

Abstract
Purpose:
Clinical sequencing emerging in healthcare may result in secondary findings (SFs).
Methods:
Seventy-four of 6,240 (1.2%) participants who underwent genome or exome sequencing through the Clinical Sequencing Exploratory Research (CSER) Consortium received one or more SFs from the original ACMG-recommended 56 gene-condition pair list; we assessed clinical and psychosocial actions.
Results:
The overall adjusted prevalence of SFs in the ACMG 56 genes across the CSER consortium was 1.7%. Initially 32% of the family histories were positive, and post disclosure, this increased to 48%. The average cost of follow-up medical actions per finding up to a 1-year period was $128 (observed, range: $0-$678) and $421 (recommended, range: $141-$1114). Case reports revealed variability in the frequency of and follow-up on medical recommendations patients received associated with each SF gene-condition pair. Participants did not report adverse psychosocial impact associated with receiving SFs; this was corroborated by 18 participant (or parent) interviews. All interviewed participants shared findings with relatives and reported that relatives did not pursue additional testing or care.
Conclusion:
Our results suggest that disclosure of SFs shows little to no adverse impact on participants and adds only modestly to near term healthcare costs; additional studies are needed to confirm these findings.
Keywords: genomic sequencing, secondary findings, healthcare resource utilization
INTRODUCTION
Clinical sequencing raises potential for discovery of a secondary finding (SF)1, a result not related to test indication. The American College of Medical Genetics and Genomics (ACMG) initially recommended analysis and disclosure of 56 medically actionable gene-condition pairs in clinical sequencing contexts2; now updated to 59 pairs.3 Estimates of the SFs yield in different settings have been reported for opportunistic screening.4–9 The original recommendations and others have argued for ongoing analysis to better understand myriad provider and patient implications of this policy2,10, but few studies have evaluated healthcare actions and associated healthcare costs, or impact on patients of these SF disclosures.8
Expectations that SF disclosure has potential to prevent serious morbidity and mortality and extend life expectancy is based largely on data collected in families in which a family member or proband presented with clinical disease, leading to cascade testing.11,12 However, data on actual clinical utility, cost impact, and cost-effectiveness of disclosing SFs are limited.13–15 Additionally, little is known about participant response to SFs psychologically, medically, or regarding communication with relatives. Investigation of individuals who have received medically actionable SFs from sequencing is essential to understand implications of SFs.
The National Institutes of Health (NIH)-funded Clinical Sequencing Exploratory Research (CSER) Consortium16 was established in 2011 to identify and generate evidence to address challenges in applying genomic sequencing to clinical care settings. CSER provides an opportunity to study SFs across diverse clinical settings, where genomic sequencing is likely to be widely adopted in clinical practice. Prior analyses showed providers associated with CSER
Consortium studies felt variants in ACMG 56 genes, such as RYR1, were important to report and act upon.17 Goals of this CSER consortium-wide analysis were to: 1) assess prevalence of SFs in study participants with a range of clinical conditions; 2) evaluate the extent to which these SFs were consistent with family history (FHx) information assessed prior and subsequently to the SF; 3) characterize additional healthcare resource utilization (HRU) attributable to SFs; and 4) describe participants’ psychological and behavioral responses to receiving SFs.
MATERIALS AND METHODS
All CSER sites developed policies and frameworks for SF disclosure.18 Due to the small number of ACMG SFs in individual sites, SF data were aggregated across CSER, providing a more robust sample for analysis. While consortium members differed in the types of findings disclosed as SFs, this cross-consortium analysis retrospectively considered only the subset of 56 gene- condition pairs included in ACMG’s initial recommendations.
