC50 value of 0.36 μM. Interestingly, a mixture of (+)-pruniflorones T (33) and U (34) (50% of each) was found to be potently active, showing an IC50 value of 0.11 μM (Table 2).52 These results imply that, for these compounds, the normal caged unit is critically important, and the neo-caged and rearranged caged xanthones may synergize reciprocally their cancer cell cytotoxicity.
A synthetic caged xanthone, epicochinchinoxanthone (36) (Figure 4), showed cytotoxicity toward A549 non-small cell lung, HepG2 liver, MCF-7 breast, and BGC-823 gastric human cancer cells, and such activities were not changed greatly when the C-2 hydroxy group of 36 was esterified by chloromethyl methyl ether, from which epicochinchinoxanthone-3-ether (37) was produced. However, both the selectivity toward these cancer cell lines and the cytotoxicity toward BGC-823 cells were improved by further introducing a 17,18-epoxy group [17,18-epoxy-epicochinchinoxanthone-3-ether (38)] or a 17,18-dihydroxy group [17,18-dihydroxy-epicochinchinoxanthone-3-ether (39)] into the molecule (37).53 In contrast, the cytotoxicity toward A549, BGC-823, HepG2, and MCF-7 cells was not altered greatly when the C-19 methyl group of 37 was changed to a primary alcohol [19-hydroxy-epicochinchinoxanthone-3-ether (40)] or a formyl group [19-carbonyl-epicochinchinoxanthone-3-ether (41)]. However, the activity against A549 cells was improved by substitution of a bromine atom at the C-19 position of 37 [19-bromo-epicochinchinoxanthone-3-ether (42)] or at the C-2 position of 40 [2-bromo-19-hydroxy-epicochinchinoxanthone-3-ether (43)] (Figure 4 and Table 2).53 These results indicate that all of the C-2 and C-17–C-19 positions are important for a given caged xanthone to mediate its cytotoxicity toward human cancer cells, but a prenyl group connected at either the C-2 or C-4 position seems unnecessary in this regard.
Figure 4.
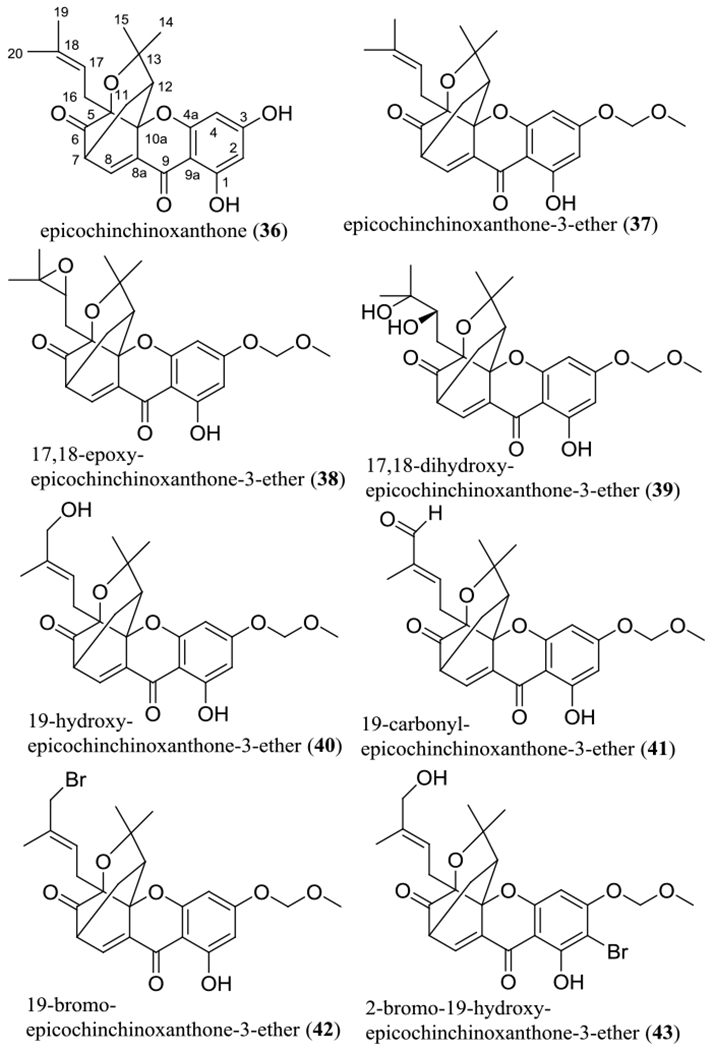
Structures of single di-prenylated synthetic caged xanthones (36–43).
For those C-4 prenylated caged xanthones isolated from the leaves of Garcinia bracteata C.Y. Wu ex Y.H. Li, inclusion of a furan ring in the C-4 prenyl group resulted in the cytotoxicity being decreased slightly, as indicated by the activities against HL-60 and K562 human leukemic cells observed for epiisobractatin (44), 13-hydroxyisobractatin (45), bractatin (46), and 3-O-methyobractatin (47) (Figure 5 and Table 2).54 Compounds 46 and 47 showed more potent activities than 44 and 45 toward both HL-60 and K562 cells, but similar potencies were observed for 44 and 45 and for 46 and 47. These biological data obtained indicate that neither introducing a primary hydroxy group at the C-13 position nor methylation of the C-2 hydroxy group made any major impact on the resultant cytotoxicity of these caged xanthones (Figure 5 and Table 2).54
Figure 5.
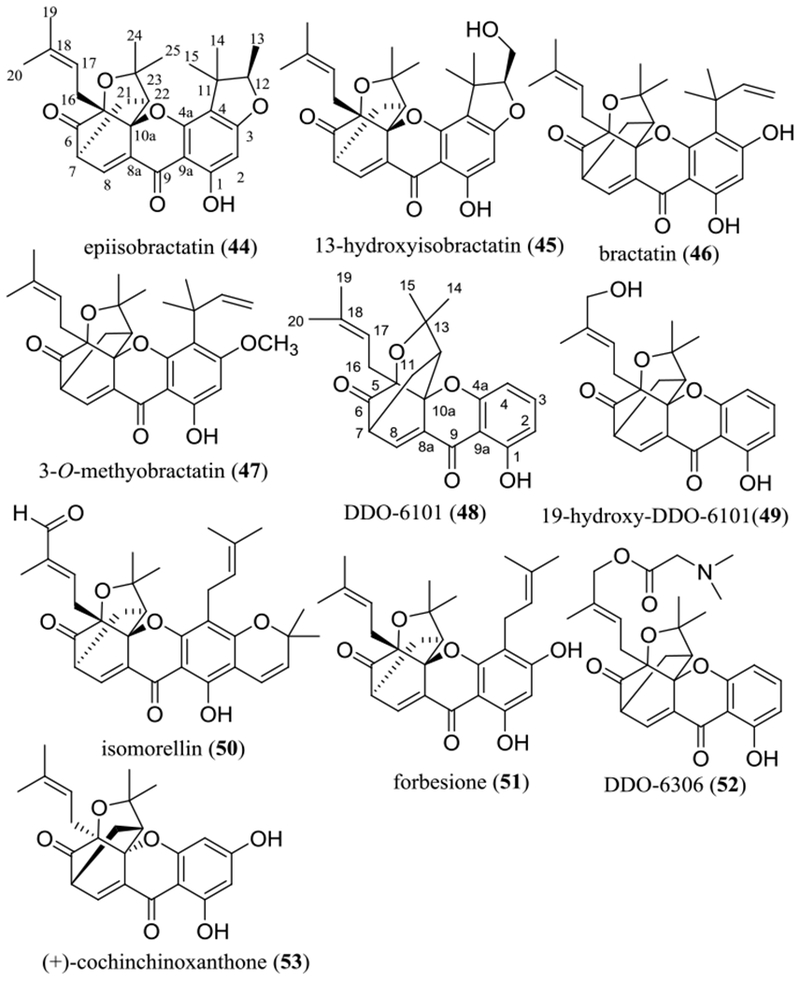
Structures of selected naturally occurring and synthetic caged xanthones 44–53 showing potential anticancer activity.
A simple caged xanthone containing only a caged 5,7-diprenyl group unit, namely, DDO-6101 (48), was found to inhibit the ATPase activity of heat shock protein 90 (Hsp90), and this activity was improved greatly when a primary hydroxy group was introduced at the C-19 position [19-hydroxy-DDO-6101 (49)] (Figure 5 and Table 2).55 This indicates the promise of further development of new anticancer agents based on caged xanthanes that target Hsp90.55
Two additional caged xanthones, isomorellin (50) and forbesione (51) (Figure 5 and Table 2), have been reported for their cytotoxicity toward KKU-100, KKU-M139, and KKU-M156 human cholangiocarcinoma (CCA) cells. These compounds induced CCA cell apoptosis and synergized with doxorubicin in inhibition of CCA cell growth and induction of apoptosis. Thus, they were suggested for potential use as adjunct treatments with chemotherapy for Opisthorchis viverrini-associated advanced cholangiocarcinoma.56
In an in vivo hollow fiber assay conducted at University of Illinois at Chicago, the cytotoxic (–)-morellic acid (28) was found to be inactive, when immunodeficient NCr nu/nu mice implanted with HT-29 cells were treated (i.p.) with this compound daily at the highest dose of 20 mg/kg for four days.51 However, another investigation has shown that this compound displayed a discernible antiangiogenic activity in an in vivo zebrafish model when a transgenic zebrafish line, Tg(flk1:EGFP), was treated with 28 at a concentration of 4 μM for 24 h.57 Also, cholangiocarcinoma growth was inhibited, when six- to eight-week-old male Syrian hamsters inoculated with CCA Ham-1 cells were treated (orally) daily with the caged xanthone forbesione (51) (50 mg/kg) for a month.58
To aid the development of a caged xanthone as a possible new anticancer agent, both the drug-like properties and cytotoxicity toward HepG2 human hepatoma cells of DDO-6101 (48) were improved by introducing a 2-(dimethylamino)acetoxy group at the C-19 position [DDO-6306 (52)] (Figure 5 and Table 2). Liver tumor growth was inhibited when mice inoculated with HepG2 cells were treated (i.v.) with DDO-6306 (52) (10, 20 or 40 mg/kg, twice daily) for eight days.59
Mechanistic investigations have shown that caged xanthones mediate their antitumor activity through multiple mechanisms, including modulation of JNK, NF-κB, p53 and PARP.50 For example, isomorellin (50) was found to inhibit cholangiocarcinoma cell growth through the p53 and NF-κB-signaling pathways,60 and (+)-cochinchinoxanthone (53) was found to show cytotoxicity toward HT-29 human colon cancer cells29 and to inhibit protein tyrosine phosphatase 1B (PTP1B).61
Among the caged xanthones, gambogic acid (54) has been developed as a promising anticancer agent and reached cancer clinical trials. It has been characterized as the major bioactive component of gamboge, the dried resin of Garcinia hanburyi Hook.f., with the structure determined completely by analysis of its NMR spectroscopic and single crystal X-ray diffraction data and electronic circular dichroism spectra.62
Gambogic acid (54) has been found to be cytotoxic toward various human cancer cell lines,63 and two of its analogues, 18-hydroxyepigambogic acid (55) and 19-hydroxyepigambogic acid (56), were also found to suppress NCI-H1650 human bronchoalveolar carcinoma cell growth.64 A preliminary structure-activity relationship study showed that substitution of a hydroxy group at either C-18 (55) or C-19 (56), opening the pyran ring connected at the C-2 and C-3 positions [deoxygambogenin (57), isogambogenin (58), and isogambogenic acid (59)], or changing the C-24 methyl group of 57 to a formyl group or a carboxylic acid unit did not change the activity greatly (Figure 6 and Table 3).64 However, saturation of the C-8 double bond and introducing a methoxy group at the C-7 position resulted in the activity being decreased slightly, as indicated by the cytotoxicity against HL-60 human leukemia cells of gambogic acid (54) (IC50 0.17 μM) and its derivative, gambogoic acid (60) (IC50 0.83 μM) (Figure 6 and Table 3).65 This evidence indicates that the tricyclic xanthone skeleton, the caged prenyl group, the α,β-unsaturated carbonyl moiety, and the hydroxy group connected at the C-1 position all are important for gambogic acid (54) to mediate its cytotoxicity toward human cancer cells.66
Figure 6.
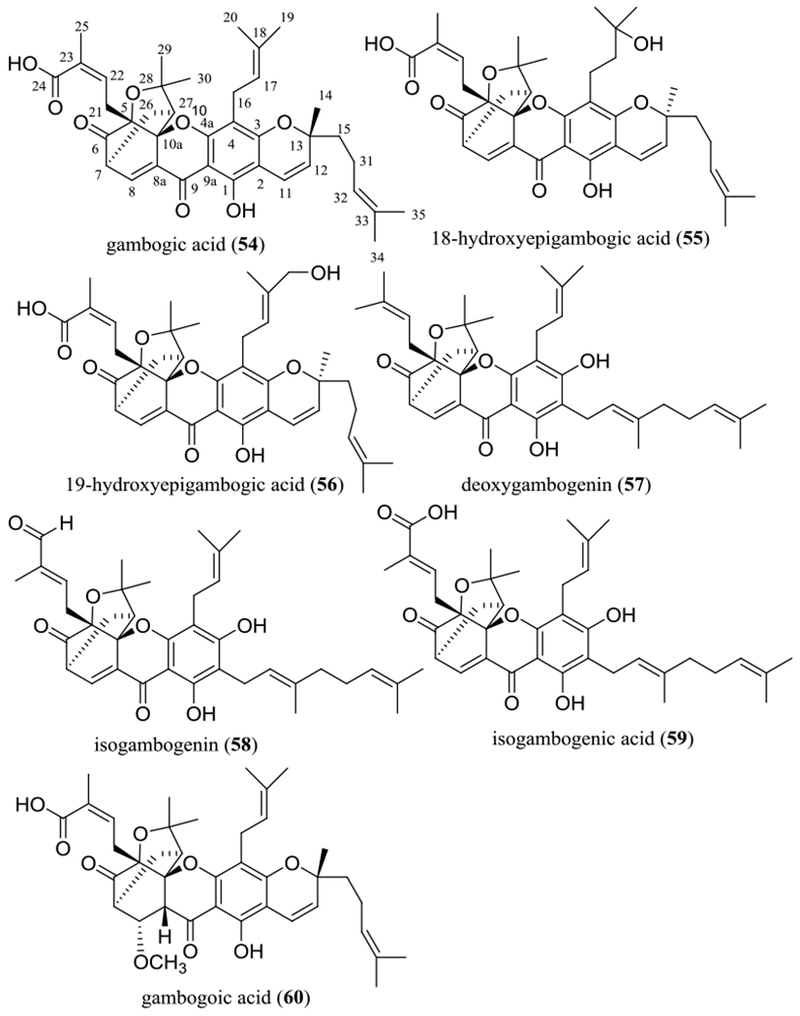
Structures of gambogic acid (54) and selected analogues (55–60) showing potential anticancer activity.
Table 3.
Potential Antitumor Activity of Gambogic Acid and Biflavonoids Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 54 | gambogic acid | Garcinia hanburyi (resin) | 0.48a 0.17b |
62,65 |
| 55 | 18-hydroxyepigambogic acid | library from Garcinia genus | 0.95c | 64 |
| 56 | 19-hydroxyepigambogic acid | library from Garcinia genus | 0.62c |
64 |
| 57 | deoxygambogenin | library from Garcinia genus | 1.1c |
64 |
| 58 | isogambogenin | library from Garcinia genus | 0.97c |
64 |
| 59 | isogambogenic acid | library from Garcinia genus | 1.4c |
64 |
| 60 | gambogoic acid | derived from 54 | 0.83b |
65 |
| 61 | (+)-morelloflavone | Garcinia lateriflora (stem bark) | >10a 1.3d |
51 |
| 62 | (±)-morelloflavone | Garcinia brasiliensis (fruit pericarps) | 10.5e 3.8f 9.6g |
87 |
| 63 | (±)-7,4′,7″,3‴,4‴-penta-O-acetylmorelloflavone | derived from 62 | 0.60e 21.1f 537g |
87 |
| 64 | (±)-7,4′,7″,3‴,4‴-penta-O-methylmorelloflavone | derived from 62 | 15.4e 1.6f 235g |
87 |
| 65 | (±)-7,4′,7″,3‴,4‴-penta-O-butanoylmorelloflavone | derived from 62 | 15.7e 19.8f 8.1g |
87 |
| 66 | amentoflavone | Selaginella pachystachys (leaves) | >10h | 88 |
| 67 | (–)-calycopterone | Calycopteris floribunda (flowers) | 0.4i 0.24a |
89 |
| 68 | (–)-isocalycopterone | C. floribunda | 0.3i | 89 |
| 69 | (–)-4-demethylcalycopterone | C. floribunda | 0.3i | 89 |
| 70 | 4′,5-dihydroxy-3,6,7,3′-tetramethoxyflavone | C. floribunda | >10i >10a |
89 |
| 71 | (–)-abiesinol A (71) | Abies sachalinensis (stem bark) | 2.1j | 90 |
| 72 | (–)-abiesinol C (72) | A. sachalinensis | 2.1j | 90 |
| 73 | (–)-abiesinol E (73) | A. sachalinensis | 2.0j | 90 |
HT-29 human colon cancer cell line (μM).
