

Abstract
Functional paragangliomas are rare neuroendocrine tumours that secrete catecholamines and are infrequently found in the mediastinum. We report a case of a young male with symptoms of catecholamine excess and a personal and family history of the paraganglioma predisposing succinate dehydrogenase subunit B mutation. The lesion had anatomical intrapericardial juxtaposition to important cardiac anatomy and posed the significant challenge of dissection at surgery. The lesion was successfully resected via sternotomy on cardiopulmonary bypass and confirmed histopathologically as paraganglioma. Intrapericardial paraganglioma is rare and treatment is difficult and time critical considering the proximity of cardiac anatomy as well as malignant potential.
Keywords: Paraganglioma, cardiothoracic surgery, succinate dehydrogenase, succinate dehydrogenase subunit B, intrapericardial
Introduction
Catecholamine-secreting paragangliomas (PGLs) are rare neoplasms of neuroendocrine origin. Overall, less than 2% of PGLs are mediastinal.1,2 There have been less than 200 cases of intrapericardial PGLs reported in the literature.3
It is now understood that up to 50% of PGL is familial.4 Several genetic conditions are associated with PGL/phaeochromocytoma including the multiple endocrine neoplasia type 2 syndromes, von Hippel–Lindau disease, neurofibromatosis, and the succinate dehydrogenase germline mutations (SDHx). Mutation in succinate dehydrogenase predisposes patients to both non-functioning and catecholamine-secreting PGL and phaeochromocytoma through a mechanism involving induction of hypoxia-inducible factor. Primary cardiac PGL associated with succinate dehydrogenase subunit B (SDHB) mutation has been described before, with four reported cases.5 While most tumours are benign, they do have malignant potential1 (especially those associated with succinate dehydrogenase subunit A (SDHA) and SDHB mutations) and secretory tumours expose patients to the risks of catecholamine excess.
Case
We present a single case of a 34-year-old male referred to our service from the endocrinology department after discovery of a mediastinal mass. The patient had a family history of SDHB mutation, with two cousins having undergone carotid body PGL resection. The patient first presented to the endocrinology clinic after familial cascade genetic screening diagnosed an SDHB mutation. Over the previous several years, he had experienced daily palpitations, headache, diaphoresis, anxiety, and central chest pain. These symptoms would present unexpectedly, were unrelated to exertion, and were short lasting. He had presented multiple times to the emergency department with chest pain, with electrocardiographic, troponin studies, and stress testing being unremarkable. The patient otherwise had no past medical history.
On examination, the patient was normotensive with a regular heart rate. Plasma metanephrines were elevated and testing undertaken 2 months prior to surgery revealed that noradrenaline and dopamine were markedly elevated with normetanephrine at 3587 pmol/L (<900 pmol/L) and 3-methoxytyramine 1075 pmol/L (<110 pmol/L).
Initial investigation with F-fluorodeoxyglucose (FDG) positron emission tomography (PET) discovered an intense uptake in the left lateral aspect of the aortic root inferior to the pulmonary artery (Figure 1(a)). This was further clarified using contrast-enhanced computed tomography and cardiac magnetic resonance imaging (MRI; Figure 1(b) and (c)). These revealed a 35 mm × 34 mm × 29 mm mass adjacent to the aortic root posterior to the pulmonary artery. MRI was suggestive that this lesion was not invading the myocardium. Whole-body metaiodobenzylguanidine (mIBG) scintigraphy did not reveal any metabolic lesion suspicious for metastasis. Transthoracic echocardiography was negative for impedance of this lesion on cardiac function or great vessel flow.
Figure 1.

(a) FDG positron emission tomography images showing avid tumour in the mediastinum. (b) Contrast-enhanced computed tomography of the thorax. (c) T2-weighted magnetic resonance image coronal section, illustrating a close relationship to the pulmonary artery. Arrows indicate tumour.
Preoperative medical stabilisation was achieved 1 month preoperatively using phenoxybenzamine 30 mg three times daily as alpha blockade and subsequently beta blockade with metoprolol 25 mg twice daily was added 1 week prior to surgery. At surgery, the patient underwent a midline sternotomy. The tumour was unable to be easily or safely accessed with the heart full and ejecting. The distal ascending aorta and the right atrium were cannulated and the patient was placed on cardiopulmonary bypass and cooled to 32°C, and after aortic cross clamping the heart was arrested with tepid blood antegrade cardioplegia. Findings were of a 3.5 cm × 3.5 cm × 3 cm firm spherical mass located between the aortic root and pulmonary artery. Inferiorly, there was a close relation to the left main coronary artery and the circumflex artery, with the right pulmonary artery superiorly. The aortic cross clamp and cardiopulmonary bypass times were 27 and 79 min, respectively (Figure 2).