Definition of secondary finding
For this analysis, a SF was defined as a pathogenic (P) or likely pathogenic (LP) variant (with the exception of including one variant of uncertain significance favoring a pathogenic interpretation (VUS-FP) in TNNT2, Technical Appendix A) in genes whose associated phenotypes are unrelated to the test indication, and that were included on the original ACMG-recommended 56 gene list. We maximized overlap in disclosed variants among the sites by focusing on the ACMG 56 gene list. Individual site protocols were established before the publication of the ACMG 56 gene list; thus, some sites only disclosed a subset of the ACMG 56 list and others disclosed findings from larger gene lists.18 A public resource provides a list of medically actionable genes that comprises included genes from all CSER sites plus additional NHGRI-funded consortia, with acknowledgement that this list is a range; not all sites report on this expanded list (https://www.genome.gov/27560596/secondary-findings-reportable-bv-nhgri-genomic-medicine-research-programs/). Sites sequencing tumor tissue for somatic mutations elected to disclose findings related to hereditary cancer genes as primary findings, and not as SFs. Some variant classifications changed during the course of the project, due to new information or reclassification according to the 2015 ACMG/AMP guidelines.19 Variants reclassified from VUS to P or LP during the course of CSER projects were not considered here. Similarly, some variants that were initially classified as P/LP were downgraded; except for the FHx analysis, these variants are still included in this analysis since they were initially reported as P/LP to the participant and potentially acted upon. Of note, the original (Green et al)2 and updated (Kalia et al)3 ACMG SF recommendations used terms ‘known pathogenic’ and ‘expected pathogenic’ in considering which variants to return. CSER sites typically disclosed variants as P and LP, consistent with ACMG/Association for Molecular Pathology (AMP) recommendations for interpreting pathogenicity of sequencing variants (Richards et al).19 A total of 75 SF results in the ACMG 56 genes (67 unique variants) were disclosed to 74 participants.
CSER sites and aggregation of SF data
Participating CSER projects included: Baylor College of Medicine’s Baylor Advancing Sequencing into Childhood Cancer Care (BASIC3) study20, Brigham and Women’s Hospital and Harvard Medical School’s MedSeq Project21, Children’s Hospital of Philadelphia’s PediSeq study, Dana-Farber Cancer Institute and Broad Institute’s (DFCI) CanSeq study, Kaiser Permanente’s NextGen study22, the HudsonAlpha Genomic Diagnoses for Children with Developmental Delay project23, NHGRI’s ClinSeq® study24, the University of North Carolina’s NCGENES study25, and the University of Washington’s New EXome Technology in (NEXT) Medicine study26. Sites gathered and shared their data with local Institutional Review Board (IRB) approval; local IRB established approval for re-contact and interview of those receiving ACMG SFs. Sequencing, variant interpretation, and result disclosure were performed at individual CSER sites and combined for post-hoc analyses.
Gene-specific prevalence analyses
Participants in whom ACMG 56 variants would have been returned as primary findings (e.g., for hereditary cancer predisposition or cardiovascular phenotypes) were excluded from contributing to a gene-specific denominator related to their disease phenotype. However, these individuals did contribute to the denominator for other genes unrelated to their condition. Sites conducting trio analysis only contributed the parents to the analysis in order to avoid over-counting a secondary finding that was transmitted to the child. We aggregated all participants to whom variants in any of the ACMG 56 genes were disclosed as SFs. Sites conducted independent reanalysis of the variants that were disclosed, identifying those subsequently downgraded to VUS. Gene-specific prevalence estimates were then generated using the variants that remained as P or LP and denominators that included all participants that would have been eligible for SFs in each gene.
Of 6,240 participants (70% European ancestry) who underwent genomic sequencing, up to 3,499 participants were eligible for gene-specific analyses. We then aggregated gene-specific prevalence values to obtain an adjusted overall prevalence. We compared reported population prevalence to our specific gene-condition prevalence.
Data on participant phenotypes, family history, and subsequent healthcare utilization
We developed a case report form to retrospectively capture specific data associated with each SF case and attributable to the SF, including participant and variant information, whether SF was previously known, participant diagnoses and symptoms, the disclosure process, and recommended and observed healthcare and lifestyle actions (Supplementary Material, Technical Appendix B). Participants receiving SF results that were subsequently downgraded were excluded. The phenotyping questions addressed availability of FHx information available before disclosure that helped inform result interpretation or obtained at the time of SF disclosure, whether re-phenotyping of the proband related to the SF occurred, and whether the SF prompted collection of additional FHx information. When available, personal (proband) history and FHx were assessed before and after SF disclosure. A FHx was designated as positive if it included individuals with either a diagnosis of the condition or manifestations of the condition that were consistent with the SF gene-condition pair. A post-SF personal or FHx was designated as positive if one or more individuals were described as having the diagnosis or a compatible symptom who had not been so designated on the prior FHx.