HL-60 human leukemia cell line (μM).
NCI-H1650 human bronchoalveolar carcinoma cell line (μM).
Proteasome inhibition (μM).
Papain protein inhibition (μM).
Trypsin protein inhibition (μM).
Cruzain protein inhibition (μM).
HeLa human cervical cancer cell line (μM).
Col2 human colon cancer cell line (μg/mL).
IR value measured at concentration of 0.35 μM.
Gambogic acid (54) was also found to reverse multidrug resistance in several human cancer cell lines. It inhibited both the activity and expression of P-glycoprotein to sensitize doxorubicin-resistant breast cancer cells to doxorubicin treatment.67 The resistance of K562/A02 human leukemia multidrug resistant cells to daunorubicin was reversed when the cells were treated with a gambogic acid drug delivery system, in which gambogic acid (54) was linked with a polyethylene glycol-polylactic-co-glycolic acid-poly-l-lysine (PEG-PLGA-PLL) moiety.68 Also, gambogic acid (54) inhibited proliferation of both 5-fluorouracil-sensitive and -resistant colorectal cancer cells.69
Cisplatin-resistant tumor growth was found to be suppressed in four- to six-week-old BALB/c female nude mice that were inoculated with A375/CDDP human melanoma cisplatin-resistant cells and treated (i.p.) with cisplatin (5 mg/kg) with or without gambogic acid (54) (1 mg/kg, every 3 days) for 15 days. In the resulting tumor tissues, miR-199a-3p expression was increased, but expression of the zinc finger E-box binding homeobox (ZEB1), an important regulator of tumor development, was decreased. This indicates that gambogic acid (54) inhibits melanoma growth through miR-199a-3p–ZEB1 signaling.70
The antitumor efficacy and mechanisms of action of gambogic acid (54), including its cytotoxicity toward various human cancer cell lines and in vivo antitumor activity targeting angiogenesis, apoptosis, topo IIα, NF-κB, and telomerase, have been summarized in a recent review article.63 Its additional Hsp90 and proteasome molecular targets are discussed in the following two paragraphs.
The 90 kDa heat shock protein (Hsp90) is a molecular chaperone that functions with a cohort of co-chaperones to facilitate the folding, activation, and stabilization of numerous client proteins, and it has emerged as a target for discovery of new anticancer agents. In a high-throughput screening campaign for selected natural products, gambogic acid (54) was identified as a potential Hsp90 inhibitor. Molecular docking experiments indicated that this compound binds to Hsp90 at a site distinct from the ATP binding pocket, and thus it could be a non-competitive inhibitor of the Hsp90 N-terminal domain.71 Further investigations using a series of Hsp90 deletion mutants and molecular docking showed that gambogic acid (54) is an Hsp90β-specific Hsp90 inhibitor. It binds selectively to a site in the middle domain of Hsp90β, a druggable binding site distinct from both the N-terminal domain and the C-terminal domain of Hsp90, and represents a lead for discovery of new anticancer drugs with a novel mechanism of Hsp90 inhibition.72
In addition, gambogic acid (54) was found to inhibit proteasome and cell growth in both SU-DHL-2 activated and SU-DHL-4 germinal center B-cell-like diffuse large B-cell human lymphoma cells.73 It inhibited lymphoma growth when nude BALB/c mice inoculated with SU-DHL-4 and SU-DHL-2 cells were treated (i.p.) with this compound (3 mg/kg, every two days) for 13 days.73 In an in vitro study, the combination treatment of gambogic acid (54) with the proteasome inhibitors MG132 was found to result in discernible synergistic effects in K562 human leukemia and H22 mouse hepatocarcinoma cells.74 Thus, gambogic acid (54) and bortezomib (a clinically used proteasome inhibitor) were found to induce cytotoxicity synergistically in cultured HepG2 human and H22 mouse hepatoma cells. However, gambogic acid (54) did not act synergistically with bortezomib to inhibit tumor growth in either H22 allograft or HepG2 xenograft tumors propagated in immunodeficient mice.75
A phase I clinical trial for the safety, maximal tolerated dose (MTD), and dose limiting toxicity (DLT) of intravenously injected gambogic acid (54) in cancer patients was conducted in the People’s Republic of China, and a dose regimen of 45 mg/m2 (daily for five days or every other day for five days) in a cycle every three to four weeks has been developed for subsequent phase II testing.76 A phase IIa clinical trial to compare the efficacy and safety of different dosage schedules of gambogic acid (54) in patients with non-small-cell lung, colon, and renal cancers was completed, and a phase IIb clinical trial was suggested for gambogic acid (54) to further assess the safety and efficacy of its intravenous (45 mg/m2) administration on days one to five of a two-week cycle.77
To improve on the aqueous solubility of gambogic acid (54), a positively charged PEGylated liposomal (GAL) formulation has been prepared, which showed an inhibitory effect on the proliferation of MDA-MB-231 triple-negative human breast cancer cells. This formulation (GAL) was found to suppress MDA-MB-231 xenograft tumor growth significantly more potently than the parent compound (54). In these in vivo investigations, six-week-old female Nu/Nu mice harboring MDA-MB-231 xenografts were treated (i.v.) with gambogic acid (54) (10 mg/kg) or GAL (10 mg/kg) six times at two-day intervals. Relative to controls, tumor growth was suppressed 1.31-fold by gambogic acid (54) and 2.0-fold by GAL.78 In addition, a titanium dioxide (TiO2)-coated gold nanorod (GNR/TiO2) has been used as a carrier of gambogic acid (54), and this nanorod complex was reported to be much more effective in inhibiting the proliferation of U-87 MG human glioblastoma cells than free gambogic acid (54).79
Tumor-targeted delivery is now considered crucial for new anticancer drug development. Nanostructured lipid carriers (NLC) decorated with cell-penetrating peptides have shown great potential for targeting tumors. A gambogic acid (54)-loaded nanostructured lipid carrier (GA-NLC) was modified with two kinds of cell penetrating peptides, the novel linear-peptide RGERPPR (yielding GA-NLC-RGE) and a cyclic peptide CRGDRGPDC (yielding GA-NLC-RGD). The GA-NLC-RGE formulation exhibited significantly more potent cytotoxicity against MDA-MB-231 cells compared with GA-NLC. Furthermore, in a murine xenograft model, the GA-NLC-RGE formulation inhibited MDA-MB-231 tumor growth more efficiently than other formulations. This indicates that the RGERPPR-modified NLC is a potential vehicle for the development of gambogic acid (54) as a potential breast cancer chemotherapy.80
Recently, a passive-targeting gambogic acid (54) delivery system, CS/PLGA-GA particles that exhibit a uniform size of approximately 7 μm, were prepared and evaluated in an orthotopic Lewis lung carcinoma mouse model. This formulation showed effective restoration of histomorphological abnormalities in the lungs, prolongation of survival time, and inhibition of tumor metastasis to the liver. Thus, this new formulation may represent an improved approach for lung cancer therapy.81
All cytotoxic benzoxanthones, prenylated xanthones, the normal xanthone, euxanthone (27), and the caged xanthones listed in Tables 1–3 contain an α,β-unsaturated carbonyl group across the C-8, C-8a, and C-9 positions and a hydrogen-bonded hydroxy group at the C-1 position, suggesting that these functional groups are important for mediation of their cytotoxicity. Comparison of the cytotoxicity of these different types of xanthones (Tables 1–3) shows that the cytotoxic potency increases in the sequence benzoxanthones, prenylated xanthones, and caged xanthones. This suggests that the caged prenyl group is preferred to more structurally simple xanthone compounds to inhibit human cancer cell growth. Mechanistically, xanthones exhibit their activity by inducing apoptosis through mitochondrial/NF-κB signaling, but inhibition of the Hsp90 protein has been reported only for the caged xanthones. This indicates, therefore, that the members of varied structural classes of the xanthones work through different mechanisms of action.
BIFLAVONOIDS
Plant-derived biflavonoids (dimeric flavonoids with two mono-flavonoid units linked through a direct C-C bond or an oxygen bridge) are distributed mainly in the gymnosperms and angiosperms and show a variety of biological activities. The potential anticancer activity of these dimers has attracted attention in cancer research, owing to their multiple mechanisms of action and cancer cell selectivity.82
In our previously mentioned study on the stem bark of Garcinia lateriflora (Clusiaceae), collected in Indonesia, several biflavonoids including (+)-morelloflavone (61) (Figure 7) were characterized as non-cytotoxic but potent proteasome inhibitors.51
Figure 7.
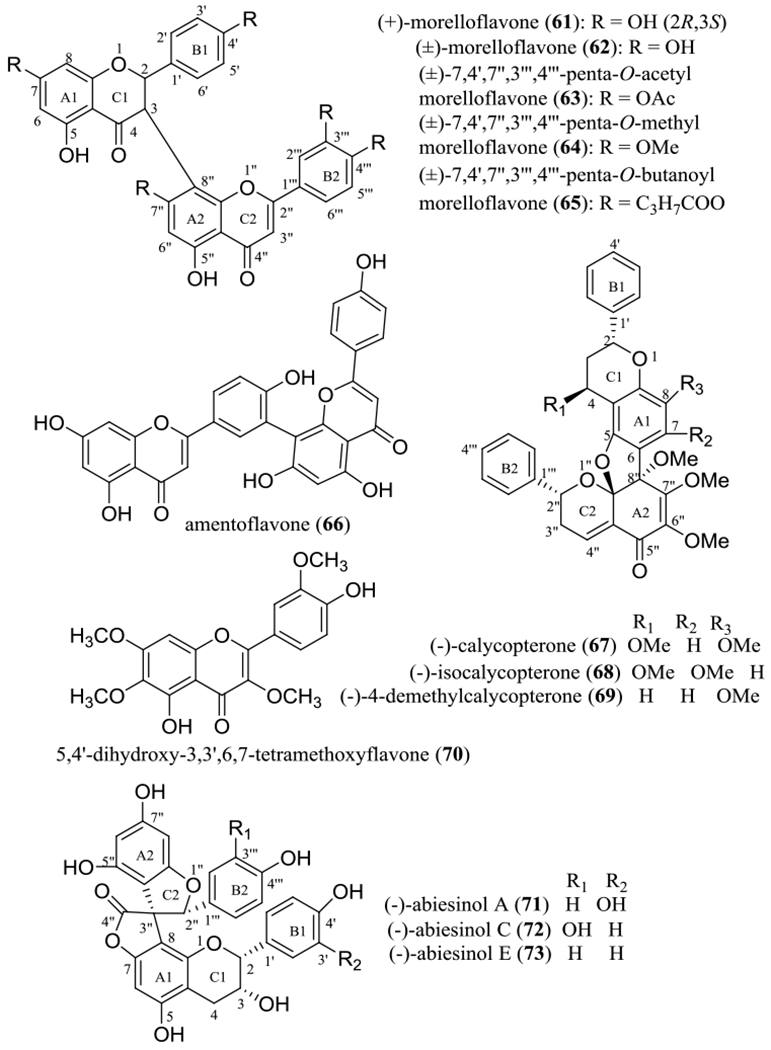
Structures of biflavonoids (61, 62, 66–69, and 71–73) and their semi-synthetic derivatives (63–65) and a monomer (70) showing potential anticancer activity.
(+)-Morelloflavone (61) was isolated and identified originally from the seeds of Garcinia morella (Gaertn.) Desr. (Clusiaceae) as the first flavanone-(3–8″)-flavone type of biflavonoid. The conformation of the flavanone-(3–8″)-flavone dimers was investigated by analysis of variable temperature NMR spectra and computational calculations, using a semi-empirical AM1 method, and the 2R,3S absolute configuration has been determined for (+)-morelloflavone (61) by analysis of its electronic circular dichroism (ECD) spectrum.83
(+)-Morelloflavone (61) was found inactive (IC50 >10 μM) when tested in a cell viability assay toward the HT-29 human colon cancer cell line.51 In addition, it was non-cytotoxic toward both MCF-7 human breast cancer and U87 human glioma cells at a concentration up to 80 μM84,85 and toward PC-3 human prostate cancer cells at a concentration up to 100 μM.86 However, (+)-morelloflavone (61) was found to effectively inhibit tumor angiogenesis. Hence, it inhibited vascular endothelial growth factor (VEGF)-induced cell proliferation, migration, invasion, and capillary-like tube formation of primary cultured human umbilical vascular endothelial cells, microvessel sprouting of endothelial cells, and the formation of new blood microvessels induced by VEGF in a mouse Matrigel plug assay. Both prostate tumor growth and angiogenesis were found to be suppressed in several combined immunodeficient (SCID) male mice inoculated with PC-3 human prostate cancer cells and treated (s.c.) daily with (+)-morelloflavone (61) (8 mg/kg) for 15 days.86
Mechanistically, (+)-morelloflavone (61) showed an inhibitory effect on the proteasome,51 and it effects its antitumor and anti-angiogenesis activities by targeting the activation of Rho-GTPases and ERK signaling pathways through inhibition of the activation of both RhoA and Rac1 GTPases.86
The racemic form of (+)-morelloflavone (61), (±)-morelloflavone (62), isolated from the fruit pericarps of Garcinia brasiliensis Mart., exhibited inhibitory activity against the cysteine proteases papain (IC50 10.5 μM) and cruzain (IC50 9.6 μM) and the serine protease trypsin (IC50 3.8 μM).87 To improve on this activity, three semi-synthetic derivatives of (±)-morelloflavone (62) were synthesized, including (±)-7,4’,7’’,3’’’,4’’’-penta-O-acetylmorelloflavone (63), (±)-7,4’,7’’,3’’’,4’’’-penta-O-methylmorelloflavone (64), and (±)-7,4’,7’’,3’’’,4’’’-penta-O-butanoylmorelloflavone (65) (Figure 7). Compound 63 showed an enhanced inhibitory activity against papain (IC50 0.60 μM) but decreased activity toward cruzain (IC50 537 μM) and trypsin (IC50 21.1 μM), while 64 showed an increased inhibitory effect on trypsin (IC50 1.6 μM) but had decreased activities toward papain (IC50 15.4 μM) and cruzain (IC50 235 μM). In contrast, compound 65 slightly improved on the inhibition of cruzain (IC50 8.1 μM) but exhibited weakened activity toward papain (IC50 15.7 μM) and trypsin (IC50 19.8 μM) (Table 3).87 These data indicate that penta-acetylation and penta-methylation may enhance the ability of (±)-morelloflavone (62) in inhibiting the proteins papain and trypsin, respectively, but pentabutyrylation had limited impact on the resultant modulation of these proteins.87
Additional examples of biflavonoids of plant origin have been reported for their potential anticancer activity. For example, amentoflavone (66) was found to mediate its antitumor potential through multiple mechanisms, including induction of cancer cell apoptosis, inhibition of fatty acid synthase (FASN), down-regulation of human epidermal growth factor receptor 2 (HER2) protein and mRNA, and up-regulation of polyoma enhancer activator 3 (PEA3).88
In our earlier collaborative National Cooperative Drug Discovery Groups (NCDDG) project, three calycopterone biflavonoids containing a reduced C-4 carbonyl group, namely, (–)-calycopterone (67), (–)-isocalycopterone (68), and (–)-4-demethylcalycopterone (69) (Figure 7), were isolated by Drs. Wall and Wani and their co-workers at Research Triangle Institute in North Carolina from the flowers of Calycopteris floribunda Lamk. ex Poir. (syn. Getonia floribunda Roxb.) (Combretaceae), collected in Thailand. When tested against the A431 epidermoid, BC1 breast, Col2 colon, Lul lung, KB (HeLa) epidermoid, and KB-V1 vinblastine-resistant KB (HeLa) human cancer cell lines, as well the human U373 glioblastoma and HT fibrosarcoma and the P-388 murine leukemia cell lines, all these biflavonoids showed potent cytotoxicity, with ED50 values being in the range 0.2–9.6 μg/mL (0.33–15.6 μM) (Table 3).89 Investigation of the structures and cytotoxicity indicated that these compounds did not show specific selectivity toward most of the cancer cell lines tested, and the activity did not change greatly by altering the location of methoxy group. Also, several dimeric flavonoids showed more potent activity against A431, BC1, Col2, KB (HeLa), U373, and P-388 cells than their monomer, 5,4’-dihydroxy-3,6,7,3’-tetramethoxyflavone (70), which did not show any activity toward any of these cell lines [ED50 >20 μg/mL (>53.5 μM)] (Table 3).89 However, this methylated monomeric flavonoid (70) exhibited promising activities against KB (HeLa)-V1 cells [ED50 0.8 μg/mL (2.14 μM)],89 suggesting its selective antitumor potential and its sensitizing effects against the vinblastine-resistant KB (HeLa) cell line used.
