

Abstract
Up to 80% of injuries sustained by U.S. soldiers in Operation Enduring Freedom and Operation Iraqi Freedom were the result of blast exposure from improvised explosive devices. Some soldiers experience multiple blasts while on duty, and it has been suggested that symptoms of repetitive blast are similar to those that follow multiple non-blast concussions, such as sport-related concussion. Despite the interest in the effects of repetitive blast exposure, it remains unknown whether an initial blast renders the brain more vulnerable to subsequent exposure, resulting in a synergistic injury response. To investigate the effect of multiple primary blasts on the brain, organotypic hippocampal slice cultures were exposed to single or repetitive (two or three total) primary blasts of varying intensities. Long-term potentiation was significantly reduced following two Level 2 (92.7 kPa, 1.4 msec, 38.5 kPa·msec) blasts delivered 24 h apart without altering basal evoked response. This deficit persisted when the interval between injuries was increased to 72 h but not when the interval was extended to 144 h. The repeated blast exposure with a 24 h interval increased microglia staining and activation significantly but did not significantly increase cell death or damage axons, dendrites, or principal cell layers. Lack of overt structural damage and change in basal stimulated neuron response suggest that injury from repetitive primary blast exposure may specifically affect long-term potentiation. Our studies suggest repetitive primary blasts can exacerbate injury dependent on the injury severity and interval between exposures.
Key words: : blast, brain, glia neuron, plasticity
Introduction
More than 44,000 soldiers have sustained traumatic brain injury (TBI) between 2000 and 2014, and nearly 86% of these men and women have sustained mild traumatic brain injury (mTBI).1 Blast injury is considered the “signature injury” of recent military conflicts.2–4 As many as 80% of all injuries sustained by US soldiers in Operation Enduring Freedom and Operation Iraqi Freedom were caused by improvised explosive devices.5,6 Some soldiers experience multiple blast exposures while on active duty, including breachers, who regularly use explosives to penetrate perimeters such as locked buildings.7 During training, breachers can experience as many as 100 explosive detonations over 5 days of practical training.7 Anecdotal symptoms reported by breachers following repetitive blast have been referred to informally as “breacher's brain” and are similar to those of people who have suffered multiple non-blast concussions, such as sport-related concussion.7,8
There is some controversy in the literature as to whether or not repetitive blast exposure even results in injury, let alone progressive neurodegeneration. In vivo models suggest that repetitive blast may produce persistent neuromotor dysfunction, axonal injury, learning deficits, and increased oxidative stress.9–11 However, in two in vitro studies using the same shock tube injury model and cell line, repetitive blast had conflicting results. In the first study, viability of human neuroblastoma cells was in fact improved and production of inflammatory cyclophilin A was decreased after repetitive blast exposure, suggesting repetitive blast improves outcome. In the second study, neuroblastoma permeability, indicating injury, was increased by repetitive blast.12–13 These in vivo and in vitro blast studies employed complex models of blast, loading the samples with both primary blast (shock wave loading) and tertiary blast (acceleration/deceleration or inertial loading) mechanisms, which may have contributed to these conflicting results.
Past studies suggest multiple mTBI from impact loading of the head can cause progressive brain injury.14–28 Both clinical and experimental studies of repetitive mTBI from inertial loading not associated with blast, such as sport-related mTBI, suggest that an initial mTBI begins a period of increased likelihood for subsequent brain injuries and heightened vulnerability to subsequent injury.15,20,29–31 However, the injury biomechanics of sports concussion differ from those of primary blast. Primary blast exposure deforms brain tissue with less than 10% strain but with associated strain rates of 100 to 1000 sec−1, whereas inertial loading deforms the brain in excess of 5% strain with associated strain rates from 5 to 50 sec−1.32 Despite these differences in causal biomechanics, breachers and other soldiers exposed to repetitive primary blast without concomitant rapid acceleration or impact loading of the head are reporting symptoms of progressive cognitive dysfunction, much like athletes with head injuries.7,8 It is still unknown whether primary blast exposure in isolation increases vulnerability of the brain to subsequent exposure, neurodegeneration, and brain dysfunction.
To more directly determine if an initial isolated primary blast predisposes the brain to greater damage from a subsequent exposure, we used an in vitro primary blast injury model developed in previous studies and exposed organotypic hippocampal slice cultures (OHSCs) to repeated primary blast exposures. Our results suggest that low-level, real-world primary blast-loading conditions may predispose the hippocampus to greater injury upon exposure to a subsequent blast. Depending on the exposure level and time interval between exposures, delivery of two primary blasts significantly and synergistically reduced long-term potentiation (LTP). This loss of LTP was accompanied by an increase in microglia activation. Current guidelines for soldier treatment may need further preclinical and clinical evaluation to consider the effects of repetitive exposure to primary blast.
Methods
Organotypic hippocampal slice cultures
All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee. OHSCs were generated as previously described.33–35 Hippocampi were excised from P8-11 Sprague-Dawley rat pups. The hippocampus was sectioned into 400-μm thick slices and grown on porous Millipore Millicell cell culture inserts (Millipore, Billerica, MA).33–35 Every 24–72 h, half of the culture medium was replaced with full-serum medium (50% Minimum Essential Medium, 25% Hank's Balanced Salt Solution, 25% heat inactivated horse serum, 2 μM L-glutamine, 25 mM D-glucose, and 10 mM HEPES; Sigma, St. Louis, MO).33–35 Cultures were maintained for 11–14 days prior to experimentation.
Blast injury
Blast injury methods have been described in detail.32–35 In brief, a shock wave was generated with a 76 mm diameter aluminum shock tube with an adjustable-length driver section (25 mm and 190.5 mm lengths were used for the current study) pressurized with helium or nitrogen and a 1240-mm long driven section.32–35 This shock tube model was characterized previously and provides a range of real-world primary blast-loading conditions.32–35 The range of peak pressures used in this study was 93 to 424 kPa, the range of durations was 0.25 to 2.3 msec, and the range of impulses was 9.2 to 248 kPa·msec (Table 1).33,35 Experimental pressure histories closely matched a Friedlander waveform, which represents a shock wave pressure history in the open field.32,35,36
Table 1.
In-Air and In-Fluid Parameters of Primary Blast Exposure
| In-air parameters | In-fluid parameters | |||||
|---|---|---|---|---|---|---|
| Level | Peak pressure (kPa) | Duration (msec) | Impulse (kPa·msec) | Peak pressure (kPa) | Duration (msec) | Impulse (kPa·msec) |
| 1 | 106 ± 2.2 | 0.25 ± 0.001 | 9.2 ± 1.6 | 134 ± 1.9 | 1.5 ± 0.01 | 88.8 ± 0.02 |
| 2 | 92.7 ± 2.6 | 1.4 ± 0.01 | 38.5 ± 0.7 | 270.1 ± 15 | 2.6 ± 0.2 | 295.1 ± 58 |
| 4 | 336 ± 8.3 | 0.89 ± 0.01 | 86.5 ± 1.4 | 598 ± 15 | 1.85 ± 0.3 | 440 ± 13 |
| 9 | 424 ± 6.4 | 2.3 ± 0.3 | 248 ± 3.4 | 1510 ± 91 | 2.8 ± 0.1 | 1420 ± 87 |
Organotypic hippocampal slice cultures were exposed to single or repetitive primary blast of varying levels (1, 2, 4, or 9). In-air parameters were evaluated from pressure-history traces collected at the end of the shock tube without the receiver in place. In-fluid parameters were collected in the fluid of the receiver adjacent to the sample. These blast injury levels were characterized and reported in a previous publication.35
Following blast or sham exposure, the sample was immediately removed from the receiver, placed into fresh full-serum medium, and returned to the incubator.34,35,37 Samples not receiving a blast at a given time-point received a sham exposure. Most samples received two blast or sham exposures and for these studies, the inter-injury duration was either 24 (Fig. 1), 72, or 144 h. A subset of samples received three Level 9 blast or sham exposures in succession delivered within 10 min of each other.