Healthcare resource utilization (HRU) costing
We estimated near-term (up to 1-year) healthcare costs from the healthcare system perspective associated with SFs from the payer perspective by collecting data on HRU (e.g., time required to disclose the result; follow-up visits and procedures) as described below and then assigning costs to these resources. We used nationally representative Medicare reimbursement rates (2017 USD), published estimates, wage data from the Bureau of Labor Statistics, and commercial lab sources to assign costs to medical actions (Table S0). We estimated provider wages required to prepare and communicate SF results, accounting for provider type (Table S1).
We harmonized recommended and observed medical action responses by collapsing responses into discrete categories for each action (Technical Appendix C). For any response with a referral to a clinician, we assumed a clinic visit with that specific clinician was associated with type of action recommended (i.e., cardiology visit assumed with specialized cardiology diagnostic actions, lipid panel assumed to be ordered by a general practitioner visit). We calculated a recommended and observed cost per-SF for gene-condition pairs. The recommended cost was estimated by assuming the patient received all of the recommended medical actions.
The observed cost was estimated by including only the observed medical actions, which were assumed to be attributed to the results disclosure. We calculated a SF frequency weight and applied that weight to the average cost per SF gene-condition pair to estimate an overall induced cost per ACMG SF (Table 1).
Table 1.
Near-term induced costs of ACMG SF by gene-condition
| Gene | Potential Medical Action | Potential Medical Action (no induced cost to payer) |
Estimated Per- Case Cost for ACMG 56 SF, 2017 USD |
# of Cases |
Frequency Weight of Total Cases (N=75) |
Weighted Cost | ||
|---|---|---|---|---|---|---|---|---|
| Observed | Recommended | Observed | Recommended | |||||
| BRCA1, BRCA2 | Election to undergo risk-reducing salpingo-oophorectomy, Baseline mammogram, Pelvic ultrasound, Full body skin and eye exam (for melanoma), Pancreatic cancer screening, Clinical breast exam, Digital rectal exam, Blood tests for PSA levels, CA-125 blood test, Clinical visit at specialty center (breast cancer), Primary care physician clinic visit, Genetics clinic visit | Breast self-exam, Other* | $115 | $576 | 21 | 0.3 | $32 | $161 |
| MLH1, MSH2, MSH6, PMS2 | Germline genetic testing for Lynch syndrome, Fully body skin and eye exam (for melanoma), Colonoscopy, Upper endoscopy, Urinalysis, Clinic visit with specialist, Primary care physician clinic visit, Oncologist clinic visit, Genetics clinic visit | Acquisition of medic alert bracelet, Other* | $63 | $435 | 6 | 0.1 | $5 | $35 |
| APC | Colonoscopy, Upper endoscopy, Clinic visit with specialist, Genetics clinic visit | $381 | $381 | 2 | 0.03 | $10 | $10 | |
| RET | Genetic testing for RET, Genetics clinisc visit | $0 | $503 | 1 | 0.01 | $0 | $7 | |
| SDHC | Head and neck MRI with contrast, Troponin lab test, Lipid panel, Creatinine kinase test, 24-hour urine protein test, CT of abdomen and pelvis, Comprehensive blood tests and panels, Primary care physician clinic visit, Cardiogenetics visit, Clinic visit with specialist | Other* | $678 | $1114 | 1 | 0.01 | $9 | $15 |
| FBN1, SMAD3 | Slit-lamp eye exam after fully dilating, Echocardiogram, MRI, CT angiography (chest), Cardiologist clinic visit, Genetics clinic visit | $0 | $679 | 2 | 0.03 | $0 | $18 | |
| MYBPC3, MYH7, TNNT2, TNNI3, MYL3 | Electrocardiogram, Echocardiogram, 24-hour Holter monitor, Troponin lab test, Lipid panel, Genetic testing for MYH7, Generic ACE Inhibitor (i.e., Lisinopril) prescription, Primary care physician clinic visit, Clinic visit at specialty center (hypertrophic cardiomyopathy), Cardiologist clinic visit, Cardiogenetics visit | Other* | $89 | $317 | 9 | 0.1 | $11 | $38 |
| PKP2, DSP, DSG2 | Electrocardiogram, Echocardiogram, 24-hour Holter monitor, Cardiologist clinic visit, Clinic visit at specialty center (hypertrophic cardiomyopathy) | Other* | $152 | $243 | 5 | 0.1 | $10 | $16 |
| KCNQ1, KCNH2, SCN5A | Electrocardiogram, Echocardiogram, 24-hour Holter monitor, Troponin lab test, MRI with gadolinium contrast, Exercise stress test, Familial genetic testing for Long QT syndrome, Primary care physician clinic visit, Cardiologist clinic visit, Genetics clinic visit, Cardiogenetics visit | $226 | $499 | 15 | 0.2 | $45 | $100 | |