Comparison of the activity toward HT-29 human colon cancer cells observed for (+)-morelloflavone (61, IC50 >10 μM)51 and (–)-calycopterone (67, IC50 0.24 μM)89 indicated that reducing the C-4 and C-4″ carbonyl groups, introducing a furan ring unit formed by an oxygen bridge at the C-5 and C-9’ positions, methylation of the hydroxy groups substituted in the A1, A2, and C1 rings, and the absence of hydroxy groups in the B1 and B2 rings all seem to contribute to the overall cytotoxicity of a given biflavonoid toward HT-29 cells.
In addition, several spiro-biflavonoids, (–)-abiesinols A (71), C (72), and E (73), isolated from the stem bark of Abies sachalinensis (F. Schmidt) Mast. (Pinaceae) (Figure 7), were found to exhibit potential antitumor-initiating effects that were comparable with a positive control when tested for their inhibition of the activation of NOR 1, a nitric oxide (NO) donor (Table 3).90 All these bioflavonoids showed a similar activity in this assay, indicating that the presence of a hydroxy group substituted at both the C-3’ and C-3’″ positions did not affect the resultant potency.
Thus, plant-derived biflavonoids could represent promising leads for the development of new anticancer agents, as a result of interacting with somewhat unusual molecular targets.
SESQUITERPENE LACTONES
Plant-derived sesquiterpene lactones (SQLs) have attracted wide and increasing interest as a result of their potent and selective antitumor activity mediated through disparate mechanisms. As a group, these include inhibition of the SERCA pump to increase the cytosolic Ca2+ concentrations that result in ER stress and cancer cell death, generation of iron-dependent free radicals that destroy cancer cells, regulation of the NF-κB, p53, and STAT3 signaling pathways that induce cancer cell apoptosis, and inhibition of angiogenesis and metastasis. Among these, inhibition of the activation of the NF-κB and STAT3 signaling pathway may decrease cancer cell resistance to existing chemotherapeutic agents or radiotherapy.91,92
In our continuing search on anticancer agents from higher plants, more than ten SQLs were purified and characterized as major cytotoxic components of the leaves of Piptocoma rufescens Cass. (Asteraceae), collected in the Dominican Republic, against HT-29 human colon cancer cells (Figures 8 and 9 and Table 4). These included five 8-methacryloxy and 8-isobutyroxygoyazensolide-type SQLs, (–)-goyazensolide (74), (–)-15-deoxygoyazensolide (75), (–)-2’,3’-dihydro-15-deoxygoyazensolide (76), (+)-rufescenolide A (77), and (–)-5-epiisogoyazensolide (78), three 8-angeloyloxygoyazensolide-type SQLs, (–)-lychnopholide (79), (+)-rufescenolide B (80), and (+)-4,5-dihydrolychnopholide (81), and a dimeric analogue, the 8-methacryloxygoyazensolide-type SQL, (+)-rufescenolide C (82), which was proposed to be formed through an ene-type reaction from (–)-15-deoxygoyazensolide. In addition, an inactive analogue, (+)-eremantholide C (83) (Figure 8), along with five 8-angeloyloxygermacranolides, (–)-ereglomerulide (84), (+)-rufesolide A (85), (–)-rufesolide B (86), (+)-rufesolide C (87), and (+)-rufesolide D (88) (Figure 9), were isolated from P. rufescens.93,94
Figure 8.
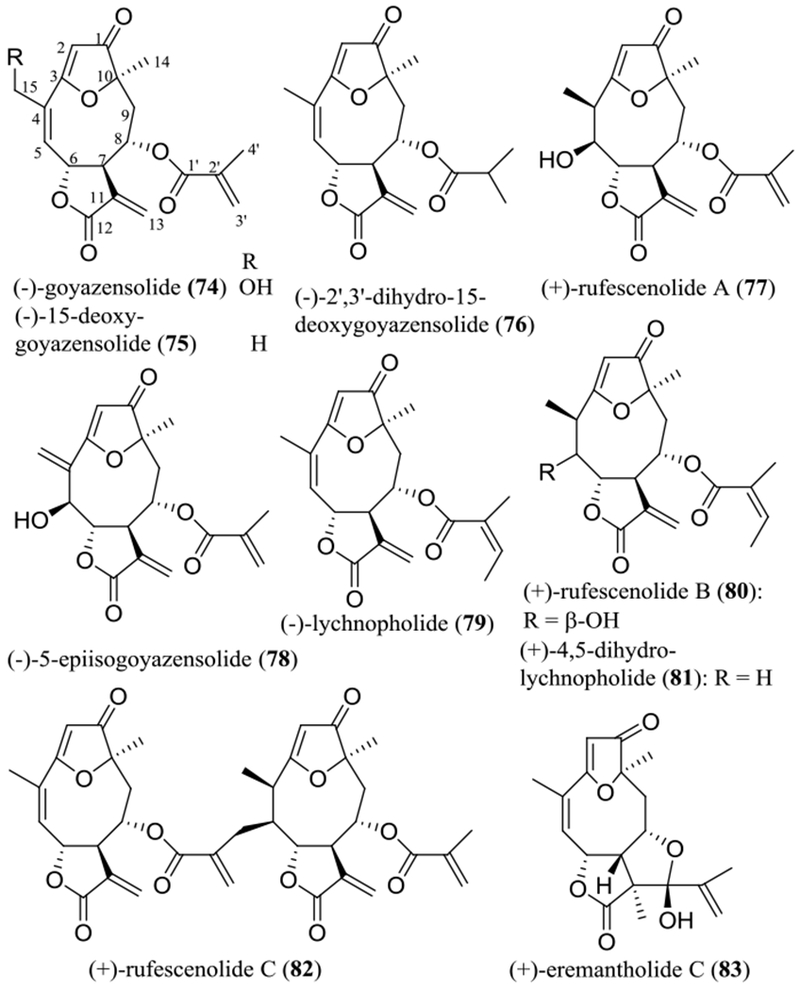
Structures of the goyazensolide-type sesquiterpene lactones (74–83) isolated from the leaves of Piptocoma rufescens.
Figure 9.
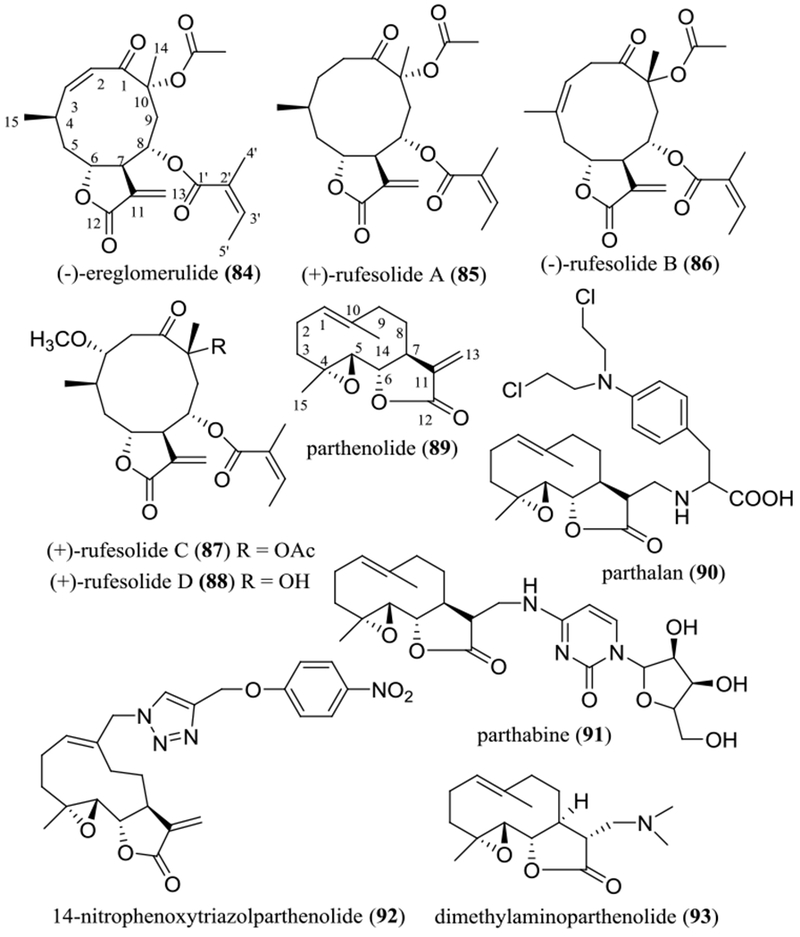
Structures of germacranolides (84–88) isolated from the leaves of Piptocoma rufescens and parthenolide (89) and its semi-synthetic derivatives (90–93).
Table 4.
Cytotoxicity of Sesquiterpene Lactones Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 74 | (–)-goyazensolide | Piptocoma rufescens (leaves) | 0.56a 0.86b 1.1c |
93 97 |
| 75 | (–)-15-deoxygoyazensolide | P. rufescens | 0.26a 1.1b 1.0c |
93 97 |
| 76 | (–)-2′,3′-dihydro-15-deoxygoyazensolide | P. rufescens | 0.58a | 93 |
| 77 | (+)-rufescenolide A | P. rufescens | 1.9a | 93 |
| 78 | (–)-5-epiisogoyazensolide | P. rufescens | 0.28a | 93 |
| 79 | (–)-lychnopholide | P. rufescens | 1.4a | 93 |
| 80 | (+)-rufescenolide B | P. rufescens | 6.6a | 93 |
| 81 | (+)-4,5-dihydrolychnopholide | P. rufescens | 0.56a | 93 |
| 82 | (+)-rufescenolide C | P. rufescens | 0.15a | 94 |
| 83 | (+)-eremantholide C | P. rufescens | >10a | 93 |
| 84 | (–)-ereglomerulide | P. rufescens | 1.2a 4.3b 4.3c |
93 97 |
| 85 | (+)-rufesolide A | P. rufescens | 3.0a 10.0b 8.0c |
93 97 |
| 86 | (–)-rufesolide B | P. rufescens | 4.0a | 93 |
| 87 | (+)-rufesolide C | P. rufescens | 1.4a | 93 |
| 88 | (+)-rufesolide D | P. rufescens | 1.0a | 93 |
| 89 | parthenolide | Michelia champaca (trunk bark) | 10.0b 7.5c >10d 7.5e 2.4f >10g 1.4h |
91 97 105 106 108 |
| 90 | parthalan | derived from 89 | 0.26d 1.8e |
105 |
| 91 | parthabine | derived from 89 | 0.24d 5.2e |
105 |
| 92 | 14-nitrophenoxytriazolparthenolide | derived from 89 | 0.43f 1.4g |
106 |
| 93 | dimethylaminoparthenolide (DMAPT) | derived from 89 | 1.7h | 108 |
HT-29 human colon cancer cell line (μM).
MOLM-13 human leukemia cell line (μM).
EOL-1 human leukemia cell line (μM).
HepG2 human hepatoma cell line (μM).
MCF-7 human breast cancer cell line (μg/mL).
HCT-116 human colon cancer cell line (μM).
A549 human lung non-small cell lung cancer cell line (μM).
Acute myelogenous leukemia (AML) cell line (μM).
(–)-Goyazensolide (74) is a germacranolide that contains a furan ring between the C-3 and C-10 positions, a carbonyl group at the C-1 position, and an ester group at the C-8 position. It was characterized initially as the main active compound from the aerial parts of Eremanthus goyazensis Sch.-Bip. (Asteraceae) against cercariae of Schistosoma mansoni, as part of an early search for schistosomicidal agents.95 Later, its planar structure was revised by single-crystal X-ray diffraction analysis, with the lactone ring being reassigned at C-6/C-7 rather than at the C-7/C-8 positions.96
The absolute configuration of (–)-goyazensolide (74) was determined as part of our joint project by analysis of its ECD and NOESY NMR spectroscopic data and by comparison of these data with the comparable values of its analogue, (+)-rufescenolide A (77), for which the absolute configuration at the C-5 position was determined using the Mosher ester method.93 This determination has been confirmed by single-crystal X-ray diffraction analysis for (–)-goyazensolide (74), with the data collected at 90 K, using Cu Kα radiation. Both the Flack parameter, refined to a value of −0.04(13), and the Hooft y parameter, determined as −0.03(4), after refinement by analysis of the Bijvoet pairs, indicated a (6R,7R,8S,10R) absolute configuration for (–)-goyazensolide (74).97
The structures and absolute configuration of compounds 75–88 were determined by analysis of their NMR and electronic circular dichroism (ECD) spectroscopic and single-crystal X-ray diffraction data and comparison of the values with those of (–)-goyazensolide (74). Investigation of the structures and ECD spectra of these compounds has confirmed that the early conclusions on the ECD spectra and the resultant configurations of SQLs are informative.98 The negative Cotton effects around 220 and 260 nm in the ECD spectra and the UV absorption maxima around 215 nm indicate a 7R absolute configuration for both goyazensolide-type SQLs and germacranolides, while the positive Cotton effects around 210 and 310 nm in the ECD spectra and the UV absorption maxima around 260 nm indicate a 10R configuration for goyazensolide-type SQLs only.93
All the SQLs isolated from P. rufescens except for (+)-eremantholide C (83, IC50 >10 μM) exhibited cytotoxicity toward HT-29 human colon cancer cells, with IC50 values in the range 0.15–6.6 μM (Table 4),93,94 and several of these compounds, including (–)-goyazensolide (74), (–)-15-deoxygoyazensolide (75), (–)-ereglomerulide (84), and (+)-rufesolide A (85) also showed inhibitory activities against MOLM-13 and EOL-1 human leukemia cells.97 In collaboration with colleagues at Nationwide Children’s Hospital, Columbus, Ohio, it was found that (–)-goyazensolide (74) inhibited Sch10545 NF2-deficient mouse schwannoma and Ben-Men-1 human benign meningioma cell growth, with IC50 values as around 1.0 μM.99
Interestingly, (–)-lychnopholide (79) showed potent activity in all of a panel of 52 human cancer cell lines [IC50 0.08–5.0 μM, except for SNB-19-CNS cells (IC50 13.8 μM)] when tested by the U.S. National Cancer Institute (NCI, USA).100 However, its structurally similar analogue, (+)-eremantholide C (83), which contains an additional furan ring formed by direct connection of the C-11 and C-1’ positions, was found to be much less potent, showing IC50 values in the range 5.0–50.0 μM [except for SNB-19-CNS cells (IC50 >100 μM)].93,100 These results suggest that the C-11/C-13 double bond, together with the C-1’ carbonyl group, is important for goyazensolide-type SQLs to mediate their activity toward human cancer cells, but the formation of an additional furan ring between the lactone and the C-8 ester units is not preferred for such activity.