FIG. 1.

Experimental paradigm for repetitive primary blast exposure with a 24 h inter-injury interval. Organotypic hippocampal slice cultures were exposed to 0, 1, or 2 primary blasts 24 h apart. Samples not receiving a primary blast at any time-point received a sham exposure. Cell death was evaluated and electrophysiological recordings were started 72 h following the second exposure time-point.
Cell death quantification
Propidium iodide (PI) fluorescence was used to quantify cell death prior to the first exposure time-point to assess culture health and 72 h following the second exposure time-point. A subset of samples was subsequently exposed to an excitotoxic injury to serve as a positive control for cell death, with these positive control cultures imaged 24 h following the excitotoxic injury (see “Excitotoxic injury” below). OHSCs were stained with PI and imaged using previously published methods.34,35,37 Following pre-injury imaging, cultures were immediately injured (Fig. 1). Cell death was determined for all regions of interest (ROIs) of OHSCs (dentate gyrus [DG], cornu ammonis 1 [CA1], and cornu ammonis 3 [CA3]), as previously described, using MetaMorph (Molecular Devices, Downingtown, PA).33–35,38,39 Any OHSC with 5% or greater cell death in any ROI at the pre-injury time-point was excluded from the study.
Excitotoxic injury
As a positive control for cell death, a subset of OHSCs were exposed to glutamate as previously described.33 These samples received two blasts (Level 4) 24 h apart and were subjected to an excitotoxic injury (10 mM glutamate in serum-free medium for 3 h) 72 h following the second blast exposure (i.e., immediately after measuring blast-induced cell death). After 3 h, the glutamate-containing medium was changed to fresh, full-serum medium. Cultures were imaged for resultant cell death 24 h following glutamate exposure.
Electrophysiological recordings
Electrophysiological function was recorded and quantified as previously described.33,34,40,41 Recordings were performed 3–5 days following the final blast or sham exposure.
Stimulus–response (S-R) curves were generated as previously described, applying a constant current, bi-polar, biphasic stimulus (100 μsec positive phase followed by a 100 μsec negative phase) of varying magnitude (0-200 μA in 10 μA increments) to electrodes located in the Schaffer collaterals (SC).33,34 The evoked response was recorded on all electrodes simultaneously, and the peak–peak response recorded for each stimulus intensity was fit to a sigmoidal curve for each electrode based on the following equation40:
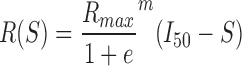 |
As described previously, Rmax represents the maximum amplitude of the evoked response, I50 represents the current necessary to generate a half-maximal response, and m is proportional to the slope of the sigmoidal fit and represents the spread in the firing threshold for the population of neurons.33,34 For S-R recordings, these three parameters were calculated for each electrode, and an average for each parameter across electrodes in each region (DG, CA3, CA1) was calculated for each slice. Data for each ROI of all OHSCs was averaged within experimental groups.
Each slice was evaluated for changes in LTP utilizing published methods.34 The baseline response was recorded for 30 min prior to LTP induction by stimulating the SC at the I50 every 60 sec and recording from all electrodes simultaneously. LTP was induced by stimulating a slice with three successive trains of 100 Hz stimulation at I50 for 1 sec with 10-sec intervals between trains. The post-induction response was recorded for 60 min, stimulating the SC at I50 every 60 sec and recording from all electrodes simultaneously. Potentiation was calculated as the difference between the average peak–peak voltage of the last 10 min of the post-induction recording and that of the last 10 min of the baseline recording normalized to the baseline recording. For calculating potentiation, data was calculated for electrodes within the CA1 only.34
Histology and immunohistochemistry
A subset of samples that received zero, one, or two Level 2 blasts were fixed with neutral buffered 10% formalin (Sigma), and dehydrated in a gradient of alcohols followed by xylene before embedding in paraffin. Samples were cut into 6-μm thick sections and mounted on slides. Paraffin was removed, and sections for histology were stained with hematoxylin and eosin (H&E; Gill's Hematoxylin 3 and Eosin Y; Thermo Fisher Scientific, Waltham, MA), dehydrated with a gradient of alcohols, and mounted for routine pathological analysis (n = 4 for each group).
Separate, adjacent sections were stained with an antibody for microtubule associated protein 2 (MAP-2; anti-MAP2 AB5622, n = 3, 1:100; Millipore) to visualize dendrites, antibodies for phosphorylated neurofilament heavy (NF-H; SMI-31, n = 3, 1:500; BioLegend, San Deigo, CA) and non-phosphorylated NF-H (SMI-32, n = 3, 1:500; BioLegend), an antibody for glial fibrillary acidic protein (GFAP) to visualize activated astrocytes (anti-GFAP Ab7260, n = 4, 1:2000; Abcam, Cambridge, MA), and an antibody for IBA1 to visualize activated microglia (anti-IBA1, n = 4, 1:400; Wako Pure Chemical Industries, Richmond, VA). Paraffin was removed. Antigen retrieval was performed for samples stained for GFAP, IBA1, SMI-31, and MAP-2 by microwaving in citrate buffer (0.01 M, pH 6.0; Fisher Scientific) for 20 min and cooling to room temperature for 30 min before being washed. Antigen retrieval was performed for samples stained for SMI-32 by warming for 25 min in a low pH retrieval solution (DAKO; Carpinteria, CA) before cooling to room temperature for 20 min. GFAP, IBA1, SMI-31, and MAP-2 samples were blocked with 10% normal goat serum (Vector Laboratories, Burlingame, CA) for 25 min and then incubated overnight at 4°C with the primary antibody. SMI-32 samples were blocked with 5% horse serum for 25 min and then incubated overnight at 4°C with the primary antibody. GFAP, IBA1, SMI-31, SMI-32, and MAP-2 samples were incubated with biotinylated anti-rabbit immunoglobulin G secondary antibody (1:200 for GFAP, IBA1, and MAP-2) or with horse anti-mouse secondary antibody (1:200 for SMI-31 and SMI-32; Vector Laboratories) for 30 min at room temperature. All samples were washed, incubated with ABC reagent (A 1:50, B 1:50) for 30 min at room temperature, washed, added to 3,3′-diaminobenzadine solution (DAKO) for 1 min (with the exception of GFAP, 30 sec), and counterstained with hematoxylin. Samples were dehydrated with a gradient of alcohols and mounted. As negative controls, additional sections received the same staining protocol without the primary antibodies.