| LDLR, APOB | Lipid panel, Statin prescription (Lipitor), Primary care physician clinic visit | $86 | $86 | 5 | 0.1 | $6 | $6 | |
| RYR1, CACNA1S | RYR1 clinical lab genetic test, Primary care physician clinic visit, Clinic visit with specialist (malignant hyperthermia) | Acquisition of medic alert bracelet, Port the risk result into the electronic medical record, Other* | $0 | $141 | 8 | 0.1 | $0 | $15 |
| TOTAL | 75 | 1.0 | ||||||
| Weighted Average Cost, 2017 USD | $128 | $421 | ||||||
Other (Included an action that involved a method of communication that could not be assigned a cost which included the following: Communication with existing cardiologist about whether further work-up was needed; Communicate with gynecologist about ovarian cancer screening; Inform future providers about result; Discussion with primary care physician (PCP) of whether male patient had enough breast tissue to undergo mammogram; ensure that OBGYN is aware of finding; Changes in medical management to occur in adulthood; discuss with family, parents of this individual; (grandparents to proband) recommended to have colonoscopies; Participant shared result with PCP; Participant disclosed results to PCP; disclosed result with living first degree relative)
Bolded text indicates medical action data used for the observed cost calculation
Of the 75 SFs, 26 (35%) did not have quantifiable data to calculate time associated with preparation for SF disclosure by the clinician and were excluded from that portion of the analysis. We also excluded one SF where preparation time included preparation for disclosure of additional SFs not eligible for this study. Data from 48 SFs were available for inclusion in the analysis of provider wages required to prepare and communicate SF results. Available data on 55 SFs (73% of total SFs) were calculated for time required to communicate the SF (via direct patient contact, e.g., in-person, phone, email).
Participant semi-structured interviews
Of the 74 participants with eligible ACMG SFs we invited, a subset of 18 agreed to participate in interviews to assess psychological and behavioral impact from receiving SF results and to complement the SF case report forms described above. For eight pediatric patients, a parent was interviewed. The interview guide was designed using a model from the NIH CCGO Secondary Findings Project (Sapp et al., submitted), with questions about health services use, insurance status, and psychological responses to the information (Technical Appendix D). A senior genetic counselor (BBB) and a qualitative researcher from Kaiser Permanente (CM) conducted telephone interviews. Two independent coders (AMS and ET) coded verbatim interview transcripts with attention to responses related to understanding use and communication of results. A Kappa score of 0.95 was calculated, indicating high reliability. Directed content analysis was conducted using the coded data.
RESULTS
Secondary finding results and characteristics
Of 6,240 participants undergoing genomic sequencing, 76 variants were identified in genes on the ACMG 56 list. One individual had two variants in RYR1, associated with malignant hyperthermia susceptibility, which was suspected to represent a complex allele in cis and was counted as one SF for the purpose of subsequent analyses. Two different variants (in TNNI3 and LDLR) reported in one individual were included in the analysis as independent SFs. Thus, a total of 75 SF were disclosed to 74 individuals across CSER and were included in these analyses. See Table S2 for list of SFs.
There were a total of 67 unique variants that accounted for 26 of the ACMG genes; 14 genes had only one variant identified and 12 had greater than one. The most common ACMG 56 genes with variants returned were associated with hereditary breast/ovarian cancer, hypertrophic cardiomyopathy, malignant hyperthermia, Lynch syndrome, familial hypercholesterolemia, and Long QT syndrome. Five unique variants in four genes (BRCA1, BRCA2, SCN5A, and RYR1) were reported more than once (Table S2). Strikingly, 14 variants in 9 genes that were initially reported as P/LP were downgraded to VUS after evaluation of new evidence, including new population data or subsequent entries in ClinVar (Table S2). After removal of VUS and accounting for gene-specific denominators, the aggregate prevalence of SFs in the ACMG 56 genes across CSER was 1.7% (Figure 1).