Consistent with these observations, all the cytotoxic sesquiterpene lactones 74–82 and 84–88 contain an α-methylene-γ-lactone ring at the C-6 and C-7 positions and an α,β-unsaturated ester group at the C-8 position, indicating the importance of these functionalities. Also, the goyazensolide-type SQLs (74–82) exhibit overall more potent activity than several germacranolides (84–88) that were obtained, indicating that either the enone O=C-C=CH2 system (C-1–C-5) or the C-3/C-10 furan ring can increase activity, as supported by their decreased cytotoxic potency toward human leukemia cells observed in the sequence (–)-goyazensolide (74)/( –)-15-deoxygoyazensolide (75), (–)-ereglomerulide (84), and (+)-rufesolide A (85)/parthenolide (89), and with a more potent activity toward the HT-29 cells observed for the dimeric (+)-rufescenolide C (82), as compared with its monomers.93,94,97 Further investigation of cytotoxicity against HT-29 human colon cancer cells of the goyazensolide-type SQLs showed that the presence of an 8-methacryloxy group is superior to an 8-angeloyloxy group, as implied by data for compounds 75 (IC50 0.26 μM) and 79 (IC50 1.4 μM) and for 77 (IC50 1.9 μM) and 80 (IC50 6.6 μM). Introducing a 5β-hydroxy group decreases activity, as indicated from the testing of compounds 80 (IC50 6.6 μM) and 81 (IC50 0.56 μM). However, substitution of the C-15 hydroxy group [74 (IC50 0.56 μM) vs. 75 (IC50 0.26 μM)] or saturation of the C-11/C-13 double bond [75 (IC50 0.26 μM) vs. 76 (IC50 0.58 μM)] seems not to affect the cytotoxic potency greatly (Figure 8 and Table 4).93
When comparing the cytotoxicity of the goyazensolide-type SQLs with those of a reference SQL, parthenolide (89), both (–)-goyazensolide (74) and (–)-15-deoxygoyazensolide (75) were found to show more potent cytotoxicity toward MOLM-13 and EOL-1 human leukemia cells. Both compounds, but not parthenolide (89), also exhibited a synergistic effect with PKC412 (midostaurin) toward these leukemia cell lines when tested in micromolar levels, which was mediated partially through inhibition of NF-κB.97 This evidence indicates that both (–)-goyazensolide (74) and (–)-15-deoxygoyazensolide (75) could be used in conjunction to improve the therapeutic efficacy of the approved anticancer drug, PKC412.
In an in vivo hollow fiber assay conducted at the University of Illinois at Chicago, cancer cell growth was inhibited significantly when the immunodeficient NCr nu/nu mice that were implanted with hollow fibers containing HT-29 human colon cancer cells and treated (i.p.) with (–)-goyazensolide (74) at a dose of 12.5 mg/kg daily for four days.101 Mechanistically, (–)-goyazensolide (74) inhibited HT-29 colon cancer cell growth in vitro and in vivo through down-regulation of IKKα and IKKβ, up-regulation of ROS levels and caspase-3 expression, and induction of cellular apoptosis by a G1 blockade in the cell cycle progression.101 In addition, (–)-goyazensolide (74) decreased the proliferation of Sch10545 and Ben-Men cells by reducing the levels of cyclins A and E, phospho-Aκt, and NF-κB, and thus it was proposed as being of interest for further biological evaluation against neurofibromatosis type 2 (NF2)-associated schwannomas and meningiomas.99
Parthenolide (89) is an epoxylated germacranolide isolated from sources including Chrysanthemum parthenium (L.) Bernh. [syn. Tanacetum parthenium (L.) Sch.-Bip] (Asteraceae) and Michelia champaca L. (syn. Magnolia champaca Baill. ex Pierre) (Magnoliaceae).91 As summarized in a previous review article, this SQL shows cytotoxicity against various human cancer cells, and it inhibits selectively cancer stem cell (CSC) growth in leukemia and solid tumors. It also exhibits promising antitumor efficacy mediated through inhibition of the NF-κB pathway.91A recent investigation showed that parthenolide (89) was cytotoxic toward Panc-1 and BxPC3 human pancreatic cancer cells. It increased the percentage of autophagic cells and the expression levels of p62/SQSTM1, Beclin 1, and LC3II in Panc-1 cells, indicating that this compound inhibits pancreatic cancer cell growth by autophagy-mediated apoptosis.102 Also, parthenolide (89) was proposed to have promising immunotherapeutic properties, due to its inhibitory effects on anti-CD3-induced mobilization of intracellular Ca2+ in Jurkat T cells. Thus, this compound was suggested to selectively inhibit the initial phases of T cell receptor (TCR) activation, a molecular target for the treatment of T cell-mediated inflammatory and autoimmune diseases.103
Parthenolide (89) was found to bind to ribosomal protein L10 (RPL10) and to reduce the T-cell factor (TCF)/lymphoid enhancer factor (LEF)1 protein levels, and thereby to inhibit the Wnt/β-catenin signaling pathway. In addition, parthenolide (89) targets and decreases levels of RPL10, which leads to reduced protein levels of p65 and IKKγ, contributing, at least in part, to its NF-κB inhibitory activity. Thus, parthenolide (89) functions as a Wnt/β-catenin signaling inhibitor targeting RPL10 to block TCF4/LEF1 synthesis, indicating the possibility of the development of this compound for the treatment of Wnt-mediated cancers.104
To improve on the potency of parthenolide (89), two analogues, parthalan (90) and parthabine (91), have been prepared. Both substances (90 and 91) were more cytotoxic than parthenolide (89) and the standard anticancer control drugs, cytarabine and melphalan, against human MCF-7 breast cancer, HepG2 hepatoma, and LNCaP prostate cancer cells.105 In addition, a series of melampomagnolide B-triazole conjugates has been synthesized and evaluated for their cytotoxicity toward a small panel of human cancer cell lines. Of these compounds, 14-nitrophenoxytriazolparthenolide (92) showed improved activity when compared with the parent compound, parthenolide (89).106
In a phase I dose escalation study, the plasma parthenolide concentrations of patients administered by low doses of parthenolide (89) were unable to be detected in a high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry (HPLC-APCI-MS), which was caused probably by its poor bioavailability.107 To overcome this problem, several water-soluble aminoparthenolides have been synthesized and tested toward primary acute myeloid leukemia (AML) cells, of which dimethylaminoparthenolide (DMAPT or LC-1, 93) was found to be potently cytotoxic.108 The potential anticancer activity of DMAPT (93) has been reviewed previously.91 For example, it exhibited inhibitory activity toward the CWR22Rv1 androgen-independent and PC3 human prostate cancer cell lines, as well as the corresponding tumor xenografts, through generating reactive oxygen species (ROS) and inhibition of NF-κB activity.109
Radiation therapy (RT) is a critical treatment mode used for advanced non-small cell lung carcinoma (NSCLC), but its effectiveness is limited by concomitant normal tissue damage. RT causes lesions characterized by DNA double-strand breaks (DSBs), which initiate multiple repair pathways, including homologous recombination (HR) and non-homologous end joining (NHEJ).110 In addition, inhibition of the NF-κB pathway has been proposed as a main mechanism of radiosensitization.110 DMAPT (93) was found to decrease NF-κB activity and to abrogate IR-induced NF-κB activity, to reduce HR and NHEJ in NSCLC cells, and to impair the DNA DSB repair pathway. In turn, DMAPT (93) was found to radiosensitize NSCLC cells through inhibition of the NF-κB pathway and by blocking DNA DSBs with targets at HR and NHEJ.110
Following these observations, DMAPT (93) was tested for its radioprotective activity. Radiation-induced apoptosis in transgenic adenocarcinomas of mouse prostate (TRAMP) tumor tissue was found to be enhanced, but the normal tissues was protected from high-dose-radiation-induced apoptosis when 16-week-old male TRAMP mice were treated orally with DMAPT (93) (100 mg/kg, three times a week) for one week. This indicates that DMAPT (93) is a promising radioprotector of normal tissues or cells for possible use in conjunction with radiotherapy for prostate cancer.111
As discussed above, parthenolide (89) has been investigated thoroughly for its potent and promising antitumor activity, and it is regarded as a representative of a new class of leukemia stem cells (LSC)-targeted anticancer drugs, owing to its selective induction of human leukemia cell apoptosis through inhibition of NF-κB, proapoptotic activation of p53, and increasing ROS production.112 In our collaborative investigation, both goyazensolide (74) and 15-deoxygoyazensolide (75) showed an enhanced cytotoxicity toward MOLM-13 and EOL-1 human leukemia cells when compared with parthenolide (89), and these compounds exhibited a synergistic effect with midostaurin against both MOLM-13 and EOL-1 cells.97 Thus, in addition to parthenolide (89), goyazensolide-type SQLs may be promising compounds for the development of new antileukemic agents.
ARYLNAPHTHALENE LIGNAN LACTONES
Arylnaphthalene lignan lactones are isolated mainly from the plant families Acanthaceae, Lamiaceae, Linaceae, Myristicaceae, Phyllanthaceae, and Rutaceae, and many of these compounds have been reported for their potent cytotoxicity toward human cancer cells.113 As part of our collaborative work, several arylnaphthalene lignan lactones, including (–)-phyllanthusmins A–E (94–98) and (–)-cleistanthin B (99) (Figure 10), were isolated from different plant parts of Phyllanthus poilanei Beille (Phyllanthaceae) collected in Vietnam.113 A known analogue, (+)-acutissimalignan A (100) has been characterized from the aerial parts of Phyllanthus songboiensis N.N. Thin collected in Vietnam.114 Also, two analogues, diphyllin (101) and (–)-2’-acetylphyllanthusmin D (102), were prepared from (–)-phyllanthusmin C (96).113
Figure 10.
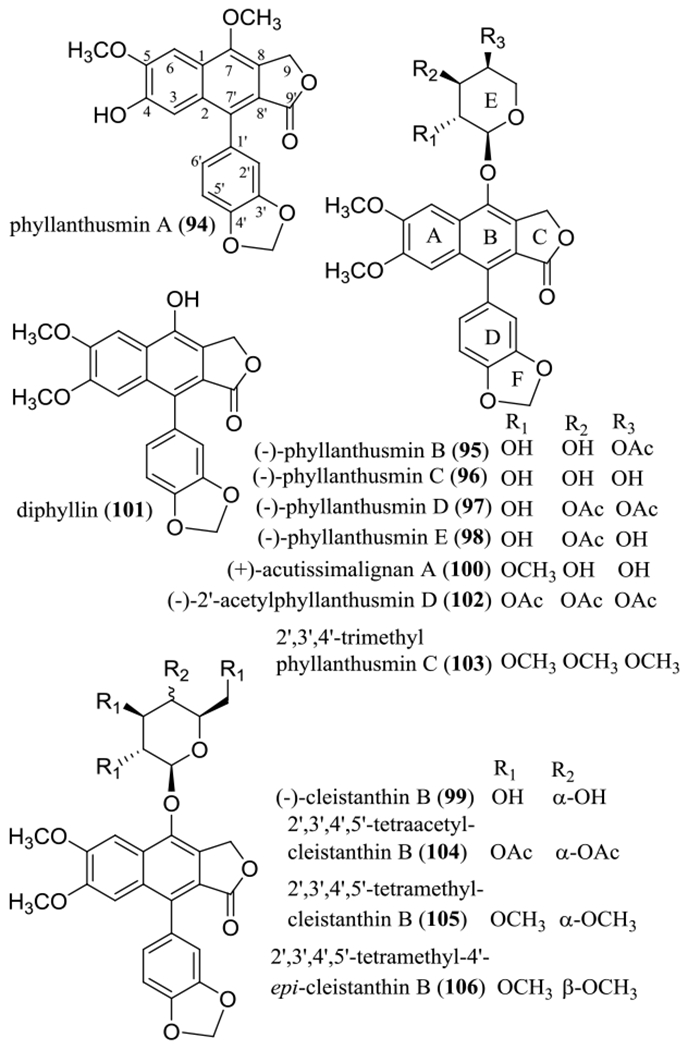
Structures of the arylnaphthalene lignan lactones isolated from different plant parts of Phyllanthus poilanei (94–99) and the aerial parts of Phyllanthus songboiensis (100) and their selected semi-synthetic derivatives (101–106).
Most of these lignans obtained were active when evaluated against HT-29 human colon cancer cells, and (–)-phyllanthusmins C and D (96 and 97) also exhibited selective activity against HT-29 cells, as indicated by their non-cytotoxicity observed against CCD-112CoN human normal colon cells.113 The activity toward HT-29 cells was found to decrease in the sequence 100, 102, 97, 95/98, 96, 101, and 94/99 (Table 5), indicating that a methyl or acetyl substituted 7α-l-arabinose unit is important for this activity. For example, (–)-phyllanthusmin C (96, IC50 3.2 μM) was more active than diphyllin (101, IC50 7.6 μM) and (–)-cleistanthin B (99, IC50 >10 μM), indicating that a C-7 α-l-arabinose unit contributes to this activity more than a C-7 β-D-glucose unit. The presence of one or more lipophilic acetyl or methyl groups linked to the arabinose residue improved the cytotoxicity of the lignan lactone glycosides, as indicated by comparing data for (–)-phyllanthusmin C (96, IC50 3.2 μM), (+)-acutissimalignan A (100, IC50 0.019 μM), and (–)-2’-acetylphyllanthusmin D (102, IC50 0.11 μM). However, the location of the acetyl group at C-3″ or C-4″ contributed equally to this activity, as implied by data obtained for (–)-phyllanthusmin B (95, IC50 1.8 μM) and (–)-phyllanthusmin E (98, IC50 1.8 μM). Based on a comparison of phyllanthusmin A (94, IC50 >10 μM) and diphyllin (101, IC50 7.6 μM) (Table 5), all of the C-4 and C-5 methoxy groups and the C-7 hydroxy group seem to be required for diphyllin (101) to mediate its cytotoxicity.113
Table 5.
Cytotoxicity of Lignan Lactones Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 94 | phyllanthusmin A | Phyllanthus poilanei (different plant parts) | >10a | 113 |
| 95 | (–)-phyllanthusmin B | P. poilanei | 1.8a | 113 |
| 96 | (–)-phyllanthusmin C | P. poilanei | 3.2a, | 113 |
| 97 | (–)-phyllanthusmin D | P. poilanei | 0.17a | 113 |
| 98 | (–)-phyllanthusmin E | P. poilanei | 1.8a | 113 |
| 99 | (–)-cleistanthin B | P. poilanei | >10a | 113 |
| 100 | (+)-acutissimalignan A | Phyllanthus songboiensis (aerial parts) | 0.019a | 114 |
| 101 | diphyllin | derived from 96 | 7.6a | 113 |
| 102 | (–)-2’-acetylphyllanthusmin D | derived from 96 | 0.11a
0.14a |
113 115 |
| 103 | 2′,3′,4′-trimethylphyllanthusmin C | derived from 96 | 0.13a | 115 |
| 104 | 2′,3′,4′,5′-tetraacetylcleistanthin B | derived from 99 | 0.043a | 115 |
| 105 | 2′,3′,4′,5′-tetramethyl-cleistanthin B | derived from 99 | 0.05b | 116 |
| 106 | 2′,3′,4′,5′-tetramethyl-4′-epi-cleistanthin B | derived from 101 | 0.01b | 116 |
| 107 | unnamed | synthetic compound | >10a | 115 |
| 108 | unnamed | synthetic compound | 1.4a | 115 |
| 109 | unnamed | synthetic compound | 0.47a | 115 |
| 110 | justicidin A | Justicia ciliata (whole plants) | 0.0018c | 117 |
| 111 | chinensinaphthol | J. ciliata | >10c | 117 |
| 112 | taiwanin C | Phyllanthus acutissima (aerial parts) | >5d >5e |
118 |
| 113 | isogadian | P. acutissima | >5d >5e |
118 |
| 114 | PHY34 | derived from 101 | 0.043a 0.0042f 0.0040g |
121 |
| 115 | podophyllotoxin | Bursera fagaroides (bark) | 0.015b | 126 |
| 116 | etoposide | derived from 115 | >10a | 113 127 |
HT-29 human colon cancer cell line (μM).
A549 human lung non-small cell lung cancer cell line (μM).