Samples were analyzed semi-quantitatively by an individual blinded to the identity of the sections. To evaluate H&E stained sections, a 4-point rating scale of 0-3 was devised to assess (0, none; 1, rare; 2, occasional; 3, frequent) pathological findings, such as shrunken neurons, vacuolization, neuronal loss, and dark neurons. For MAP-2 immunohistochemistry, a rating scale of 0-3 corresponded to intensity and consistency of dendritic staining (0, uniform staining; 1, patchy loss of staining; 2, extensive loss of staining; 3, complete loss of staining). Samples stained with SMI-31 for phosphorylated NF-H were visually inspected for loss of axons, axonal swellings and discontinuities, and graded on a 4-point scale of 0-3 (0, uniform staining; 1, patchy loss of staining; 2, extensive loss of staining; 3, complete loss of staining). Samples stained with SMI-32 for non-phosphorylated NF-H were visually inspected for presence of non-phosphorylated and injured axons and graded on a scale of 0-3 (0, uniform staining; 1, patchy loss of staining and beading; 2, extensive loss of staining and significant beading; 3, complete loss of staining). Relative presence of microglia and macrophages was evaluated and graded on a scale of 0-3 (0, no IBA1 expression; 1, minimal number of IBA1 positive cells; 2, moderate number of IBA1 positive cells with varying presence of activated microglia [amoeboid shape] and macrophages; 3, high number of IBA1 positive cells with large number of activated microglia and macrophages). Presence of activated astrocytes was evaluated with GFAP and graded on a scale of 0-3 (0, no GFAP expression; 1, minimal number of GFAP positive astrocytes; 2, moderate number of GFAP positive astrocytes; 3, large number of GFAP positive astrocytes).
Statistical analysis
Statistical significance among LTP responses for varying repetitive injuries (Fig. 2) was first determined with a one-way analysis of variance (ANOVA) followed by a post hoc Bonferroni analysis (SPSS v. 22; IBM, Armonk, NY). For statistical analysis of S-R parameters, a one-way ANOVA was used to determine significance among groups (sham, single, double) for a given parameter (I50, Rmax, m) followed by a post hoc Bonferroni analysis for each ROI separately (Fig. 3). Statistical significance among cell death responses for varying repetitive injuries was determined with a one-way ANOVA followed by a post hoc Bonferroni analysis for each ROI separately (Fig. 4). A one-way ANOVA was used to determine significance among groups (sham, single, double) for semi-quantification of H&E, SMI-31, SMI-32, MAP-2, GFAP, and IBA1 separately (Fig. 5 and Fig. 6), followed by a Dunnett post hoc analysis with the sham-injured group as the control condition for comparisons. Statistical analysis of the extended interval LTP data (Fig. 7) was performed with a one-way ANOVA to determine significance among groups and a Bonferroni post hoc analysis to determine significance. Significance was set for all calculations as p < 0.05.
FIG. 2.

Repetitive primary blast altered long-term potentiation (LTP). The percent increase in potentiation of evoked response following induction of LTP was calculated for samples receiving 0 (Sham), 1 (Single), or 2 (Double) primary blasts of varying Levels (1, 2, or 4). LTP was significantly reduced for the single and double Level 4 blast groups, compared with sham. LTP was not significantly reduced for the single and repetitive Level 1 blast groups. LTP was not reduced significantly for a single Level 2 blast, but LTP was significantly reduced for two Level 2 blasts delivered 24 h apart (n ≥ 5; ± standard error of the mean; *p < 0.05).
FIG. 3.

Stimulus-response parameters after repetitive Level 2 primary blast with a 24 h inter-injury interval. There was no significant change in (A) the I50 parameter, (B) the Rmax parameter, or (C) the m parameter following single or repetitive Level 2 primary blast (n ≥ 5; ± standard error of the mean; not significant; dentate gyrus [DG], cornu ammonis 3 [CA3], cornu ammonis 1 [CA1]).
FIG. 4.

Cell death following single and repetitive primary blast injury. Cell death did not increase significantly following single or repetitive primary blast alone (n ≥ 9; ± standard error of the mean; not significant) in any region of interest (ROI; dentate gyrus [DG], cornu ammonis 3 [CA3], cornu ammonis 1 [CA1]). Cell death increased substantially and significantly in all ROI in the positive control group (Level 4 + Glutamate; *p < 0.05).
FIG. 5.

Histological and immunohistochemical evaluation of neurons, axons, and dendrites in organotypic hippocampal slice cultures (OHSCs) following exposure to single and repetitive Level 2 primary blast. (A-C) Hematoxylin and eosin (H&E) staining of OHSCs receiving the sham (A, scale bar = 1 mm), a single Level 2 blast (B), or two Level 2 blasts 24 h apart (C) revealed intact principal cell body layers. (D-F) SMI-31 staining of OHSCs receiving the sham (D), a single Level 2 blast (E), or two Level 2 blasts 24 h apart (F) showed the majority of axons were undisturbed by primary blast. (G-I) SMI-32 staining of OHSCs receiving the sham (G), a single Level 2 blast (H), or two Level 2 blasts 24 h apart (I) revealed minimal damage to axons. (J-L) Microtubule associated protein 2 (MAP-2) staining of OHSCs receiving the sham (J), a single Level 2 blast (K), or two Level 2 blasts 24 h apart (L) showed the majority of dendrites were undisturbed by primary blast. Color image is available online at www.liebertpub.com/neu
FIG. 6.

Immunohistochemical evaluation of organotypic hippocampal slice culture (OHSC) glial response following exposure to single and repetitive Level 2 primary blast. (A-C) glial fibrillary acidic protein (GFAP) staining of OHSCs receiving the sham (A, scale bar = 1 mm), a single Level 2 blast (B), or two Level 2 blasts 24 h apart (C) did not reveal an increase in astrogliosis. (D-F) IBA1 staining of OHSCs receiving the sham (D), a single Level 2 blast (E), or two Level 2 blasts 24 h apart (F) suggested an increase in activated microglia following primary blast. (G-I) Under higher magnification (scale bar = 100 μm) of the indicated regions in D-F, activated microglia (arrows) were observed in a region of the cornu ammonis 1 (CA1) following sham (G), single (H), and repetitive (I) injury. Semi-quantification of the samples (J) suggests there was a nearly significant increase in microglia response to a single primary blast (p = 0.058) and a significant increase in microglia response to the double primary blast exposure as compared to sham (n = 4; ± standard error of the mean; *p < 0.05). Color image is available online at www.liebertpub.com/neu
FIG. 7.

The duration of heightened vulnerability to subsequent primary blast may be 72 to 144 h long. (A) The interval between exposures was extended from 24 h, shown in Figure 4 and here, to 72 h (A, middle) and 144 h (A, right; n ≥ 5; ± standard error of the mean [SEM]; *p < 0.05, compared with sham 24 h and sham 144 h intervals). Potentiation was still significantly depressed for the 72 h “Double Level 2” group but was not for the 144 h “Double Level 2” group. (B) Cell death for sham, single and repetitive Level 2 groups with the 24 h interval, shown in Figure 4 and here, were compared to that of the extended inter-injury interval groups. On average, cell death was below 5% in all regions of interest for all blast- and sham-injured groups (n ≥ 6; ± standard error of the mean; not significant).