Figure 1.
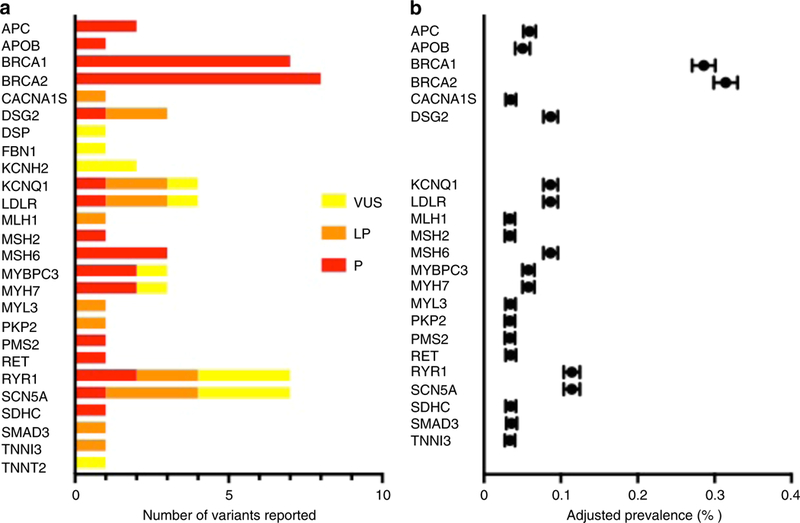
A) Unique variants reported following re-classification, which included downgraded variants. B) Gene-specific adjusted prevalence, which excluded downgraded variants originally disclosed as pathogenic or likely pathogenic. VUS: variant of uncertain significance LP: likely pathogenic P: pathogenic
Family history known prior to results disclosure and following SF results disclosure
For the FHx evaluations, we focused on the 62 individuals whose variants were P/LP after our re-evaluation (i.e., were not downgraded). Of these SF, four were in participants for whom no FHx information was obtained (Figure 2). Of the remaining 58 SFs with an initial FHx, 20 were initially positive and 38 initially negative for a symptom or diagnosis of the condition associated with the SF. Of the 20 who were positive, 18 had a post-SF FHx and four (22%) of those identified additional family members with compatible symptoms or diagnoses. Of the 38 who were initially negative, all had post-SF family histories and 8 (27%) of those were designated as positive. The total yield of positive family histories therefore changed from 20/58 to 28/58, or 34% to 48%.
Figure 2.
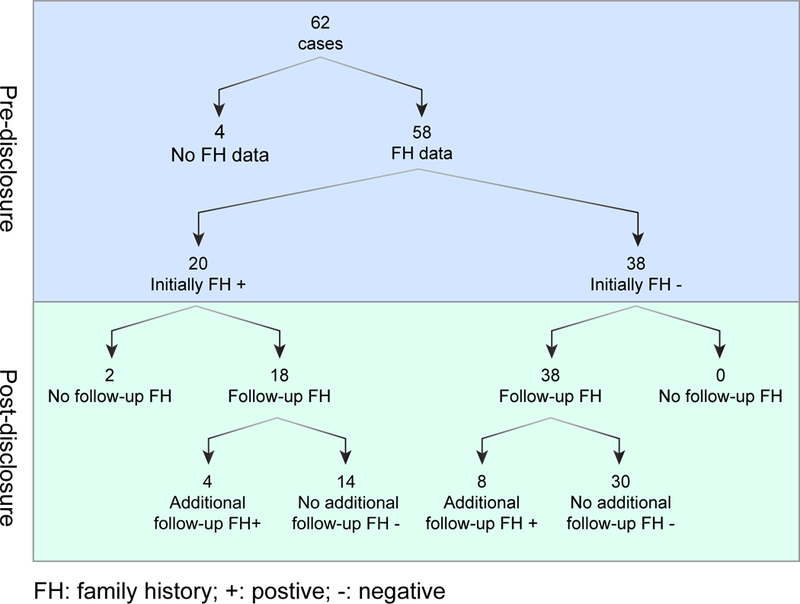
Family history known prior to result disclosure and following SF results disclosure
Healthcare resource utilization
For the purposes of the following analyses, we used all 75 SFs that were disclosed. Data on length of time associated with provider preparation of the disclosed SF was available for 48 SFs (64% of total SFs), yielding an average time of 67 minutes required for preparation (range: 8–160 minutes) and average cost was $68 (range: $5-$166) in provider wages. The average time required for communication was 31 minutes (range 3 to 101 minutes), associated with an average cost of provider wages of $31 (range: $3-$105). The average recommended cost attributable to the 75 SFs up to 1-year post-result return was $421 (range $141 to $1114). The average observed cost was $128 (range: $0 to $678) (Table 1). However, five SFs were disclosed at 6 months or less prior to our analysis; thus, we did not have a year of data. Four SFs (5%) were disclosed for a pediatric participant. Any associated recommended medical actions were based on adults and did not apply; these were not included in the recommended or observed cost estimate. Assigned costs per medical action and labor wages by provider are displayed in Table S0 and Table S1, respectively. There was variability in the frequency of and follow-up on medical recommendations associated across each SF gene-condition pair (Table S3).