HT-3 human cervical carcinoma cell line (μg/mL).
KB human oral epidermoid carcinoma cell line (μg/mL).
P-388 murine leukemia cell line (μg/mL).
OVCAR3 human ovarian cancer cell line (μM).
OVCAR8 human ovarian cancer cell line (μM).
Following these observations, series of acetylated or methylated diphyllin glycosides showing more potent cytotoxicity than (–)-phyllanthusmin C or D (96 or 97) toward HT-29 human colon cancer cells has been synthesized, including (–)-2’-acetylphyllanthusmin D (102, IC50 0.11 μM), 2’,3’,4’-trimethylphyllanthusmin C (103, IC50 0.13 μM), and 2’,3’,4’,5’-tetraacetylcleistanthin B (104, IC50 0.043 μM) (Figure 10, Table 5).113,115 Compound 103 was also found to show potent cytotoxicity toward K/VP.5 VP-16-resistant K562 human immortalized myelogenous leukemia cells (IC50 1.3 μM),115 indicating that this methylated arylnaphthalene lignan lactone could be a promising lead to combat the problem of cancer cell resistance to etoposide.
Members of another series of acetylated, methylated, and ethylated diphyllin glycosides have been synthesized, with their cytotoxicity evaluated toward HCT-116 human colon, RM-1 murine prostate, A549 human lung cancer cells, as well A549T human lung paclitaxel-resistant carcinoma cells.116 All these synthetic compounds showed selective activities among the cancer cell lines tested, and 2’,3’,4’,5’-tetramethyl-cleistanthin B (105) and 2’,3’,4’,5’-tetramethyl-4’-epi-cleistanthin B (106) were found to be the most potent against A549 cells, showing IC50 values of 50 and 10 nM, respectively.116 Interestingly, compound 105 exhibited topoisomerase II (topo II) inhibitory activity, but 106 did not. A further in vitro tubulin polymerization study showed that both compounds exhibited antimicrotubule activity in a paclitaxel-like mode,116 indicating that the diphyllin glycosides target topo II and/or tubulin, and the saccharide unit may function critically for binding to these molecular targets.
To test the importance of the lactone unit, compounds 107–109 were synthesized and evaluated for their cytotoxicity toward HT-29 cells (Figure 11 and Table 5).115 The activity was found to be decreased in the sequence 102 (IC50 0.14 μM), 109 (IC50 0.47 μM), 108 (IC50 1.4 μM), and 107 (IC50 >10 μM) (Table 5), suggesting that the lactone unit is important. Opening the lactone ring system (107) resulted in a loss of activity, and either changing the C-9’ carbonyl group to a methylene group or exchanging the C-9’ carbonyl group and the C-9 methylene group decreased the cytotoxic potency of the acetylated arylnaphthalene lignan lactone glycoside 102.115
Figure 11.
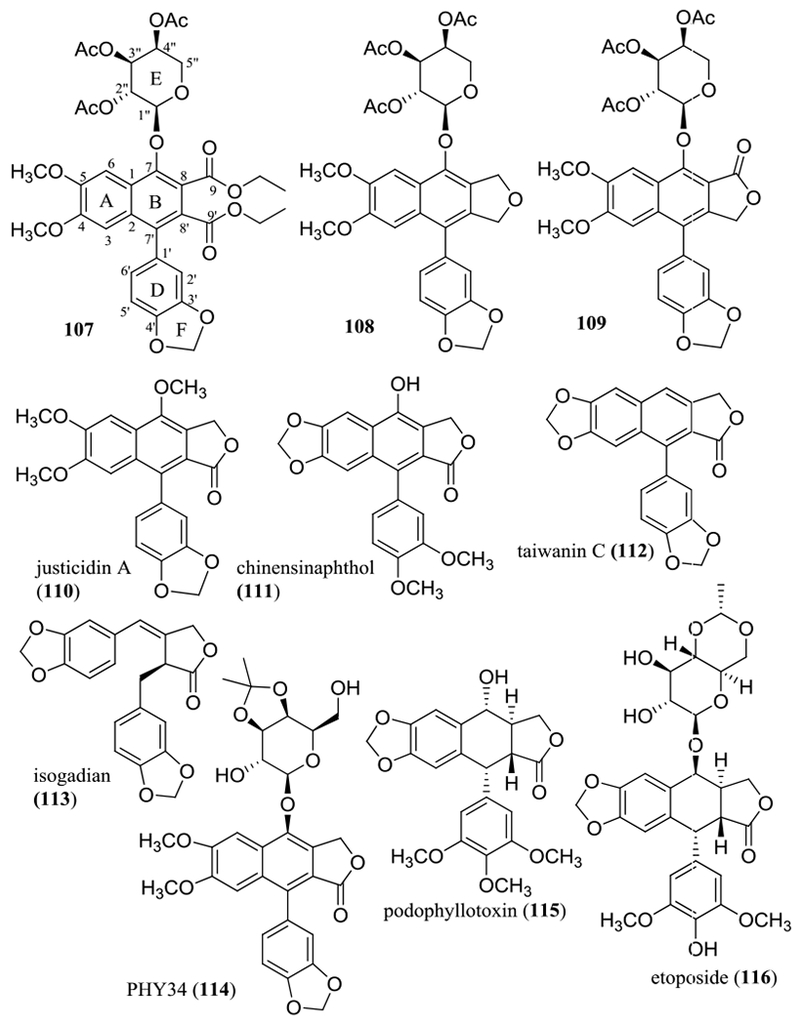
Structures of selected naturally occurring and semi-synthetic arylnaphthalene lignan lactone derivatives (107–114) and podophyllotoxin (115) and etoposide (116).
The importance of the C-4 and C-5 methoxy groups and the C-7 hydroxy group has been supported by cytotoxicity against a small panel of human cancer cell lines observed for justicidin A (110) and chinensinaphthol (111), with both compounds being isolated from the whole plants of Justicia ciliata (Yamamoto) Hsieh & Huang (syn. J. hayatai Yamamoto) (Acanthaceae) (Figure 11). Justicidin A (110) was found to be highly cytotoxic, with IC50 values being in the range 1.8–7.4 ng/mL (4.6–18.8 nM), but chinensinaphthol (111), which contains a 4,5-methylenedioxy unit and a 7-hydroxy group, was found to be inactive [IC50 >10 μg/mL (>26.3 μM)].117 Consistent with these observations, both taiwanin C (112), an analogue of 111 without a hydroxy group at the C-7 position, and isogadian (113), an analogue of 112 with cleaved ring B, isolated from the aerial parts of Phyllanthus acutissimus Miq. (Phyllanthaceae), were non-cytotoxic against a small panel of human cancer cell lines (Figure 11 and Table 5).118
Following these SAR observations, the cytotoxic potency of arylnaphthalene lignan lactone glycosides was found to be enhanced by acetylation of the glycose unit or introducing a cyclic lipophilic group at this unit.119,120 After much experimentation, a promising diphyllin-β-D-galactoside analogue, namely, PHY34 (114), has been prepared at Ohio State University by cyclization of a 2″,3″-dihydroxy group of galactose unit through acetal formation. This synthetic derivative showed highly cytotoxic activity toward human HT-29 colon (IC50 43.3 nM) and OVCAR3 (IC50 4.2 nM) and OVCAR8 (IC50 4.0 nM) ovarian cancer cells (IC50 values observed for paclitaxel toward these cell lines were 10.6, 10.7, and 8.2 nM, respectively) (Table 5).121
In an in vivo hollow fiber assay conducted at the University of Illinois at Chicago, (–)-phyllanthusmin D (97) exhibited potential antitumor activity when immunodeficient NCr nu/nu mice were implanted with HT-29 cells and treated (i.p.) with this compound daily at doses of 10.0, 15.0, or 20.0 mg/kg for four days.113 In this same in vivo assay, the growth of all HT-29, OVCAR3, and OVCAR8 cancer cells was found to be inhibited when mice were treated (i.p.) with PHY34 (114) daily at a dose of 0.75 mg/kg for four days.121 Also, in an ovarian tumor xenograft model conducted at the University of Illinois at Chicago, a significant inhibition of tumor growth was observed when six- to eight-week-old NCr nu/nu athymic female mice inoculated with OVCAR8-RFP cells (OVCAR8 cells expressing red fluorescent protein) were treated (i.p.) with PHY34 (114) (0.75 mg/kg, once daily, three times per week) for three weeks.121
Mechanistically, (–)-phyllanthusmin D (97) induces cancer cell apoptosis through activation of caspase-3, but it did not inhibit DNA topoisomerase IIα activity.113 Interestingly, PHY34 (114) was found to exert its cytotoxic effects in human ovarian cancer cells by inhibiting autophagy at a late stage, and such autophagy inhibition was found to be necessary for PHY34 (114) to induce ovarian cancer cell apoptosis.121 Mutations in autophagy genes can cause genetic diseases such as cancer, and autophagy contributes to cancer as both a tumor survival promoter and a tumor suppressor.122,123 PHY34 (114) inhibited autophagy in human ovarian cancer cells and thus shows promise for the development of ovarian cancer chemotherapeutic agent targeting autophagy.121
It is worthy of note that, when investigated collaboratively at the Wexner Medical Center of The Ohio State University, (–)-phyllanthusmin C (96) was found to induce interferon-γ (IFN-γ) production by human natural killer (NK) cells, with or without the presence of interleukin-12 (IL-12). It stimulated IFN-γ production in both human CD56bright and CD56dim NK cell subsets through upregulation of toll-like receptor (TLR)-mediated NF-κB signaling, but it induced little T cell IFN-γ production or NK cell cytotoxicity.124 NK cells are large granular lymphocytes derived from CD34+ hematopoietic progenitor cells that could kill a target cell “naturally” and function as a critical component of innate immunity in the host cancer immune surveillance network.125 NK cells contribute to the control of cancer mainly through lysis of cancer cells by CD56dim NK cells and release of IFN-γ by CD56bright NK cells. (–)-Phyllanthusmin C (96) enhanced IFN-γ production in both CD56bright and CD56dim NK subsets, with or without IL-12, indicating that this compound could be translationally promising for the development of new agents to treat cancer targeting NK cells.124 It also provides a new mechanism of enhancing NK cell immunosurveillance in the search for new anticancer agents from natural products.
Podophyllotoxin (115), an arytetralin lignan structurally similar to phyllanthusmin A (94), showed potent cytotoxicity toward human A2780 ovarian (IC50 18 nM) and A549 lung non-small cell lung (IC50 15 nM) (Table 5) cancer cells. Based on this lead compound, the anticancer drug etoposide (VP-16) (116) has been developed.4,126,127 However, side effects including myelosuppression and the development of secondary leukemia linked to topo II inhibitory activity have been observed for this standard clinical agent (116).128 Thus, it is desirable to discover new etoposide-related agents to treat cancer by diverse mechanisms of action. Significantly, (–)-phyllanthusmins C (96) and D (97) and PHY34 (114) were found to mediate their antitumor activity through stimulation of human NK cell activity,124 induction of cell apoptosis by activation of caspase-3,113 and inhibition of autophagy,121 respectively. Thus, these arylnaphthalene lignan lactones (96, 97, and 114) are promising leads for the development of new alternatives to etoposide (116).
PHYTOSTEROLS
As mentioned above, adaptive and innate immune NK cells play a critical role in the host cancer immunosurveillance and immunoediting networks and have been proposed as a potential candidate for adoptive cellular therapy, for use as a stand-alone therapy or an adjunct to hematopoietic stem cell transplants and novel genetic engineered NK cells with the improved antitumor activity.129 In our search for anticancer agents from tropical plants, a previously known phytosterol glycoside ester, namely, (–)-β-sitosterol-3-O-β-D-(6-O-palmitoyl)glucopyranoside (117), was isolated and identified from the aerial parts of Phyllanthus songboiensis collected in Vietnam (Figure 12).114 This compound was found to be non-cytotoxic against HT-29 human colon cancer cells (IC50 >10 μM) (Table 6), but it was active when tested for NK stimulatory effects at the Wexner Medical Center of The Ohio State University. Human NK cells treated by interleukin-12 (IL-12) plus 117 produced more IFN-γ than those treated by IL-12 alone, indicating that 117 is able to induce NK cells to produce more IFN-γ in the presence of IL-12.114
Figure 12.
Structures of sitosterol derivative 117 isolated from the aerial parts of Phyllanthus songboiensis and its analogues (118 and 119) showing human NK cell stimulatory activity.
Table 6.
Cytotoxicity of Steroids Isolated from Selected Tropical Plants and Their Analogues
| no. | compound | source [species (part)] | IC50 | ref. |
|---|---|---|---|---|
| 117 | (–)-β-sitosterol-3-O-β-d-(6-O-palmitoyl)glucopyranoside | Phyllanthus songboiensis (aerial parts) | >10a | 114 |
| 118 | (–)-β-sitosterol-3-O-β-d-glucopyranoside | Cratoxylum cochinchinense (stems) | >10a | 29 |
| 119 | β-sitosterol |
Lysimachia japonica (aerial parts) Duguetia glabriuscula (leaves and stem bark) |
>10b 4.7c |
130 131 |
| 120 | (+)-strebloside | Streblus asper (stem bark) | 0.17a 0.11d 0.09e 0.09f 0.13g 0.46h 0.54i 0.09j |
138 139 |
| 121 | (+)-19-hydroxykamaloside | S. asper | 0.16a | 138 |
| 122 | (+)-5-hydroxyasperoside | S. asper | 0.69a | 138 |
| 123 | (+)-3-O-β-d-fucopyranosylperiplogenin | S. asper | 0.09a | 138 |
| 124 | strophanthidin | Antiaris toxicaria (trunk bark) | 0.61b 0.11k 0.38l |
142 |
| 125 | convallatoxin | A. toxicaria | 0.014b 0.0027k 0.0065l |
142 |
| 126 | calotropagenin | Asclepias curassavica (whole plants) | 0.48m | 143 |
| 127 | calotropin | A. curassavica | 0.03m | 143 |
| 128 | 3-O-β-d-glucopyranosyl-(1→4)-6-deoxy-β-d-allopyranosyl-17β-hydroxyuzarigenin | Asclepias syriaca (above-ground biomass) | >10n >10o >10p |
144 |
| 129 | 3-O-β-d-glucopyranosyl-(1→4)-6-deoxy-β-d-allopyranosyluzarigenin | A. syriaca | 5.3n 1.8o 0.59p |
144 |
| 130 | 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyldigitoxigenin | Thevetia peruviana (fruits) | 0.40q 0.42r 0.62s |
145 |
| 131 | 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyluzarigenin | T. peruviana | 2.7q 3.4r 4.5s |
145 |
| 132 | 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyl-19-oxo-cannogenin | T. peruviana | >10q >10r >10s |
145 |
| 133 | digoxin | Digitalis lanata (leaves) | 0.38a 0.024n |
138 141 151 |
| 134 | digitoxin | Digitalis purpurea (leaves) | 0.068a 0.010n |
139 140 141 |
| 135 | oleandrin | Nerium indicum (leaves) | 0.014t | 162 |
HT-29 human colon cancer cell line (μM).
KB human oral epidermoid carcinoma cell line (μg/mL).
Hep-2 human laryngeal cancer cell line (μg/mL).
H1299 human lung cancer cell line (μM).
Kasumi-1 human leukemia cell line (μM).
MV4-11 human leukemia cell line (μM).
OVCAR3 human ovarian cancer cell line (μM).
OVCAR4 human ovarian cancer cell line (μM).
OVCAR5 human ovarian cancer cell line (μM).
OVCAR8 human ovarian cancer cell line (μM).
A549 human lung non-small cell lung cancer cell line (μg/mL).
MCF-7 human breast cancer cell line (μg/mL).
DU-145 human prostate cancer cell line (μM).
MCF-7 human breast cancer cell line (μM).
T47D human breast cancer cell line (μM).
Hs578T human breast cancer cell line (μM).
P15 human lung cancer cell line (μM).
SW1990 human pancreatic cancer cell line (μM).
MCG-803 human gastric cancer cell line (μM).
HPB-ALL human T-cell leukemia cell line (μM).