Results
Long-term potentiation was significantly depressed following repetitive primary blast without altering basal evoked response
Potentiation was roughly 64% in the CA1 in sham-injured samples (Fig. 2). LTP decreased non-significantly following both a single and double Level 1 primary blast with a 24-h interval between exposures. LTP was not significantly reduced following a single Level 2 primary blast exposure, whereas it was significantly reduced, compared with sham, after two exposures of Level 2 primary blast delivered 24 h apart. LTP decreased significantly following both a single and double Level 4 primary blast with a 24-h interval between exposures, compared with the sham.
There were no statistically significant changes in any S-R parameter (I50, Rmax, m) in any of the three ROI (DG, CA3, CA1) after sham, single, or repetitive Level 2 blast exposures applied with a 24 h interval between injuries (Fig. 3).
Cell death was not increased by single or repetitive primary blast
Cell death increased minimally (< 5%) in all ROIs after sham injury (Fig. 4). Cell death increased minimally following single and repetitive primary blast (Levels 1, 2, 4; Table 1) with an inter-injury interval of 24 h. Given that multiple primary blasts (Levels 1, 2, 4) did not result in significant cell death, we investigated the potential for repetitive exposure to our highest primary blast level (Level 9) to cause cell death. Three Level 9 blasts delivered within 10 min did not increase cell death, either. However, cell death increased substantially and significantly following glutamate exposure (after blast), which served as the positive control (double level 4+ glutamate; > 37% cell death in all ROIs).
Single and repetitive primary blast did not significantly alter histology or structure of axons or dendrites
Samples stained with H&E were evaluated for loss of neurons, vacuolization, and dark or shrunken neurons. No histological damage was observed in samples receiving the sham, single Level 2, or repetitive Level 2 blasts with the 24 h interval (representative images shown in Fig. 5). Staining with MAP-2 and SMI-31 was uniform and did not suggest a significant loss of dendrites or axons, respectively (Fig. 5D-F and 5J-L). Staining with SMI-32 was consistent with neuronal cell bodies and dendrites in the hippocampus and did not show appreciable differences between groups, suggesting no de-phosphorylation of NF-H or axonal damage (Fig. 5G-I).
Microglia activation was increased after Level 2 primary blast exposure; however, astroglial response was unaffected by blast exposure
Samples stained with GFAP did not show any significant differences in activated astrocytes with either single or repetitive primary blast, compared with the sham (Fig. 6A-C). The number of IBA1 positive cells (microglia and macrophages) was increased by Level 2 primary blast exposure (Fig. 6D-I). This response was significantly higher for the double injury group, compared with the sham (Fig. 6J).
Heightened vulnerability to subsequent primary blast persisted for at least 72 h
Increasing the interval to 72 h did not mitigate the deficit in LTP, which was still significantly depressed by both Level 2 primary blast exposures (Fig. 7A). When the interval was increased to 144 h, potentiation was decreased but was not significantly lower than sham from the 24-h interval study or its own time-matched sham (Fig. 7A; sham 144 h interval). LTP was not decreased when the inter-sham interval was extended to 144 h, indicating that the extended experimental duration did not negatively impact induction of LTP. Similarly, cell death did not increase with the extended duration of the experiment. All samples appeared otherwise healthy, with less than 5% cell death in all ROIs (Fig. 7B).
Discussion
This study is the first to report that an initial primary blast (Level 2) leads to a period of heightened vulnerability to subsequent primary blast injury. These studies suggest repetitive primary blast may exacerbate brain injury, resulting in significant microglia activation and LTP deficits; however, this LTP deficit may be overcome by increasing the interval between exposures. We report that heightened vulnerability following Level 2 primary blast may last as long as 72 h but does not exceed 144 h. Further, repetitive injury may specifically alter long-term plasticity given that there was no significant loss of cells, overt structural damage to axons or dendrites, or altered basal evoked function.
Clinical reports and in vivo experimental studies suggest that cell loss, dendritic damage, and axonopathy can be caused by blast.42–46 However, given the difficulty of delivering isolated primary blast in vivo without confounding effects of rapid head motion and the complexity of human exposure and medical histories, it is unclear whether to attribute these pathologies to primary blast or potentially confounding effects of rapid head motion, which is well-known to injure the brain.36 This is the first study to show repetitive exposure to isolated primary blast can alter LTP without significant changes in axon or dendrite structure or loss of cells. In addition, basal evoked response was not changed with single nor repetitive primary blast. Previous work from our laboratory has evaluated OHSC cell death and electrophysiological functional tolerance, and we reported a single primary blast even at the highest level tested (Level 9) can reduce LTP without altering short-term synaptic plasticity, increasing cell death, or changing basal evoked response.33,34 Taken together, these studies suggest deficits in LTP from primary blast may be specific to long-term neuronal plasticity and not the result of overt physical damage to axons and dendrites, loss of neurons, or damage to pre-synaptic signaling.
Despite the intense interest in the mechanisms and consequences of primary blast TBI, there remains relatively little data in the literature on specific impairments to the hippocampal circuitry from primary blast. In other studies, LTP measured in acute mouse hippocampal slices was reduced by a single in vivo blast (167 kPa·msec) out to 4 weeks following exposure accompanied by astrogliosis, axonal injury, and cell loss.42 Outside of our own work,34 to our knowledge, this is the only evidence of LTP deficits after blast exposure, but these results are difficult to interpret further because the head was allowed to accelerate/decelerate during the blast exposure, raising the possibility that these deficits were at least in part due to brain deformation caused by head motion.41 To this end, these LTP deficits disappeared when the head was restrained during blast exposure.42 Inertial loading resulting in rapid head acceleration and deformation of brain is well-known to reduce potentiation in vivo and can be accompanied by cell loss and microglia activation.47–52
Other in vitro studies with OHSCs have reported significant reductions in LTP following barotrauma; however, cell loss was not evaluated.53–57 Additionally, there is precedent for observing deficits in neural plasticity after inertial injury without significant cell loss, as reported in the current study after primary blast. One study reported reduced capacity for experience-dependent plasticity in rats following fluid percussion injury (FPI) without neuron loss, and a second study reported deficits in working memory in rats subjected to mild and moderate FPI for at least 15 days following exposure without neuron loss.58,59
In the current study, the biomechanical threshold for a single primary blast to reduce LTP was between Level 2 and Level 4 (Table 1). However, this threshold is lower when considering the effects of multiple blast exposures, as exposure to Level 2 primary blast loading 24 h following the first exposure lead to significant impairment in long-term neural plasticity (Fig. 2). Ours is the first study to report that a second primary blast (Level 2) within a certain period resulted in LTP deficits that were synergistic or more than twice that of the single exposure. Given these changes, we considered how the levels of primary blast used in this study (Table 1), compared with real-world loading conditions.33 Using proposed scaling relationships for blast exposure, we observe a Level 1 primary blast is comparable to exposure to a M49A6 60 mm mortar round at a 0.25 to 2 m stand-off distance, and a Level 9 primary blast is comparable to exposure to a M118 bomb at a 10 to 32 m stand-off distance.33 For comparison, breachers may use up to 4.5 kg of composition C-4 explosive (cyclotrimethylene-trinitramine) to gain entry to a reinforced concrete wall. In the free-field, 2.7 kg of C-4 at a 5 m stand-off distance produces a shock wave with a 96 kPa peak pressure and 4.6 msec duration (CONWEP), which is a much longer duration and thus higher impulse than our Level 2 exposure (92.7 kPa, 1.4 msec, 38.5 kPa·msec).60
For all blast levels used in this in vitro study, both the unscaled in-air parameters and these parameters scaled for rat-brain mass according to Bowen's relationships are consistent with real-world loading conditions for human's exposed to blast.33,61,62 Compared with the in-air shock wave, the intracranial pressure (ICP) wave has a slower rise-time and may be otherwise modified as it transitions through media with varying properties and reflections are introduced (see Table 1).32,33.35,63 In an in vivo study, exposure to a 70 kPa overpressure, 7 msec duration shock wave generated by a shock tube produced an ICP history with a rise-time 0.5−1 msec longer than that of the shock wave input, a peak pressure of 60–145 kPa, and a 7.0–7.5 msec duration.64 Peak pressure was modified greatly by animal mass and head orientation relative to the direction of shock wave propagation. In our experimental set-up, the pressure history recorded at the sample in the receiver closely mimicked an ICP history following primary blast exposure.33,35 Our in vitro results, when combined with limited human volunteer data, detailed reconstructions, and extensive computational modeling, will yield a much more informed estimate of the tolerance to primary blast exposure, which has implications for designing equipment to better protect soldiers from blast-induced brain injury.