Participant interviews
Eighteen participants (or their parent, for eight pediatric participants) were interviewed regarding their experiences receiving SFs and the ways they sought additional clinical care and communicated these findings to relatives. For 15 (83%) interviewees, their SF was unexpected. Surprise led to a variety of responses ranging from “glancing over it” or ignoring the results, to feeling scared and working hard to avoid thinking about them. Additionally, interviewees described these responses largely moderating with time. A major theme was the importance of the life context in which participants learned their SF. They described working to make sense of their results while managing other life circumstances. Parents described exhaustion and a need to reserve their resources to care for their child. When asked directly, no participant voiced regret about learning their SF. None of the interviewees reported pursuit of health care services beyond recommendations based on their SF. Rather, there was under-compliance with recommended care (e.g., by not following up with a provider). Thirteen (72%) interviewees met with a clinician; seven with their primary care provider (PCP); and six met with a specialist as recommended by the SF disclosing team (Table S4). All 18 reported their SF to one or more first degree relatives. Some reported their results to multiple relatives (Table 2). Three interviewees reported changing their health and/or life insurance upon learning their SF (Table S5). Seventeen participants expected that their, or their child’s, SF was recorded in the medical record, although this was not confirmed. Additional exemplifying quotes and reactions are reported in Table S6.
Table 2.
Interviewed participants and subsequent family communication regarding medical actions.
| Gene | Participant # | Immediate family | Extended family | Family follow up with doctor |
Family tested |
Quote |
|---|---|---|---|---|---|---|
| BRCA1 | 14 | Brother, parents, sister | Uncles, female cousins (2) | Female cousins (2), sister | ||
| BRCA1 | 15 | Brother, mother, father | No | “So, I brought this up to family members. And I have to say that they weren’t like super receptive.” | ||
| BRCA1 | 16 | Wife | No | |||
| BRCA2 | 8 | Daughter, wife | Daughter, wife | |||
| BRCA2 | 11 | Parents | Aunt, all cousins on that side | No | No | |
| BRCA2 | 10 | Father, three sisters, | No | No | “They (siblings) didn’t really seem moved by the information.” | |
| MSH6 | 12 (adopted) | Mother, Step mother | No | 12 (adopted) | ||
| APC | 17 | Parents, sister | No | No | ||
| RET(MEN2A) | 6 | Daughter, two sisters | Daughter, one sister | |||
| TNNT2 | 4 | Husband, children, parents, brother | No | No | ||
| DSP | 3 | Wife, Father, sister | No | No | ||
| DSG2 | 5 | Sister | No | No | ||
| DSG2 | 9 | Parents | Uncles, grandparents | No | No | |
| DSG2 | 5 | Sister | No | No | ||
| KCNQ1 | 1 | Brother, wife, mother | Unsure | Unsure | “…family doc did not recommend anybody else in the family be tested for Long QT.” | |
| KCNQ1 | 2 | Brother, sister | No | No | ||
| SCN5A | 7 | Daughter, parents, brother, sisters | No | Plan to test daughter | ||
| SCN5A | 13 | Husband, children, sisters, father | No | |||
| APOB | 18 | Daughters | Nieces, Nephews | Unsure | Unsure | “I plan to follow up with them again” |
DISCUSSION
Across CSER, ACMG-recommended SF results were disclosed to 74 of 6,240 participants. Our SF adjusted prevalence (1.7%) is similar to, albeit slightly lower than, previous estimates that included both P and LP variants, which included genes not part of the ACMG 56 list, e.g., ClinSeq, the DiscovEHR cohort.6,9,27 The combined consortium estimates are averaged across those of individual sites; any differences are likely due to an expanded list of possible SFs.28 Furthermore, the consortium work was able to investigate different scientific questions. Reevaluation of the cases post-SF disclosure provided 1.5 times more positive FHx information of disorders associated with the SF, compared to the pre-SF FHx (Figure 2). The average recommended cost induced by an ACMG SF up to a 1-year period, assuming all patients followed recommended actions, was $421. A central theme from the interviews was that impact of the SF was dependent on life context of the participant upon receiving the SF.