Similarly, (–)-β-sitosterol-3-O-β-D-glucopyranoside (118) isolated from the stems of Cratoxylum cochinchinense, collected in Vietnam, was found to be inactive toward HT-29 human colon cancer cells (IC50 >10 μM).29 β-Sitosterol (119) isolated from the aerial parts of Lysimachia japonica Thunb. (Primulaceae) and the leaves and stem bark of Duguetia glabriuscula R.E. Fr. (Annonaceae) did not show any activity [IC50 >100 μg/mL (>241.5 μM)] toward KB (HeLa) human epidermoid carcinoma cells and exhibited weak activity [IC50 4.7 μg/mL (11.4 μM) toward Hep-2 human laryngeal cancer cells.130,131 However, these two phytosterols and their mixture increased T-cell proliferation and T-cell secretion of IL-2 and IFN-γ in the presence of phytohaemagglutinin (PHA), and they also enhanced human NK cell activity.132
A liposomal β-sitosterol (LS) formulation composed of egg yolk phosphatidylcholine has been prepared, and its antitumor activity has been evaluated. When five-week-old C57BL/6 male mice injected with B16-BL6 melanoma cells were treated orally with LS (4 μmol/mouse, daily, seven times), NK cell activity was increased after a one-week treatment, and lung tumor metastasis was prevented after a two-week treatment. This suggests that the lung tumor metastasis inhibitory activity of LS resulted from enhancing immune surveillance activity in mice.133
The signaling interferon proteins are made and released by host cells in response to the presence of pathogens, of which IFN-γ is essential for both innate and adaptive immune responses in the clearance of viral infections and for the host defense against malignant transformation.125,134 In addition, IFN-α can directly inhibit normal or cancer cell proliferation, and the administration of IFN-α has a curative therapeutic action on cancer patients. Thus, this cytokine (IFN-α) has been proposed as an immune adjuvant for the development of a cancer vaccine to be used in cancer immunotherapy.135
Increasing research efforts and clinical studies show that NK cells are effective in cancer treatment, and compounds that enhance NK cell activity are of considerable interest. Thus far, several natural products have been investigated for their NK cell stimulatory activity, including curcumin, genistein, and resveratrol.136 Thus, the documentation of NK cell stimulatory activity for β-sitosterol (119) and its glucosides β-sitosterol-(6-O-palmitoyl)glucopyranoside (117) and (–)-β-sitosterol-3-O-β-D-glucopyranoside (118), could be valuable in the future design of effective cancer chemotherapy targeting NK cells.
CARDIAC GLYCOSIDES
Cardiac glycosides (CGs) derived mainly from the Apocynaceae, Asclepiadaceae, Asparagaceae, Crassulaceae, Moraceae, and Plantaginaceae plant families, have attracted wide interest, not only due to their long-established therapeutic use in treating congestive heart disease, but also for their potential anticancer and antiviral effects as a result of targeting Na+/K+-ATPase.137
Several new and known cardiac glycosides, including (+)-strebloside (120), (+)-19-hydroxykamaloside (121), (+)-5-hydroxyasperoside (122), and (+)-3-O-β-D-fucopyranosylperiplogenin (123), were identified as the major cytotoxic components against HT-29 human colon cancer cells from the stem bark of Streblus asper Lour. (Moraceae), collected in Vietnam (Figure 13, Table 6).138 Among these compounds, (+)-strebloside (120) showed potent activity toward HT-29 cells, but was not found to be cytotoxic toward CCD-112CoN normal human colon cells. It also exhibited a broad spectrum of cytotoxicity toward both human solid tumor and leukemic cells, including H1299 lung and OVCAR3, 4, 5, and 8 ovarian cancer cells and Kasumi-1 and MV4–11 human leukemia cells.138,139
Figure 13.
Structures of cytotoxic cardiac glycosides (120–123) isolated from the stem bark of Streblus asper and their analogues (124 and 125).
A preliminary SAR study showed that all of the C-3–C-5, C-14, and C-19 positions are important for CGs to mediate their cytotoxicity toward human cancer cells.138 For example, CGs are found frequently to be more active than their corresponding aglycones, as indicated by the cytotoxicity toward A549 human lung cancer cells observed for strophanthidin [124, IC50 0.11 μg/mL (0.27 μM)] and convallatoxin [125, IC50 0.0027 μg/mL (0.0067 μM)] (Figure 13 and Table 6).140–142 This type of activity was increased when the carbohydrate moiety was connected to both C-2 and C-3 through a 1,4-dioxane moiety, as implied by the activity against DU-145 human prostate cancer cells of calotropagenin (126, IC50 0.48 μM) and calotropin (127, IC50 0.03 μM).143
However, cytotoxicity was abolished when a hydroxy group is introduced at the C-17 position, as shown by 3-O-β-d-glucopyranosyl-(1→4)-6-deoxy-β-d-allopyranosyl-17β-hydroxyuzarigenin (128) and 3-O-β-d-glucopyranosyl-(1→4)-6-deoxy-β-d-allopyranosyluzarigenin (129), of which both compounds were isolated from the above-ground biomass (stems, leaves, and flowers) of Asclepias syriaca L. (Apocynaceae).144 Of these compounds, 129 was cytotoxic toward a small panel of human cancer cell lines (IC50 0.59–5.3 μM), but 128 did not show any discernible activity (IC50 >10 μM) against all the human cancer cell lines tested (Figure 14 and Table 6).144 While the configuration of the C-5 position seems not to be of great importance, cytotoxicity was not observed when the 19-methyl group was changed to a carboxylic acid unit. Thus, both 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyldigitoxigenin (130, IC50 0.40–0.62 μM) and 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyluzarigenin (131, IC50 2.7–4.5 μM) showed activity against a panel of human cancer cell lines, but 3-O-β-d-glucopyranosyl-(1→4)-α-l-thevelosyl-19-oxo-cannogenin (132, IC50 >10 μM) did not (Figure 14 and Table 6).145 Compounds 130–132 were all isolated from the fruits of Thevetia peruviana (Pers.) K. Schum. [syn. Cascabela thevetia (L.) Lippold] (Apocynaceae).145
Figure 14.
Structures of cardiac glycoside with potential anticancer activity (126, 127, and 129–131) and their inactive analogues (128 and 132).
In an in vivo hollow fiber assay conducted at the University of Illinois at Chicago, the relative cancer cell survival values from treatment with (+)-strebloside (120) at 5.0 mg/kg or higher doses were significantly lower than those from treatment with the vehicle control when immunodeficient NCr nu/nu mice implanted with hollow fibers containing MDA-MB-231 or OVCAR3 cells were treated (i.p.) daily with (+)-strebloside (120) at doses of 1.0, 5.0, 10.0, 15.0, or 30.0 mg/kg for four days. No gross toxicities were observed for any of the animals in this in vivo study.138 A follow-up mechanistic investigation conducted by our group indicated that (+)-strebloside (120) binds to and inhibits Na+/K+-ATPase, and it inhibited ovarian cancer cell growth through cancer cell apoptosis induction from the blocked cell cycle progression at the G2-phase and induced PARP cleavage. In addition, this compound inhibited potently mutant p53 expression through the induction of ERK pathways and inhibited NF-κB activation.139
Several other CGs were found to inhibit Na+/K+-ATPase and exhibited selective anticancer effects on STK11 (a major mediator of lung cancer progression) mutant lung cancer cell lines, and thus these CGs were regarded as a potential targeted therapy for STK11 mutant lung cancer.146 The 3-glycose unit, the 20,22-α.β-unsaturated double bond, and a non-cyclized 1,19-dihydroxy group have been profiled as being important for cardiac glycosides to inhibit Na+/K+ transport.147 Also, CGs have been documented as inducing immunogenic cell death, which bodes well for their potential clinical use as anticancer agents.148,149 Thus far, several cardiac glycosides have been tested in preclinical or phase I clinical trials for the cancer treatment, including digoxin (133), digitoxin (134), and oleandrin (135) (Figure 15 and Table 6).150
Figure 15.
Structures of the antineoplastic cardiac glycosides, digoxin (133), digitoxin (139), and oleandrin (135).
Digoxin (133), isolated originally from Digitalis lanata Ehrh. (Plantaginaceae), has been long used to treat congestive heart failure through inhibition of the α subunits of Na+/K+-ATPase.151,152 Recent studies showed that digoxin (133) is cytotoxic toward Raji and NAMALWA human Burkitt’s lymphoma and K562 and THP-1 human leukemia cells.153,154 Tumor growth was inhibited when four-week-old male BALB/c nude mice bearing Raji lymphoma xenografts were treated (i.p.) daily with digoxin (133) (2 mg/kg) for three weeks.153 Also, leukemia growth was suppressed synergistically by a combination treatment of digoxin (133) and the unfolded protein response signaling inhibitor, GSK2606414, when six-week-old female nude mice inoculated with K562 cells were treated (i.p) with digoxin (133) (2 mg/kg), GSK2606414 (50 mg/kg), or digoxin (2 mg/kg) plus GSK2606414 (50 mg/kg) every three days for four weeks.154
Mechanistically, the antitumor activity of digoxin (133) was proposed to be mediated by induction of Raji lymphoma cell apoptosis through inhibition of NF-κB activation and induction of the activation of unfolded protein response signaling in K562 and THP-1 human leukemia cell.153,154
In a stage-two prostate cancer risk study conducted in 47,884 men who were followed up over ten years, the use of digoxin (133) was found to be associated with a lower risk of prostate cancer.155 The life expectancy of cancer patients was also improved by digoxin (133) through inducing immunogenic cell death.148
Digitoxin (134) is another important cardiac glycoside isolated from Digitalis purpurea L. (Plantaginaceae) that showed potent cytotoxicity toward human cancer cells.140,156 Interestingly, this compound showed more potent cytotoxicity than digoxin (133) toward a small panel of human cancer cell lines, indicating the significance of the C-12 hydroxy group.138–141 Several case studies for patients with cardiac disease who were treated with digitoxin (134) suggested that an intake of this drug increased the occurrence of leukemias or lymphomas. However, an internal dose-response analysis showed that a lower risk for leukemia and kidney or urinary cancers occurred in the patients with a higher plasma concentration of digitoxin (134). Thus, this therapeutic agent may be promising for the treatment of leukemias and kidney and urinary cancers.157
It is worth noting that oleandrin (135), a cardiac glycoside isolated from Nerium indicum L. (syn. N. oleander L.) (Apocynaceae), with a deoxyarabinose unit linked at the C-3 position and an acetyloxy group substituted at the C-6 position, was cytotoxic toward various human cancer cell lines, including breast, cervical, colon, glioma, hepatoma, lung, melanoma, oral, ovarian, pancreas, and prostate solid tumor and leukemia cells, with targets determined as NF-κB, mitochondrial disruption, and cancer cell autophagy.158,159 The tumor size was reduced when SCID or C57BL/6 mice inoculated with human U87MG, U251, GBM19, or murine GL261 glioma cells were treated (i.p.) daily with oleandrin (135) (0.03, 0.3, 3 mg/kg) for one week. The survival time of the tumor-bearing mice was increased when mice were treated daily (i.p.) with this compound (0.3 mg/kg) for 100 days.160 In a recent study, oleandrin (135) was found to inhibit the proliferation and invasion of human U2OS bone and SaOS-2 sapiens osteosarcoma cells by suppressing the Wnt/β-catenin signaling pathway,161 and its cytotoxicity against HPB-ALL human T-cell leukemia cells was mediated by inhibition of the Notch signaling pathway.162 Based on its potent antitumor efficacy, a standardized Nerium oleander plant-based botanical preparation, Anvirzel™, containing the main cardiac glycoside, oleandrin (135), polysaccharides, and proteins, has reached a cancer clinical trial, but this was withdrawn later in 2015, without any new trials on this formulation being reported thus far.163
A major challenge for the development of cardiac glycoside-like anticancer drugs is their narrow therapeutic index. For example, digoxin (133) showed a non-toxic dose (i.p.) of 0.7 mg/kg/day in mice,164 while digitoxin (134) exhibited an oral LD50 value of 10‒80 mg/kg in mice.165 Another critical problem is the limited selective cytotoxicity and the resistance to rodent cells of these compounds. In a cellular basis study, all cardiac glycosides and their genins tested exhibited more than 100-fold higher toxicity towards cultured human and monkey cells than the mouse, Syrian hamster, and Chinese hamster cell lines, and the selectivity toward the human cancer cells versus human non-malignant cells of several cardiac glycosides is not great.166,167 Owing to these disadvantages, at least in part, no cardiac glycoside has been developed as a new anticancer drugs thus far.
CONCLUSIONS
In our current search for anticancer agents from the secondary metabolites of tropical plants, natural products representing the benzoxanthone-type prenylated flavonoid, prenylated xanthone, caged xanthone, biflavonoid, sesquiterpene lactone, arylnaphthalene lignan lactone, phytosterol glycoside, and cardiac glycoside compound classes have been identified as promising leads. Among these compounds, α-mangostin (10), (–)-goyazensolide (74), (–)-phyllanthusmin D (97), and (+)-strebloside (120) showed potent in vitro cytotoxicity and promising in vivo antitumor efficacy. Gambogic acid (54) has reached cancer clinical trials in the People’s Republic of China, with several novel formulations developed.76–81 The absolute configuration of gambogic acid (54) was determined by our collaborative team.62 Other compounds isolated, including (+)-morelloflavone (61), (–)-phyllanthusmin C (96), (–)-β-sitosterol-3-O-β-d-(6-O-palmitoyl)glucopyranoside (117), and (–)-β-sitosterol-3-O-β-d-glucopyranoside (118) have shown very limited cytotoxicity toward human cancer cells, but they exhibited interesting target-specific anticancer potentials for further investigations. Therefore, higher plants remain a viable source for discovering compounds with chemical diversity and novel mechanisms to combat cancer, as evidenced by their contributions to the more than ten novel natural products and their derivatives approved as new anticancer drugs in the last decade.168
ACKNOWLEDGEMENTS
The laboratory work described in this review article was supported by grant P01 CA125066, funded by the National Cancer Institute, NIH, Bethesda, MD, USA. We are grateful for our taxonomic collaborators for their kind corporation with plant collections, inclusive of Dr. Ricardo García (Dominican Republic), the late Dr. Leonardus B. S. Kardono and the late Dr. Soedarsono Riswan (Indonesia), and Dr. Tran Ngoc Ninh (Vietnam). Also, we wish to thank many present and former faculty and staff colleagues, postdoctoral associates, and graduate and undergraduate students who have participated in this collaborative project. Finally, special thanks are due to Drs. Chunhua Yuan, David J. Hart, David M. Lucas, Hee-Byung Chai, Jack C. Yalowich, Jianhua Yu, Judith C. Gallucci, and Harinantenaina Liva Rakotondraibe (Ohio State University, Columbus, OH, USA), Dr. Long-Sheng Chang (Nationwide Children’s Hospital, Columbus, OH), and Dr. Xiaozhuo Chen (Ohio University, Athens, OH, USA) for their constructive contributions.
Footnotes
Dedicated to Dr. Rachel Mata of the National Autonomous University of Mexico, Mexico City, Mexico, and to Dr. Barbara N. Timmermann of the University of Kansas for their pioneering work on bioactive natural products
The authors declare no competing financial interest.