Activation of glia is commonly observed after blast exposure, which is also a consequence of FPI or controlled cortical impact injury.47,65 In one in vivo study in which rats were exposed to detonation of an explosive charge within a tube (154 kPa, 1.7 msec) resulting in a complex blast exposure, microglia activation (stained with OX-42) increased in the hippocampus 2 days following exposure and returned to sham levels 7 days following exposure.66 This same study reported an increase in GFAP staining for astrocytes in the hippocampus over the course of 21 days following blast (240 kPa, 2 msec) through the mossy fiber region and at astrocytic end-feet surrounding capillaries,66 suggesting compromise of the blood–brain barrier (BBB). In a separate study utilizing an in vivo explosive blast model, microglia activation (stained with OX-42) increased as early as 1 day and persisted out to 28 days following blast exposure (202 dB or 240 kPa peak pressure and 2 msec duration), and increased microglia, macrophage, and lymphocyte staining (OX-18 and OX-6) was observed with evidence of subarachnoid hemorrhage between 1 and 14 days following exposure.45 Hippocampal microglia also were activated after exposure of rodents to blast generated from an air-driven shock tube (120 kPa) accompanied by BBB breakdown.67 An in vivo study reported microglia activation (IBA1) without increased astrogliosis (GFAP) in rats 7 days following low-level exposure (69 kPa, 5.5 msec), which was similar to our finding, albeit for a different level blast in vitro.68 Together, these studies suggest that blast increases microglia activation, as we also report. However, given the complexities of in vivo blast loading, the contribution from primary versus tertiary loading mechanisms could not be separated, as was possible in our study. Additionally, opening of the BBB, which may occur in vivo and may have occurred in those studies, can independently result in gliosis.45,66,67,69 However, we observed activation of microglia by a pressure impulse in the absence of BBB opening. Characterization of microglia in OHSCs suggests that the functional characteristics of these in vitro microglia resemble those of microglia in the in vivo environment.70,71 Microglia reactivity in response to sustained barotrauma (15 or 30 mm Hg for 24 h) in vitro suggests mechanical stress can activate microglia inflammatory responses without exposure to blood serum constituents that accompanies BBB compromise.72 Ours is the first study to report that repetitive isolated primary blast exposure alone can increase microglia activation. Microglia may play a part in hippocampal plasticity and synaptic pruning.73,74 In the mouse primary visual cortex, it was observed that increased microglia contact with synaptic elements resulted in a prolonged reduction in size or elimination of dendritic spines.75 Loss of dendritic spines or abnormalities can negatively impact LTP.76 Further studies are necessary to elucidate the potential influence of activated microglia on LTP deficits following trauma.
There are limitations to consider when interpreting the results of our study. Here, we have investigated the effect of repetitive primary blast exposure to OHSCs grown in vitro and subsequently exposed to injury without the surrounding brain-skull complex or the rest of the body. As previously described, our motivation was to investigate primary blast in isolation, necessitating the in vitro approach.32,42,77,78 Some soldiers may experience multiple primary blasts of varying intensity, inter-injury interval, complexity, and repetition while on active duty or in practical training.7 Here, we have principally focused on investigating the effects of two primary blasts with simple pressure histories with a 24-h inter-injury interval for three different levels of primary blast. These levels correspond to real-world blast-loading conditions, but may not represent the most common exposures or even those that can be withstood without concomitant head motion in the real world. We chose to investigate a simplified and highly controlled set of repetitive injury paradigms given the wide range of parameters that could be modulated. We also chose to investigate a simplified subset of primary blast loading that mimics the Friedlander waveform given the unknown potential for more complex pressure-histories to cause injury. Additionally, it is unclear how in vitro electrophysiological measures relate to in vivo behavioral measures or clinically reported symptoms.42,79 Soldiers who experience single or repetitive primary blast loading report difficulty thinking, confusion, retrograde amnesia, amnesia, difficulty concentrating, and other concussion-like symptoms.7,80,81 Although LTP is considered an in vitro correlate for learning and memory, the LTP deficits observed in our study cannot be directly related to learning and memory deficits experienced by soldiers exposed to blast.
The effects of repetitive concussions and mTBI from sport-related injury have been studied both clinically and experimentally, suggesting that an initial mTBI increases brain vulnerability to subsequent mTBI.15,18,20,28–30,82–86 While this result is similar to those here, it is important to highlight that the biomechanics of sport-related TBI and primary blast loading are dissimilar, and for this reason interpreting our data in the context of sports injury would be inappropriate.32,36 Here, we report that the period of time during which OHSCs may experience heightened vulnerability to primary blast may be more than 72 h but fewer than 144 h in duration. Understanding the time course of blast-induced injury may be useful for future experimental studies investigating safe rest periods following blast exposure, which could ultimately inform medical guidelines for the soldier who is exposed to blast. Translating our findings to safe rest-periods for humans, however, is an area for future preclinical and clinical studies.