Figure 2.
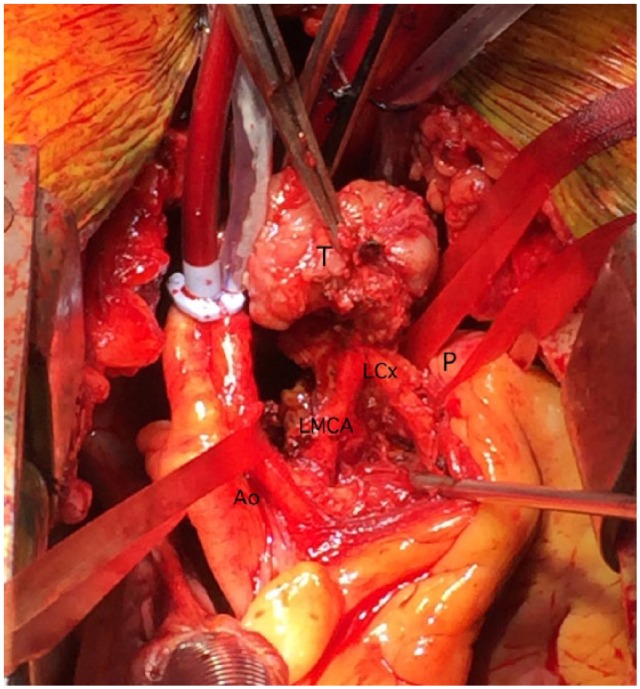
Partially excised tumour, showing close adherence to the left circumflex artery (LCx). The aorta (Ao) and pulmonary artery (P) have been retracted. Left main coronary artery (LMCA).
Pathological examination was typical of PGL. Macroscopically, the specimen consisted of a 30 mm × 30 mm × 30 mm tan-coloured nodule weighing 14 g (Figure 3(a)). The tumour cells were positive for synaptophysin chromogranin and S-100 markers. Mitotic activity was low; however, intracytoplasmic hyaline globules were not apparent. The capsule was seen to be infiltrated by tumour cells (Figure 3(b)).
Figure 3.

(a) Macroscopic photograph of the tumour. (b) Haematoxylin and eosin (40× magnification)-stained section showing tumour extending through its capsule. (c) 100× Magnified H&E stain highlighting a markedly atypical cell. (d) Immunohistochemical analysis revealing positivity for synaptophysin stain (100× magnification).
The patient’s recovery was unremarkable and he was discharged from hospital on the 5th postoperative day. At surgical follow-up, the patient reported improved symptomatology. Repeat plasma metanephrines and chromogranin A had normalised from their preoperative values and have remained as such 18 months postoperative. Further FDG and DOTA-TATE (tyrosine-3-octreotate) PET at 12 months post resection did not show recurrence of mediastinal disease, nor any lesion suspicious for metastasis.
Discussion
PGLs arise from neural crest tissue and the majority are benign.3 This rare neoplasm represents about 1%–3% of cardiac tumours.6 They are classified as being either functional or non-functional and patients will present with either symptoms of catecholamine excess or mass effect on surrounding structures.7 As is the case with this patient, they may also present from genetic screening investigations. Aside from the presence of malignant appearing tissue in areas where paraganglion tissue is usually absent, there exists no standardised criterion for the differentiation of benign from malignant PGL.8
Diagnosis of intrapericardial PGL can be challenging and various biochemical and imaging techniques have been used. Plasma metanephrine and normetanephrine are the metabolites of adrenaline and noradrenaline, respectively, and have been used in diagnosis of catecholamine-producing tumours. 3-Methoxytyramine is the breakdown product of dopamine and, as a diagnostic test, has been shown to modestly increase sensitivity of diagnosis of PGL.9 FDG PET has a role in identifying these tumours.10 Increasingly, 68-Gallium somatostatin analogue PET scanning such as DOTA-TATE is being used for PGL, with superior sensitivity over FDG scans in these lesions due to their expression of somatostatin receptors.11,12 Cardiac MRI plays an important role in delineating invasion into the myocardium. Coronary angiography can also be employed to rule out compression or invasion of the coronary circulation, as well as assist in surgical planning.