REFERENCES
- (1).Torre LA; Bray F; Siegel RL; Ferlay J; Lortet-Tieuent J; Jemal A CA Cancer J. Clin. 2015, 65, 87–108. [DOI] [PubMed] [Google Scholar]
- (2).Siegel RL; Miller KD; Jemal A CA Cancer J. Clin. 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- (3).Newman DJ; Cragg GM J. Nat. Prod. 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- (4).Cragg GM; Kingston DGI; Newman DJ Anticancer Agents from Natural Products, Second Edition; CRC Press/Taylor and Francis Group: Boca Raton, FL, 2012. [Google Scholar]
- (5).Butler MS; Robertson AAB; Cooper MA Nat. Prod. Rep. 2014, 31, 1612–1661. [DOI] [PubMed] [Google Scholar]
- (6).Cragg GM, Grothaus PG; Newman DJ Chem. Rev. 2009, 109, 3012–3043. [DOI] [PubMed] [Google Scholar]
- (7).Rosshandler Y; Shen AQ; Cortes J; Khoury HJ Exp. Rev. Hematol. 2016, 9, 419–424. [DOI] [PubMed] [Google Scholar]
- (8).Silverman JA; Dietcher SR Cancer Chemother. Pharmacol. 2013, 71, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hare JI; Lammers T; Ashford MB; Puri S; Storm G; Barry ST Adv. Drug Deliv. Rev. 2017, 108, 25–38. [DOI] [PubMed] [Google Scholar]
- (10).Zhang H OncoTargets Ther. 2016, 9, 3001–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kinghorn AD; Carcache de Blanco EJ; Lucas DM; Rakotondraibe HL; Orjala J; Soejarto DD; Oberlies NH; Pearce CJ; Wani MC; Stockwell BR; Burdette JE; Swanson SM; Fuchs JR; Phelps MA; Xu L; Zhang X; Shen YY Anticancer Res. 2016, 36, 5623–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kinghorn AD; Carcache de Blanco EJ; Chai H-B; Orjala J; Farnsworth NR; Soejarto DD; Oberlies NH; Wani MC; Kroll DJ; Pearce CJ; Swanson SM; Kramer RA; Rose WC; Fairchild CR; Vite GD; Emanuel S; Jarjoura D; Cope FO Pure Appl. Chem. 2009, 81, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mi Q; Pezzuto JM; Farnsworth NR; Wani MC; Kinghorn AD; Swanson SM J. Nat. Prod. 2009, 72, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Orjala J, Oberlies NH; Pearce CJ; Swanson SM; Kinghorn AD In Bioactive Compounds from Natural Sources Second Edition: Natural Products as Lead Compounds in Drug Discovery; Tringali C, Ed.; Taylor & Francis: London, 2012; pp 37–63. [Google Scholar]
- (15).Pearce CJ; Lantvit DD; Shen Q; Jarjoura D; Zhang X; Oberlies NH; Kroll DJ; Wani MC; Orjala J; Soejarto DD; Farnsworth NR; Carcache de Blanco EJ; Fuchs JR; Kinghorn AD; Swanson SM In Methods in Molecular Biology, vol. 944 Fungal Secondary Metabolism: Methods and Protocols; Keller NP; Turner G, Eds.; Humana Press/Springer: Totowa, NJ, 2012; pp. 267–277. [DOI] [PubMed] [Google Scholar]
- (16).Bueno Pérez L, Still PC; Naman CB; Ren Y; Pan L; Chai H-B; Carcache de Blanco EJ; Ninh TN; Thanh BV; Swanson SM; Soejarto DD; Kinghorn AD Phytochem. Rev. 2014, 13, 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Henkin JM; Ren Y; Soejarto DD; Kinghorn AD In Progress in the Chemistry of Organic Natural Products; Kinghorn AD; Falk H; Gibbons S; Kobayashi J; Asakawa Y; Liu J-K, Eds.; Springer International Publishing AG: Cham, Switzerland, 2018; Vol. 107, pp 1–94. [DOI] [PubMed] [Google Scholar]
- (18).Ren Y; Kardono LBS; Riswan S; Chai H-B; Farnsworth NR; Soejarto DD; Carcache de Blanco EJ; Kinghorn AD J. Nat. Prod. 2010, 73, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hano Y; Yamagami Y; Kobayashi M; Isohata R; Nomura T Heterocycles 1990, 31, 877–882. [Google Scholar]
- (20).Suhartati T; Achmad SA; Aimi N; Hakim EH; Kitajima M; Takayama H; Takeya K Fitoterapia 2001, 72, 912–918. [DOI] [PubMed] [Google Scholar]
- (21).Abd Rahman M; Ramli F; Karimian H; Dehghan F; Nordin N; Mohd Ali H; Mohan S; Hashim NM PLoS One 2016, 11, e0151466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Etti IC; Abdullah R; Kadir A; Hashim NM; Yeap SK; Imam MU; Ramli F; Malami I; Lam KL; Etti U; Waziri P; Rahman M PLoS One 2017, 12, e0182357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Etti IC; Rasedee A; Hashim NM; Abdul AB; Kadir A; Yeap SK; Waziri P; Malami I; Lim KL; Etti CJ Drug Des. Dev. Ther. 2017, 11, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Singh A; Kaur N; Sharma S; Bedi PMS J. Chem. Pharm. Res. 2016, 8, 75–131. [Google Scholar]
- (25).Chin Y-W; Kinghorn AD Mini-Rev. Org. Chem. 2008, 5, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kao D; Henkin JM; Soejarto DD; Kinghorn AD; Oberlies NH Phytochemistry Lett. 2018, 28, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang K-J; Gu Q-L; Yang K; Ming X-J; Wang J-X Planta Med. 2017, 83, 188–202. [DOI] [PubMed] [Google Scholar]
- (28).Han A-R; Kim J-A; Lantvit DD; Kardono LBS; Riswan S; Chai H; Carcache de Blanco EJ; Farnsworth NR; Swanson SM; Kinghorn AD J. Nat. Prod. 2009, 72, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ren Y; Matthew S; Lantvit DD; Ninh TN; Chai H; Fuchs JR; Soejarto DD; Carcache de Blanco EJ; Swanson SM; Kinghorn AD J. Nat. Prod. 2011, 74, 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Suksamrarn S; Komutiban O; Ratananukul P; Chimnoi N; Lartpornmatulee N; Suksamrarn A Chem. Pharm. Bull. 2006, 54, 301–305. [DOI] [PubMed] [Google Scholar]
- (31).Teh SS; Ee GCL; Mah SH; Lim YM; Ahmad Z Molecules 2013, 18, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chang H-F; Yang L-L Molecules 2012, 17, 8010–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhu L; Liu X; Li D; Sun S; Wang Y; Sun X Biomed. Pharmacother. 2018, 103, 708–718. [DOI] [PubMed] [Google Scholar]
- (34).Kaomongkolgit R; Yiemwattana I Trop. J. Pharm. Res. 2016, 15, 2063–2070. [Google Scholar]
- (35).Yuan J; Wu Y; Lu G Oncol. Lett. 2013, 5, 1958–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pérez-Rojas JM; González-Macías R; González-Cortes J; Jurado R; Pedraza-Chaverri J; García-López P Oxid. Med. Cell. Longevity 2016, 7981397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wu C-P; Hsiao S-H; Murakami M; Lu Y-J; Li Y-Q; Huang Y-H; Hung T-H; Ambudkar SV; Wu Y-S Mol. Pharmaceutics 2017, 14, 2805–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gutierrez-Orozco F; Chitchumroonchokchai C; Lesinski GB; Suksamrarn S; Failla ML J. Agric. Food Chem. 2013, 61, 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chitchumroonchokchai C; Thomas-Ahner JM; Li J; Riedl KM; Nontakham J; Suksumrarn S; Clinton SK; Kinghorn AD; Failla ML Mol. Nutr. Food Res. 2013, 57, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hsieh S-C; Huang M-H; Cheng C-W; Hung J-H; Yang S-F; Hsieh Y-H Apoptosis 2013, 18, 1548–1560. [DOI] [PubMed] [Google Scholar]
- (41).Hafeez BB; Mustafa A; Fischer JW; Singh A; Zhong W; Shekhani MO; Meske L; Havighurst T; Kim KM; Verma AK Antioxid. Redox Signal. 2014, 21, 682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Johnson JJ; Petiwala SM; Syed DN; Rasmussen JT; Adhami VM; Siddiqui IA; Kohl AM; Mukhtar H Carcinogenesis 2012, 33, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lee HN; Jang HY; Kim HJ; Shin SA; Choo GS; Park YS; Kim SK; Jung JY Int. J. Mol. Med. 2016, 37, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ibrahim MY; Hashim NM; Mohan S; Abdulla MA; Abdelwahab SI; Arbab IA; Yahayu M; Ali LZ; Ishag OE Arab. J. Chem. 2015, 8, 129–137. [Google Scholar]
- (45).Li L; Han A-R; Kinghorn AD; Frye RF; Derendorf H; Butterweck V Planta Med. 2013, 79, 646–653. [DOI] [PubMed] [Google Scholar]
- (46).Chen J-J; Long Z-J; Xu D-F; Xiao R-Z; Liu L-L; Xu Z-F; Qiu SX; Lin D-J; Liu Q Leuk. Lymphoma 2014, 55, 628–638. [DOI] [PubMed] [Google Scholar]
- (47).Watanapokasin R; Jarinthanan F; Nakamura Y; Sawasjirakij N; Jaratrungtawee A; Suksamrarn S World J. Gastroenterol. 2011, 17, 2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gutierrez-Orozco F; Thomas-Ahner JM; Berman-Booty LD; Galley JD; Chitchumroonchokchai C; Mace T; Suksamrarn S; Bailey MT; Clinton SK; Lesinski GB; Failla ML Mol. Nutr. Food Res. 2014, 58, 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Anantachoke N; Tuchinda P; Kuhakarn C; Pohmakotr M; Reutrakul V Pharm. Biol. 2012, 50, 78–91. [DOI] [PubMed] [Google Scholar]
- (50).Jia B; Li S; Hu X; Zhu G; Chen W AAPS PharmSciTech 2015, 16, 742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ren Y; Lantvit DD; Carcache de Blanco EJ; Kardono LBS; Riswan S; Chai H; Cottrell CE; Farnsworth NR; Swanson SM; Ding Y; Li X-C; Marais JPJ; Ferreira D; Kinghorn AD Tetrahedron 2010, 66, 5311–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Boonnak N; Chantrapromma S; Fun H-K; Yuenyongsawad S; Patrick BO; Maneerat W; Williams DE; Andersen RJ J. Nat. Prod. 2014, 77, 1562–1571. [DOI] [PubMed] [Google Scholar]
- (53).Miao G; Ma J; Yang K; Huang Z; Gu Q; Wang Y; Guo Q; You Q; Wang J Aust. J. Chem. 2015, 68, 872–880. [Google Scholar]
- (54).Niu S-L; Li D-H; Li X-Y; Wang Y-T; Li S-G; Bai J; Pei Y-H; Jing Y-K; Li Z-L; Hua H-MJ Nat. Prod. 2018, 81, 749–757. [DOI] [PubMed] [Google Scholar]
- (55).Xu X; Wu Y; Hu M; Li X; Gu C; You Q; Zhang X Bioorg. Med. Chem. 2016, 24, 4626–4635. [DOI] [PubMed] [Google Scholar]
- (56).Hahnvajanawong C; Wattanawongdon W; Chomvarin C; Anantachoke N; Kanthawong S; Sripa B; Reutrakul V Cancer Cell Int. 2014, 14, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yang J; He S; Li S; Zhang R; Peng A; Chen L Molecules 2013, 18, 15305–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Boueroy P; Hahnvajanawong C; Boonmars T; Saensa-Ard S; Anantachoke N; Vaeteewoottacharn K; Reutrakul V Oncol. Lett. 2016, 12, 4685–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wu Y; Hu M; Yang L; Li X; Bian J; Jiang F; Sun H; You Q; Zhang X Bioorg. Med. Chem. Lett. 2015, 25, 2584–2588. [DOI] [PubMed] [Google Scholar]
- (60).Hahnvajanawong C; Ketnimit S; Pattanapanyasat K; Anantachoke N; Sripa B; Pinmai K; Seubwai W; Reutrakul V Biol. Pharm. Bull. 2012, 35, 1914–1925. [DOI] [PubMed] [Google Scholar]
- (61).Li ZP; Lee H-H; Uddin Z; Song YH; Park KH Bioorg. Chem. 2018, 78, 39–45. [DOI] [PubMed] [Google Scholar]
- (62).Ren Y; Yuan C; Chai H-B; Ding Y; Li X-C; Ferreira D; Kinghorn AD J. Nat. Prod. 2011, 74, 460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Banik K; Harsha C; Bordoloi D; Lalduhsaki Sailo B; Sethi G; Leong HC; Arfuso F; Mishra S; Wang L; Kumar AP; Kunnumakkara AB Cancer Lett. 2018, 416, 75–86. [DOI] [PubMed] [Google Scholar]
- (64).Xu L; Lao Y; Zhao Y; Qin J; Fu W; Zhang Y; Xu H BioMed Res. Int. 2015, 2015, 910453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Han Q-B; Cheung S; Tai J; Qiao C-F; Song J-Z; Xu H-X Biol. Pharm. Bull. 2005, 28, 2335–2337. [DOI] [PubMed] [Google Scholar]
- (66).Wang X; Lu N; Yang Q; Gong D; Lin C; Zhang S; Xi M; Gao Y; Wei L; Guo Q; You Q Eur. J. Med. Chem. 2011, 46, 1280–1290. [DOI] [PubMed] [Google Scholar]
- (67).Wang S; Wang L; Chen M; Wang Y Chem. Biol. Interact. 2015, 235, 76–84. [DOI] [PubMed] [Google Scholar]
- (68).Xu P; Wang R; Li J; Ouyang J; Chen B RSC Adv. 2015, 5, 61051–61059. [Google Scholar]
- (69).Wen C; Huang L; Chen J; Lin M; Li W; Lu B; Rutnam ZJ; Iwamoto A; Wang Z; Yang X; Liu H Int. J. Oncol. 2015, 47, 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Liang L; Zhang Z; Qin X; Gao Y; Zhao P; Liu J; Zeng W Basic Clin. Pharmacol. Toxicol. 2018, 123, 692–703. [DOI] [PubMed] [Google Scholar]
- (71).Davenport J; Manjarrez JR; Peterson L; Krumm B; Blagg BSJ; Matts RL J. Nat. Prod. 2011, 74, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Yim KH; Prince TL; Qu S; Bai F; Jennings PA; Onuchic JN; Theodorakis EA; Neckers L Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4801–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Shi X; Lan X; Chen X; Zhao C; Li X; Liu S; Huang H; Liu N; Zang D; Liao Y; Zhang P; Wang X; Liu J Sci. Rep. 2015, 5, 9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Huang H; Chen D; Li S; Li X; Liu N; Lu X; Liu S; Zhao K; Zhao C; Guo H; Yang C; Zhou P; Dong X; Zhang C; Guanmei; Dou QP; Liu J, Cancer Lett. 2011, 301, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Liu N; Huang H; Xu L; Hua X; Li X; Liu S; Yang C; Zhao C; Zhao C; Li S; Dou QP; Liu J Toxicol. Lett. 2014, 224, 333–340. [DOI] [PubMed] [Google Scholar]
- (76).Zhou Z; Wang J Chin. J. New Drugs 2007, 16, 79–82. [Google Scholar]
- (77).Chi Y; Zhan X-K; Yu H; Xie G-R; Wang Z-Z; Xiao W; Wang Y-G; Xiong F-X; Hu J-F; Yang L; Cui C-X; Wang J-W Chin. Med. J. 2013, 126, 1642–1646. [PubMed] [Google Scholar]
- (78).Doddapaneni R; Patel K; Owaid IH; Singh M Drug Deliv. 2016, 23, 1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Wan H-Y; Chen J-L; Yu X-Y; Zhu X-M RSC Adv. 2017, 7, 49518–49525. [Google Scholar]
- (80).Huang R; Li J; Kebebe D; Wu Y; Zhang B; Liu Z Drug Deliv. 2018, 25, 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Yang Y; Cai H; Yuan X; Xu H; Hu Y; Rui X; Wu J; Chen J; Li J; Gao X; Yin D Mol. Pharmaceutics 2018, 15, 2007–2016. [DOI] [PubMed] [Google Scholar]
- (82).Mercader AG; Pomilio AB Anti-Cancer Agents Med. Chem. 2013, 13, 1217–1235. [DOI] [PubMed] [Google Scholar]