Acknowledgments
This work was supported by a Multi-disciplinary University Research Initiative from the Army Research Office (W911NF-10-1-05026).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Fischer H.E. (2015). A guide to U.S. military casualty statistics: Operation inherent resolve, operation new dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Available at: https://fas.org/sgp/crs/natsec/RS22452.pdf Accessed February15, 2016
- 2.Zoroya G. (2015). Key Iraq wound: brain trauma. USA Today. Available at: http://usatoday30.usatoday.com/news/nation/2005-03-03-brain-trauma-lede_x.htm Accessed February15, 2016
- 3.Elder G.A. and Cristian A. (2009). Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt. Sinai J. Med. 76, 111–118 [DOI] [PubMed] [Google Scholar]
- 4.Alvarez L. (2015) War veteran's concussions are often overlooked. New York Times. Available at: www.nytimes.com/2008/08/26/us/26tbi.html?_r=0 Accessed February15, 2016
- 5.Faul M., Xu L., Wald M.M., Coronado V.G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention: Atlanta, GA [Google Scholar]
- 6.Centers for Disease Control and Prevention. (2009). Blast injuries: traumatic brain injuries. Available at: www.brainline.org/content/2010/10/blast-injuries-traumatic-brain-injuries-from-explosions.html Accessed February15, 2016
- 7.Tate C.M., Wang K.K., Eonta S., Zhang Y., Carr W., Tortella F.C., Hayes R.L., and Kamimori G.H. (2013). Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study. J. Neurotrauma 30, 1620–1630 [DOI] [PubMed] [Google Scholar]
- 8.DeKosky S.T., Ikonomovic M.D., and Gandy S. (2010). Traumatic brain injury—football, warfare, and long-term effects. N. Engl. J. Med. 363, 1293–1296 [DOI] [PubMed] [Google Scholar]
- 9.Ahlers S.T., Vasserman-Stokes E., Shaughness M.C., Hall A.A., Shear D.A., Chavko M., McCarron R.M., and Stone J.R. (2012). Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front. Neurol. 3, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed F.A., Kamnaksh A., Kovesdi E., Long J.B., and Agoston D.V. (2013). Long-term consequences of single and multiple mild blast exposure on select physiological parameters and blood-based biomarkers. Electrophoresis 34, 2229–2233 [DOI] [PubMed] [Google Scholar]
- 11.Wang Y., Wei Y., Oguntayo S., Wilkins W., Arun P., Valiyaveettil M., Song J., Long J. B., and Nambiar M.P. (2011). Tightly coupled repetitive blast-induced traumatic brain injury: Development and characterization in mice. J. Neurotrauma 28, 2171–2183 [DOI] [PubMed] [Google Scholar]
- 12.Arun P., Abu-Taleb R., Valiyaveettil M., Wang Y., Long J.B., and Nambiar M.P. (2012). Transient changes in neuronal cell membrane permeability after blast exposure. Neuroreport 23, 342–346 [DOI] [PubMed] [Google Scholar]
- 13.Arun P., Spadaro J., John J., Gharavi R.B., Bentley T.B., and Nambiar M.P. (2011). Studies on blast traumatic brain injury using in-vitro model with shock tube. Neuroreport 22, 379–384 [DOI] [PubMed] [Google Scholar]
- 14.Guskiewicz K.M., Marshall S.W., Bailes J., McCrea M., Cantu R.C., Randolph C., and Jordan B. D. (2005). Association between recurrent concussion and late-life cognitive impairment in retired professional football players. J. Neurosurg 57, 719–726 [DOI] [PubMed] [Google Scholar]
- 15.Guskiewicz K.M., McCrea M., Marshall S.W., Cantu R.C., Randolph C., Barr W., Onate J.A., and Kelly J.P. (2003). Cumulative effects associated with recurrent concussion in collegiate football players. JAMA 290, 2549–2555 [DOI] [PubMed] [Google Scholar]
- 16.Matser J.T., Kessels A.G.H., Jordan B.D., Lezak M.D., and Troost J. (1998). Chronic traumatic brain injury in professional soccer players. J. Neurol 51, 791–796 [DOI] [PubMed] [Google Scholar]
- 17.Slemmer J. E., Matser E. J. T., De Zeeuw C. I., and Weber J. T. (2002). Repeated mild injury causes cumulative damage to hippocampal cells. Brain 125, 2699–2709 [DOI] [PubMed] [Google Scholar]
- 18.Slemmer J.E. and Weber J.T. (2005). The extent of damage following repeated injury to cultured hippocampal cells is dependent on the severity of insult and inter-injury interval. Neurobiol. Dis. 18, 421–431 [DOI] [PubMed] [Google Scholar]
- 19.DeFord S.M., Wilson M.S., Rice A.C., Clausen T., Rice L.K., Barabnova A., Bullock R., and Hamm R.J. (2002). Repeated mild brain injuries result in cognitive impairment in b6c3f1 mice. J. Neurotrauma 19, 427–438 [DOI] [PubMed] [Google Scholar]
- 20.DeRoss A.L., Adams J.E., Vane D.W., Russell S.J., Terella A.M., and Wald S.L. (2002). Multiple head injuries in rats: Effects on behavior. J. Trauma 52, 708–714 [DOI] [PubMed] [Google Scholar]
- 21.Friess S.H., Ichord R.N., Ralston J., Ryall K., Helfaer M.A., Smith C., and Margulies S.S. (2009). Repeated traumatic brain injury affects composite cognitive function in piglets. J. Neurotrauma 26, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huh J.W., Widing A.G., and Raghupathi R. (2007). Basic science; repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: A preliminary report. J. Neurotrauma 24, 15–27 [DOI] [PubMed] [Google Scholar]
- 23.Laurer H.L., Bareyre F.M., Lee V.M., Trojanowski J.Q., Longhi L., Hoover R., Grady M.S., and McIntosh T.K. (2001). Mild head injury increasing the brain's vulnerability to a second concussive impact. J. Neurosurg. 95, 859–870 [DOI] [PubMed] [Google Scholar]
- 24.Longhi L., Saatman K.E., Fujimoto S., Raghupathi R., Meaney D.F., Davis J., McMillan A., Conte V., Laurer H.L., Stein S., Stocchetti N., and McIntosh T.K. (2005). Temporal window of vulnerability to repetitive experimental concussive brain injury. J. Neurosurg. 56, 364–374 [DOI] [PubMed] [Google Scholar]
- 25.Raghupathi R., Mehr M.F., Helfaer M.A., and Margulies S.S. (2004). Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J. Neurotrauma 21, 307–316 [DOI] [PubMed] [Google Scholar]
- 26.Uryu K., Laurer H.L., McIntosh T.K., Pratico D., Martinez D., Leight S., Lee V.M., and Trojanowski J. Q. (2002). Repetitive mild brain trauma accelerates a beta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of alzheimer amyloidosis. J. Neurosci. 22, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing G., Barry E.S., Benford B., Grunberg N.E., Li H., Watson W.D., and Sharma P. (2013). Impact of repeated stress on traumatic brain injury-induced mitochondrial electron transport chain expression and behavioral responses in rats. Front. Neurol. 4, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prins M.L., Alexander D., Giza C.C., and Hovda D.A. (2013). Repeated mild traumatic brain injury: Mechanisms of cerebral vulnerability. J. Neurotrauma 30, 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giza C.C. and Hovda D.A. (2001). The neurometabolic cascade of concussion. J. Athl. Training 36, 228–235 [PMC free article] [PubMed] [Google Scholar]