SDHB mutation, as in the case highlighted, is an autosomal dominant syndrome and predisposes the individual to PGL, phaeochromocytoma, and renal cell carcinoma. Compared to the other SDHx mutations, SDHB poses a higher morbidity and mortality associated with their higher malignancy rate.13,14 A meta-analysis by Van Hulsteijn et al.15 found a malignancy rate of 17% for PGL associated with SDHB mutation. For these reasons, surgical resection remains the gold standard and in most cases requires median sternotomy and cardiopulmonary bypass due to relation of cardiac PGL to the coronary and great vessels.3
We report resection and biochemical cure of an intrapericardial PGL associated with SDHB mutation. Clear margins were not observed under pathological assessment, and histological features raised concern for malignant disease, especially in a patient with SDHB mutation syndrome. However, in this disease malignancy can only be confirmed by the presence of metastases, of which our patient has no evidence. He will require close ongoing follow-up in the future.
Acknowledgments
The authors thank Dr Vince Murdolo, specialist in anatomical pathology at the Royal Hobart Hospital for the pathological diagnosis as well as the macroscopic and microscopic photographs of the tumour.
Footnotes
Author contributions: D.A.S. and A.F.V. reviewed the literature and formulated the concept of the paper. D.A.S. wrote the manuscript, S.T. and A.H. operated on the patient and reviewed and edited the manuscript. J.R.B. was involved in assessment and perioperative management. All authors read and approved the final manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval: Our institution does not require ethical approval for reporting individual cases or case series.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent: Informed consent was obtained from the patient included in this study for the publication of this report and accompanying images. A copy of the consent form is available upon request.
ORCID iD: Dylan A Siejka  https://orcid.org/0000-0003-1080-4451
https://orcid.org/0000-0003-1080-4451
References
- 1. Michalowska I, Cwikla J, Prejbisz A, et al. Mediastinal paragangliomas related to SDHx gene mutations. Kardiochir Torakochirurgia Pol 2016; 13(3): 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. González López MT, Gonzalez SG, Garcia ES, et al. Surgical excision with left atrial reconstruction of a primary functioning retrocardiac paraganglioma. J Cardiothorac Surg 2013; 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang JG, Han J, Jiang T, et al. Cardiac paragangliomas. J Card Surg 2015; 30(1): 55–60. [DOI] [PubMed] [Google Scholar]
- 4. Tracy JC, Wein RO. Intrapericardial paraganglioma associated with succinate dehydrogenase complex subunit C mutation syndrome. Head Neck 2013; 35(8): E251–E253. [DOI] [PubMed] [Google Scholar]
- 5. Samuel N, Ejaz R, Silver J, et al. Primary mediastinal paraganglioma associated with a familial variant in the succinate dehydrogenase B subunit gene 2018; 117(2): 160–162. [DOI] [PubMed] [Google Scholar]
- 6. Martucci VL, Emaminia A, del Rivero J, et al. Succinate dehydrogenase gene mutations in cardiac paragangliomas. Am J Cardiol 2015; 115(12): 1753–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huo JL, Choi JC, DeLuna A, et al. Cardiac paraganglioma: diagnostic and surgical challenges. J Card Surg 2012; 27(2): 178–182. [DOI] [PubMed] [Google Scholar]
- 8. Feng N, Zhang WY, Wu XT. Clinicopathological analysis of paraganglioma with literature review. World J Gastroenterol 2009; 15(24): 3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rao D, Peitzsch M, Prejbisz A, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017; 177(2): 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kornaczewski ER, Pointon OP, Burgess JR. Utility of FDG-PET imaging in screening for succinate dehydrogenase B and D mutation-related lesions. Clin Endocrinol 2016; 85(2): 172–179. [DOI] [PubMed] [Google Scholar]
- 11. Taïeb D, Pacak K. New insights into the nuclear imaging phenotypes of cluster 1 pheochromocytoma and paraganglioma. Trends Endocrinol Metab 2017; 28(11): 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2015; 21(17): 3888–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. J Surg Oncol 2005; 89(3): 193–201. [DOI] [PubMed] [Google Scholar]
- 14. Khan MF, Datta S, Chisti MM, et al. Cardiac paraganglioma: clinical presentation, diagnostic approach and factors affecting short and long-term outcomes. Int J Cardiol 2013; 166(2): 315–320. [DOI] [PubMed] [Google Scholar]
- 15. Van Hulsteijn LT, Dekkers OM, Hes FJ, et al. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet 2012; 49(12): 768–776. [DOI] [PubMed] [Google Scholar]