- (83).Li X-C; Joshi AS; Tan B; ElSohly HN; Walker LA; Zjawiony JK; Ferreira D Tetrahedron 2002, 58, 8709–8717. [Google Scholar]
- (84).Taher M; Aminuddin A; Susanti D; Aminudin NI; On S; Ahmad F; Hamidon H Nat. Prod. Sci. 2016, 22, 122–128. [Google Scholar]
- (85).Li X; Ai H; Sun D; Wu T; He J; Xu Z; Ding L; Wang L Oncol. Lett. 2016, 12, 3373–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Pang X; Yi T; Yi Z; Cho SG; Qu W; Pinkaew D; Fujise K; Liu M Cancer Res. 2009, 69, 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Gontijo VS; Januário JP; Júdice W. A. de S.; Antunes AA; Cabral IR; Assis DM; Juliano MA; Camps I; Marques MJ; Viegas C Jr.; dos Santos MH J. Med. Plants Res. 2015, 9, 426–434. [Google Scholar]
- (88).Yu S; Yan H; Zhang L; Shan M; Chen P; Ding A; Li SFY Molecules 2017, 22, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Wall ME; Wani MC; Fullas F; Oswald JB; Brown DM; Santisuk T; Reutrakul V; McPhail AT; Farnsworth NR; Pezzuto JM; Kinghorn AD; Besterman JM J. Med. Chem. 1994, 37, 1465–1470. [DOI] [PubMed] [Google Scholar]
- (90).Wada S-I; Hitomi T; Tokuda H; Tanaka R Chem. Biodiversity 2010, 7, 2303–2308. [DOI] [PubMed] [Google Scholar]
- (91).Ren Y; Yu J; Kinghorn AD Curr. Med. Chem. 2016, 23, 2397–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Babaei G; Aliarab A; Abroon S; Rasmi Y; Aziz SG-G Biomed. Pharmacother. 2018, 106, 239–246. [DOI] [PubMed] [Google Scholar]
- (93).Ren Y; Muñoz Acuña U; Jiménez F; García R; Mejía M; Chai H; Gallucci JC; Farnsworth NR; Soejarto DD; Carcache de Blanco EJ; Kinghorn AD Tetrahedron 2012, 68, 2671–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Ren Y; Jiménez F; García R; Mejía M; Chai H; Farnsworth NR; Soejarto DD; Kinghorn AD Tetrahedron Lett. 2013, 54, 5457–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Vichnewski W; Sarti SJ; Gilbert B; Herz W Phytochemistry 1976, 15, 191–193. [Google Scholar]
- (96).Herz W; Goedken VL J. Org. Chem. 1982, 47, 2798–2800. [Google Scholar]
- (97).Ren Y; Gallucci JC; Li X; Chen L; Yu J; Kinghorn AD J. Nat. Prod. 2018, 81, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Waddell TG; Stoecklin W; Geissman TA Tetrahedron Lett. 1969, 1313–1316. [DOI] [PubMed] [Google Scholar]
- (99).Spear SA; Burns SS; Oblinger JL; Ren Y; Pan L; Kinghorn AD; Welling DB; Chang L-S. Otol. Neurotol. 2013, 34, 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Saúde-Guimarães DA; Raslan DS; Oliveira AB Rev. Bras. Pl. Med. 2014, 16, 275–282. [Google Scholar]
- (101).Muñoz Acuña U; Shen Q; Ren Y; Lantvit DD; Wittwer JA; Kinghorn AD; Swanson SM; Carcache de Blanco EJ Int. J. Cancer Res. 2013, 9, 36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Liu W; Wang X; Sun J; Yang Y; Li W; Song J OncoTargets Ther. 2017, 10, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Schepetkin IA; Kirpotina LN; Mitchell PT; Kishkentaeva AS; Shaimerdenova ZR; Atazhanova GA; Adekenov SM; Quinn MT Phytochemistry 2018, 146, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Zhu X; Yuan C; Tian C; Li C; Nie F; Song X; Zeng R; Wu D; Hao X; Li LJ Biol. Chem. 2018, 293, 5335–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Taleghani A; Nasseri MA; Iranshahi M Bioorg. Chem. 2017, 71, 128–134. [DOI] [PubMed] [Google Scholar]
- (106).Ding Y; Guo H; Ge W; Chen X; Li S; Wang M; Chen Y; Zhang Q Eur. J. Med. Chem. 2018, 156, 216–229. [DOI] [PubMed] [Google Scholar]
- (107).Curry III EA; Murry DJ; Yoder C; Fife K; Armstrong V; Nakshatri H; O’Connell M; Sweeney CJ Invest. New Drugs 2004, 22, 299–305. [DOI] [PubMed] [Google Scholar]
- (108).Neelakantan S; Nasim S; Guzman ML; Jordan CT; Crooks PA Bioorg. Med. Chem. Lett. 2009, 19, 4346–4349. [DOI] [PubMed] [Google Scholar]
- (109).Shanmugam R; Kusumanchi P; Cheng L; Crooks P; Neelakantan S; Matthews W; Nakshatri H; Sweeney CJ Prostate 2010, 70, 1074–1086. [DOI] [PubMed] [Google Scholar]
- (110).Deraska PV; O’Leary C; Reavis HD; Labe S; Dinh T-K; Lazaro J-B; Sweeney C; D’Andrea AD; Kozono D Cell Death Discov. 2018, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Morel KL; Ormsby RJ; Bezak E; Sweeney CJ; Sykes PJ Radiation Res. 2017, 187, 501–512. [DOI] [PubMed] [Google Scholar]
- (112).Guzman ML; Rossi RM; Karnischky L; Li X; Peterson DR; Howard DS; Jordan CT Blood 2005, 105, 4163–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Ren Y; Lantvit DD; Deng Y; Kanagasabai R; Gallucci JC; Ninh TN; Chai H-B; Soejarto DD; Fuchs JR; Yalowich JC; Yu J; Swanson SM; Kinghorn AD J. Nat. Prod. 2014, 77, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Ren Y; Yuan C; Deng Y; Kanagasabai R; Ninh TN; Tu VT; Chai H-B; Soejarto DD; Fuchs JR; Yalowich JC; Yu J; Kinghorn AD Phytochemistry 2015, 111, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Woodard JL; Huntsman AC; Patel PA; Chai H-B; Kanagasabai R; Karmahapatra S; Young AN; Ren Y; Cole MS; Herrera D; Yalowich JC; Kinghorn AD; Burdette JE; Fuchs JR Bioorg. Med. Chem. 2018, 26, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Zhao Y; Ni C; Zhang Y; Zhu L Arch. Pharm. Chem. Life Sci. 2012, 345, 622–628. [DOI] [PubMed] [Google Scholar]
- (117).Day S-H; Chiu N-Y; Won S-J; Lin C-NJ Nat. Prod. 1999, 62, 1056–1058. [DOI] [PubMed] [Google Scholar]
- (118).Tuchinda P; Kornsakulkarn J; Pohmakotr M; Kongsaeree P; Prabpai S; Yoosook C; Kasisit J; Napaswad C; Sophasan S; Reutrakul VJ Nat. Prod. 2008, 71, 655–663. [DOI] [PubMed] [Google Scholar]
- (119).Susplugas S; Hung NV; Bignon J; Thoison O; Kruczynski A; Sévenet T; Guéritte FJ Nat. Prod. 2005, 68, 734–738. [DOI] [PubMed] [Google Scholar]
- (120).Shi D-K; Zhang W; Ding N; Li M; Li Y-X Eur. J. Med. Chem. 2012, 47, 424–431. [DOI] [PubMed] [Google Scholar]
- (121).Young AN; Herrera D; Huntsman AC; Korkmaz MA; Lantvit DD; Mazumder S; Kolli S; Coss CC; King S; Wang H; Swanson SM; Kinghorn AD; Zhang X; Phelps MA; Aldrich LN; Fuchs JR; Burdette JE. Mol. Cancer Ther. 2018, 17, 2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Zhang X; Yang H; Yue S; He G; Qu S; Zhang Z; Ma B; Ding R; Peng W; Zhang H; Yang Z; Dou K; Tao K; Li X Cancer Med. 2017, 6, 1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Hanahan D; Weinberg RA Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (124).Deng Y; Chu J; Ren Y; Fan Z; Ji X; Mundy-Bosse B; Yuan S; Hughes T; Zhang J; Cheema B; Camardo AT, Xia Y; Wu L-C; Wang L-S; He X; Kinghorn AD; Li X; Caligiuri MA; Yu JJ Immunol. 2014, 193, 2994–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Caligiuri MA Blood 2008, 112, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Antúnez-Mojica M; Rodríguez-Salarichs J; Redondo-Horcajo M; León A; Barasoain I; Canales A; Cañada FJ; Jiménez-Barbero J; Alvarez L; Díaz JF J. Nat. Prod. 2016, 79, 2113–2121. [DOI] [PubMed] [Google Scholar]
- (127).Zhang X; Rakesh KP; Shantharam CS; Manukumar HM; Asiri AM; Marwani HM; Qin H-L Bioorg. Med. Chem. 2018, 26, 340–355. [DOI] [PubMed] [Google Scholar]
- (128).Ezoe S Int. J. Environ. Res. Public Health 2012, 9, 2444–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Mehta RS; Randolph B; Daher M; Rezvani K Int. J. Hematol. 2018, 107, 262–270. [DOI] [PubMed] [Google Scholar]
- (130).Arisawa M; Ohmura K; Kobayashi A; Morita N Chem. Pharm. Bull. 1989, 37, 2431–2434. [DOI] [PubMed] [Google Scholar]
- (131).Matos MFC; Leite LISP; Brustolim D; de Siqueira JM; Carollo CA; Hellmann AR; Pereira NFG; da Silva DB Fitoterapia 2006, 77, 227–229. [DOI] [PubMed] [Google Scholar]
- (132).Bouic PJD; Etsebeth S; Liebenberg RW; Albrecht CF; Pegel K; Van Jaarsveld PP Int. J. Immunopharmacool. 1996, 18, 693–700. [DOI] [PubMed] [Google Scholar]
- (133).Imanaka H; Koide H; Shimizu K; Asai T; Shimizu NK; Ishikado A; Makino T; Oku N Biol. Pharm. Bull. 2008, 31, 400–404. [DOI] [PubMed] [Google Scholar]
- (134).Dunn GP; Old LJ; Schreiber RD Immunity 2004, 21, 137–148. [DOI] [PubMed] [Google Scholar]
- (135).Belardelli F; Ferrantini M; Proietti E; Kirkwood JM Cytokine Growth Factor Rev. 2002, 13, 119–134. [DOI] [PubMed] [Google Scholar]
- (136).Thornthwaite JT; Shah H; Shah P; Respess HJ Immune Based Ther. Vaccine Antimicrob. 2012, 1, 21–52. [Google Scholar]
- (137).Patel S Biomed. Pharmacother. 2016, 84, 1036–1041. [DOI] [PubMed] [Google Scholar]
- (138).Ren Y; Chen W-L; Lantvit DD; Sass EJ; Shriwas P; Ninh TN; Chai H-B; Zhang X; Soejarto DD; Chen X; Lucas DM; Swanson SM; Burdette JE; Kinghorn AD J. Nat. Prod. 2017, 80, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Chen W-L; Ren Y; Ren J; Erxleben C; Johnson ME; Gentile S; Kinghorn AD; Swanson SM; Burdette JE J. Nat. Prod. 2017, 80, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Johansson S; Lindholm P; Gullbo J; Larsson R; Bohlin L; Claeson P Anti-Cancer Drugs 2001, 12, 475–483. [DOI] [PubMed] [Google Scholar]
- (141).López-Lázaro M; Pastor N; Azrak SS; Ayuso MJ; Austin CA; Cortés FJ Nat. Prod. 2005, 68, 1642–1645. [DOI] [PubMed] [Google Scholar]
- (142).Shi L-S; Kuo S-C; Sun H-D; Morris-Natschke SL; Lee K-H; Wu T-S Bioorg. Med. Chem. 2014, 22, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Zhang R-R; Tian H-Y; Tan Y-F; Chung T-Y; Sun X-H; Xia X; Ye W-C; Middleton DA; Fedosova N; Esmann M; Tzen JTC; Jiang R-W Org. Biomol. Chem. 2014, 12, 8919–8929. [DOI] [PubMed] [Google Scholar]
- (144).Araya JJ; Kindscher K; Timmermann BN J. Nat. Prod. 2012, 75, 400–407. [DOI] [PubMed] [Google Scholar]
- (145).Tian D-M; Cheng H-Y; Jiang M-M; Shen W-Z; Tang J-S; Yao X-SJ Nat. Prod. 2016, 79, 38–50. [DOI] [PubMed] [Google Scholar]
- (146).Kim N; Yim HY; He N; Lee C-J; Kim JH; Choi J-S; Lee HS; Kim S; Jeong E; Song M; Jeon S-M; Kim W-Y; Mills GB; Cho Y-Y; Yoon S Sci. Rep. 2016, 6, 29721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Magpusao AN; Omolloh G; Johnson J; Gascón J; Peczuh MW; Fenteany G ACS Chem. Biol. 2015, 10, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Kepp O; Menger L; Vacchelli E; Adjemian S; Martins I; Ma Y; Sukkurwala AQ; Michaud M; Galluzzi L; Zitvogel L; Kroemer G Oncoimmunol. 2012, 1, 1640–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Diederich M; Muller F; Cerella C Biochem. Pharmacol. 2017, 125, 1–11. [DOI] [PubMed] [Google Scholar]
- (150).Slingerland M; Cerella C; Guchelaar HJ; Diederich M; Gelderblom H Invest. New Drugs 2013, 31, 1087–1094. [DOI] [PubMed] [Google Scholar]
- (151).Smith SJ Chem. Soc. 1930, 508–510. [Google Scholar]
- (152).Go K; Kartha G Acta Cryst. 1980, B36, 1811–1819. [Google Scholar]
- (153).Wang T; Xu P; Wang F; Zhou D; Wang R; Meng L; Wang X; Zhou M; Chen B; Ouyang J Leuk. Lymphoma 2017, 58, 1673–1685. [DOI] [PubMed] [Google Scholar]
- (154).Zhang X-H; Wang X-Y; Zhou Z-W; Bai H; Shi L; Yang Y-X; Zhou S-F; Zhang X-C BioFactors 2017, 43, 812–820. [DOI] [PubMed] [Google Scholar]
- (155).Platz EA; Yegnasubramanian S; Liu JO; Chong CR; Shim JS; Kenfield SA; Stampfer MJ; Willett WC; Giovannucci E; Nelson WG Cancer Discov. 2011, 1, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Elbaz HA; Stueckle TA; Tse W; Rojanasakul Y; Dinu CZ Exp. Hematol. Oncol. 2012, 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Haux J; Klepp O; Spigset O; Tretli S BMC Cancer 2001, 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Kumar A; De T; Mishra A; Mishra AK Pharmacog. Rev. 2013, 7, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (159).Pan L; Zhang Y; Zhao W; Zhou X; Wang C; Deng F Cancer Chemother. Pharmacol. 2017, 80, 91–100. [DOI] [PubMed] [Google Scholar]
- (160).Garofalo S; Grimaldi A; Chece G; Porzia A; Morrone S; Mainiero F; D’Alessandro G; Esposito V; Cortese B; Di Angelantonio S; Trettel F; Limatola CJ Neurosci. 2017, 37, 3926–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Ma Y; Zhu B; Liu X; Yu H; Yong L; Liu X; Shao J; Liu ZJ Exp. Clin. Cancer Res. 2015, 34, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Arai MA; Akamine R; Hayashi N; Koyano T; Kowithayakorn T; Ishibashi MJ Nat. Prod. 2018, 81, 1235–1240. [DOI] [PubMed] [Google Scholar]
- (163).Sublingual Anvirzel in Advance Non-Small Cell Lung Cancer (NSCLC). https://www.clinicaltrials.gov/ct2/show/NCT01562301?term=Anvirzel%E2%84%A2&rank=1.
- (164).Svensson A; Azarbayjani F; Bäckman U; Matsumoto T; Christofferson R Anticancer Res. 2005, 25, 207–212. [PubMed] [Google Scholar]
- (165).Goda Y; Sakai S; Nakamura T; Akiyama H; Toyoda M Shokuhin Eiseigaku Zasshi 1998, 39, 256–265. [Google Scholar]
- (166).Gupta RS; Chopra A; Stetsko DK J. Cell. Physiol. 1986, 127, 197–206. [DOI] [PubMed] [Google Scholar]
- (167).Calderón-Montaño JM; Burgos-Morón E; López-Lázaro M Oncogene 2014, 33, 2947–2948. [DOI] [PubMed] [Google Scholar]
- (168).Basmadjian C; Zhao Q; Bentouhami E; Djehal A; Nebigil CG; Johnson RA; Serova M; de Gramont A; Faivre S; Raymond E; Désaubry LG Front. Chem. 2014, 2, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]