- 30.McCrea M., Barr W., Guskiewicz K.M., Randolph C., Marshall S.W., Cantu R.C., Onate J.A., and Kelly J. P. (2005). Standard regression-based methods for measuring recovery after sport-related concussion. J. Int. Neuropsychol. Soc. 11, 58–69 [DOI] [PubMed] [Google Scholar]
- 31.Creeley C.E. (2004). Multiple episodes of mild traumatic brain injury result in impaired cognitive performance in mice. Acad. Emerg. Med. 11, 809–819 [DOI] [PubMed] [Google Scholar]
- 32.Panzer M.B., Matthews K.A., Yu A.W., Morrison B., III, Meaney D.F., and Bass C.R. (2012). A multiscale approach to blast neurotrauma modeling: Part I—development of novel test devices for in vivo and in vitro blast injury models. Front. Neurol. 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Effgen G.B., Vogel E.W., III, Lynch K.A., Lobel A., Hue C.D., Meaney D.F., Bass C.R., and Morrison B., III (2014). Isolated primary blast alters neuronal function with minimal cell death in organotypic hippocampal slice cultures. J. Neurotrauma 31, 1202–1210 [DOI] [PubMed] [Google Scholar]
- 34.Vogel E.W., III, Effgen G.B., Patel T.P., Meaney D.F., Bass C.R., and Morrison B., III (2015). Isolated primary blast inhibits long-term potentiation in organotypic hippocampal slice cultures. J. Neurotrauma 2015. December 2; Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Effgen G.B., Hue C.D., Vogel E.W., III, Panzer M. B., Meaney D.F., Bass C.R., and Morrison B., III (2012). A multiscale approach to blast neurotrauma modeling: Part II: methodology for inducing blast injury to in vitro models. Front. Neurol. 3, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bass C.R., Panzer M.B., Rafaels K.A., Wood G., Shridharani J., and Capehart B. (2012). Brain injuries from blast. Ann. Biomed. Eng. 40, 185–202 [DOI] [PubMed] [Google Scholar]
- 37.Effgen G.B., Gill E., and Morrison B. I. (2012). A model of repetitive, mild traumatic brain injury and a novel pharmacological intervention to block repetitive injury synergy. Presented at the 2012 International IRCOBI Conference on the Biomechanics of Injury, Dublin, Ireland [Google Scholar]
- 38.Morrison B., III, Cater H.L., Benham C.D., and Sundstrom L. E. (2006). An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J. Neurosci. Methods 150, 192–201 [DOI] [PubMed] [Google Scholar]
- 39.Cater H.L., Sundstrom L.E., and Morrison B., III (2006). Temporal development of hippocampal cell death is dependent on tissue strain but not strain rate. J Biomech 39, 2810–2818 [DOI] [PubMed] [Google Scholar]
- 40.Yu Z., Elkin B.S., and Morrison B., III (2009). Quantification of functional alterations after in vitro traumatic brain injury. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 1135–1138 [DOI] [PubMed]
- 41.Kang W. H. and Morrison B.I. (2014). Functional tolerance to mechanical deformation developed from organotypic hippocampal slice cultures. Biomech. Model Mechanobiol. 14, 561–575 [DOI] [PubMed] [Google Scholar]
- 42.Goldstein L.E., Fisher A.M., Tagge C.A., Zhang X.L., Velisek L., Sullivan J.A., Upreti C., Kracht J.M., Ericsson M., Wojnarowicz M.W., Goletiani C.J., Maglakelidze G.M., Casey N., Moncaster J.A., Minaeva O., Moir R.D., Nowinski C.J., Stern R.A., Cantu R.C., Geiling J., Blusztajn J.K., Wolozin B.L., Ikezu T., Stein T. D., Budson A.E., Kowall N.W., Chargin D., Sharon A., Saman S., Hall G F., Moss W.C., Cleveland R.O., Tanzi R.E., Stanton P.K., and McKee A.C. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Trans. Med. 4, 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saljo A., Bauo F., Haglid K.G., and Hansson H.A. (2000). Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J. Neurotrauma 17, 719–726 [DOI] [PubMed] [Google Scholar]
- 44.Ryu J., Horkayne-Szakaly I., Xu L., Pletnikova O., Leri F., Eberhart C., Troncoso J.C., and Koliatsos V.E. (2014). The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol. Commun. 2, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur C., Singh J., Lim M. K., Ng B. L., Yap E.P.H., and Ling E.A. (1995). The response of neurons and microglia to blast injury in the rat brain. Neuropathol. Appl. Neurobiol. 21, 369–377 [DOI] [PubMed] [Google Scholar]
- 46.Saljo A., Bao F., Jingshan S., Hamberger A., Hansson H.A., and Haglid K.G. (2002). Exposure to short-lasting impulse noise causes neuronal c-jun expression and induction of apoptosis in the adult rat brain. J. Neurotrauma 19, 985–991 [DOI] [PubMed] [Google Scholar]
- 47.Aungst S.L., Kabadi S.V., Thompson S.M., Stoica B.A., and Faden A.I. (2014). Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyazaki S., Katayama Y., Lyeth B.G., Jenkins L.W., De Witt D.S., Goldberg S.J., DNewlon P.G., and Hayes R.L. (1992). Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 585, 335–339 [DOI] [PubMed] [Google Scholar]
- 49.Sanders M.J., Sick T.J., Perez-Pinzon M.A., Dalton Dietrich W., and Green E. (2000). Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 861, 69–76 [DOI] [PubMed] [Google Scholar]
- 50.Shultz S. R., Bao F., Omana V., Chiu C., Brown A., and Cain D. P. (2012). Repeated mild lateral fluid percussion brain injury in the rat causes cumulative long-term behavioral impairments, neuroinflammation, and cortical loss in an animal model of repeated concussion. J. Neurotrauma 29, 281–94 [DOI] [PubMed] [Google Scholar]
- 51.Giza C.C. and Prins M.L. (2006). Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev. Neurosci. 28, 364–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albensi B.C. (2001). Models of brain injury and alterations in synaptic plasticity. J. Neurosci. Res. 65, 279–283 [DOI] [PubMed] [Google Scholar]
- 53.Girard J., Panizzon K., and Wallis R.A. (1996). Azelastine protects against ca1 traumatic neuronal injury in the hippocampal slice. Eur. J. Pharmacol. 300, 43–49 [DOI] [PubMed] [Google Scholar]
- 54.Panizzon K.L., Dwyer B.E., Nishimura R.N., and Wallis R.A. (1996). Neuroprotection against ca1 injury with metalloporphyrins. Neuroreport 7, 662–666 [DOI] [PubMed] [Google Scholar]
- 55.Panizzon K.L., Shin D., Frautschy S., and Wallis R.A. (1998). Neuroprotection with bcl-2 20-34 peptide against trauma. Neuroreport 9, 4131–4136 [DOI] [PubMed] [Google Scholar]
- 56.Wallis R.A. and Panizzon K.L. (1995). Felbamate neuroprotection against ca1 traumatic neuronal injury. Eur. J. Pharmacol. 294, 475–482 [DOI] [PubMed] [Google Scholar]
- 57.Wallis R.A., Panizzon K.L., and Girard J.M. (1996). Traumatic neuroprotection with inhibitors of nitric oxide and adp-ribosylation. Brain Res. 710, 169–177 [DOI] [PubMed] [Google Scholar]
- 58.Gurkoff G.G., Giza C.C., and Hovda D.A. (2006). Lateral fluid percussion injury in the developing rat causes an acute, mild behavioral dysfunction in the absence of significant cell death. Brain Res. 1077, 24–36 [DOI] [PubMed] [Google Scholar]
- 59.Lyeth B.G., Jenkins L.W., Hamm R.J., Dixon C.E., Phillips L.L., Clifton G.L., Young H.F., and Hayes R.L. (1990). Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 526, 249–258 [DOI] [PubMed] [Google Scholar]
- 60.U.S. Dept of the Army, F-22. (2007).The infantry rifle platoon and squad (FM 3-21.8). Available at: https://rdl.train.army.mil/catalog-ws/view/100.ATSC/04183AF4-34EB-47F0-BCEE-29C93432DA49-1274564010088/3-21.8/toc.htm Accessed February15, 2016
- 61.Rafaels K.A., Bass C.R., Panzer M.B., Salzar R.S., Woods W.A., Feldman S.H., Walilko T., Kent R.W., Capehart B.P., Foster J.B., Derkunt B., and Toman A. (2012). Brain injury risk from primary blast. J. Trauma 73, 895–901 [DOI] [PubMed] [Google Scholar]
- 62.Rafaels K., Bass C. R., Salzar R.S., Panzer M.B., Woods W., Feldman S., Cummings T., and Capehart B. (2015). Survival risk assessment for primary blast exposures to the head. J. Neurotrauma 28, 2319–2328 [DOI] [PubMed] [Google Scholar]
- 63.Clemedson C.J. and Pettersson H. (1956). Propagation of a high explosive air shock wave through different parts of an animal body. Am. J. Physiol. 184, 119–126 [DOI] [PubMed] [Google Scholar]
- 64.Dal Cengio A.L., Keane N.J., Bir C., Ryan A.G., Xu L., and VandeVord P.J. (2012). Head orientation affects the intracranial pressure response resulting from shock wave loading in the rat. J. Biomech. 45, 2595–2602 [DOI] [PubMed] [Google Scholar]
- 65.Evilsizor M.N., Ray-Jones H.F., Ellis T. W., Jr., Lifshitz J., and Ziebell J.M. (2015) Microglia in experimental brain injury: Implications on neuronal injury and circuit remodeling. In: Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Kobeissy F. H. (ed). CRC Press: Boca Raton, FL, pps. 9–90 [PubMed] [Google Scholar]
- 66.Saljo A., Bao F., Hamberger A., Haglid K.G. and Hansson H.A. (2001). Exposure to short-lasting impulse noise causes microglial and astroglial cell activation in the adult rat brain. Pathophysiology 8, 105–111 [DOI] [PubMed] [Google Scholar]
- 67.Readnower R.D., Chavko M., Adeeb S., Conroy M.D., Pauly J.R., McCarron R.M., and Sullivan P.G. (2010). Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 88, 3530–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sajja V.S., Ereifej E.S., and VandeVord P.J. (2014). Hippocampal vulnerability and subacute response following varied blast magnitudes. Neurosci. Lett. 570, 33–37 [DOI] [PubMed] [Google Scholar]
- 69.Nimmerjahn A., Kirchhoff F., and Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 [DOI] [PubMed] [Google Scholar]
- 70.Czapiga M. and Colton C. A. (1999). Function of microglia in organotypic hippocampal slice cultures. J. Neurosci. Res. 56, 6440651. [DOI] [PubMed] [Google Scholar]
- 71.Heailer N.P., Jarhult J.D., and Mitsch R. (1996). Resting microglial cells in vitro: analysis of morphology and adhesion molecule expression in organotypic hippocampal slice cultures. Glia 18, 319–331 [DOI] [PubMed] [Google Scholar]
- 72.Yu G., Dymond M., Yuan L., Chaturvedi L.S., Shiratsuchi H., Durairaj S., March H.M., and Basson M. (2011). Anti-inflammatory effects of propofol's effects on phagocytosis, proliferation, nitrate production, and cytokine secretion in pressure-stimulated microglial cells. J. Surg. 150, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schafer D.P. and Stevens B. (2015). Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect. Biol. 7, a020545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tremblay M.E., Stevens B., Sierra A., Wake H., Bessis A., and Nimmerjahn A. (2011). The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wake H., Moorhouse A.J., Jinno S., Kohsaka S., and Nabekura J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuste R. and Bonhoeffer T. (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Ann. Rev. Neurosci. 24, 1071–1089 [DOI] [PubMed] [Google Scholar]
- 77.Gullotti D.M., Beamer M., Panzer M.B., Chen Y.C., Patel T.P., Yu A., Jaumard N., Winkelstein B., Bass C.R., Morrison B.I., and Meaney D.F. (2014). Significant head accelerations can influence immediate neurological impairments in a murine model of blast-induced traumatic brain injury. J. Biomech. Eng. 136, 091004. [DOI] [PubMed] [Google Scholar]
- 78.Hue C.D., Cho F.S., Cao S., Nicholls R.E., Vogel E.W., III, Sibindi C., Arancia O., Bass C.R., Meaney D.F., and Morrison B., III (2015). Time course and size of blood-brain barrier opening in a mouse model of blast-induced traumatic brain injury J. Neurotrauma 2015. November 12; Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 79.Yin T.C., Britt J.K., Ready J.M., and Pieper A.A. (2014). P7c3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Reports 8, 1731–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Appelboom G., Han J., Bruce S., Szpalski C., and Connolly E.S., Jr. (2012). Clinical relevance of blast-related traumatic brain injury. Acta Neurochir. 154, 131–134 [DOI] [PubMed] [Google Scholar]
- 81.Hicks R.R., Fertig S.J., Desrocher R.E., Koroshetz W.J., and Pancrazio J.J. (2010). Neurological effects of blast injury. J. Trauma 68, 1257–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Browne K.D., Chen X.H., Meaney D.F., and Smith D.H. (2011). Mild traumatic brain injury and diffuse axonal injury in swine. J. Neurotrauma 28, 1747–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kane M.J., Hatic H., Delic V., Dennis J.S., Butler C.L., Saykally J.N., and Citron B.A. (2011). Modeling the pathobiology of repetitive traumatic brain injury in immortalized neuronal cell lines. Brain Res. 1425, 123–131 [DOI] [PubMed] [Google Scholar]
- 84.De Monte V.E., Geffen G.M., May C.R., McFarland K., Heath P., and Neralic M. (2005). The acute effects of mild traumatic brain injury on finger tapping with and without word repetition. J. Clin. Exp. Neuropsychol. 27, 224–239 [DOI] [PubMed] [Google Scholar]
- 85.Viano D.C., Hamberger A., Bolouri H., and Saljo A. (2009). Concussion in professional football: Animal model of brain injury–part 15. J. Neurosurg. 64, 1162–1173 [DOI] [PubMed] [Google Scholar]
- 86.Signoretti S., Lazzarino G., Tavazzi B., and Vagnozzi R. (2011). The pathophysiology of concussion. PM R 3, S359–S368 [DOI] [PubMed] [Google Scholar]