FHx data showed that 34% of initial family histories had diagnoses or symptoms compatible with the SF condition. Following receipt of the SF, the number of compatible family histories increased to 48%, indicating that with the SF disclosure, additional FHx was uncovered. There are a number of possible explanations for these findings. Firstly, a variable amount of time passed from collection of the initial to the second FHx; the increment of findings could be due to the passage of time. It is also possible that the SF triggered a recollection of FHx details not previously recalled, or that more targeted FHx questions were asked. Our threshold for scoring FHx was liberal, reasoning that all potentially relevant information is useful, regardless of whether it substantially supports the diagnosis of the SF or not. Our data suggest that receipt of SFs may inform FHx collection allowing for the addition of targeted FHx questions, which may uncover previously unrecognized history with clinical relevance to families.
Estimating observed cost was limited to a shorter time horizon for 7% of SFs disclosed less than six months before our analysis, therefore, what medical actions were subsequently taken is unknown. Similarly, our up to 1-year time horizon limits capture of medical action follow-up for the 5% of SFs for whom individuals were younger than the recommended age to take medical action, which impacts the observed cost estimate. Notably, average costs from this study reflect the proportion of gene-specific disclosures and costs reported for this specific patient population (Table S7), which impacted the contribution to the average recommended and observed costs. Future work may consider evaluating cost consequences for individual SF gene-condition pairs.
Medical actions differed among providers for the same SF; providers did recommend specialist referrals, which may account for appropriate medical action downstream beyond what our data captured. Variability in patient clinical history may account for SF recommendation differences. Due to the limited availability of cost and of electronic medical record (EMR) data tracking methods to ascertain accurate billing codes, we only evaluated up to a 1-year time horizon for the cost analyses. Thus, the cost for following recommendations that included, for example, visiting a specialist for follow-up in two-three years and any subsequent care management, was not captured. A longer time horizon and additional perspectives (e.g., societal) warrant future investigation.
Our findings suggest modest near-term induced costs for these recipients of ACMG 56 SFs, although they may be lower than expected given concerns about increased HRU regarding clinical sequencing.10,29 However, our findings are consistent with recent analyses from the MedSeq study, which also found low near-term costs associated with return of SFs that were not exclusive to the ACMG 56 gene list.15,30 Notably, though the MedSeq study did contribute two participants with ACMG 56 findings to this dataset, the inclusion of their ACMG 56 SFs do not substantially overlap our economic analyses.
There are several possible explanations for our results. First, this retrospective, 1-year follow-up study may have incompletely captured significant amounts of testing and evaluation that was yet-to-be-performed. Prospective studies utilizing randomization or use of a control group should ideally be performed in health systems that more completely capture HRU (e.g., closed panel health maintenance organizations, integrated delivery networks) to help ensure attribution is appropriately assigned to the observed utilization. Secondly, patients and providers may have lesser levels of concern or motivation to investigate SFs than has been widely presumed 10,31 and/or the provider and patient may harbor additional information necessary to gauge medical management of the SF. Although much has been written about how SFs will overwhelm the health care system14,32, these data suggest that patients and their providers may be just as likely to minimize findings as they are to over-diagnose patients or over-use medical services. Additional studies similar to our study design, but with larger samples and prospective inquiry of individuals with SF from the updated ACMG SF 59 gene list3 are needed to validate our observation.
From the interview transcripts, the theme for the importance of life context at SF disclosure was most poignant among parents of children undergoing cancer treatment and those caring for children with intellectual disabilities. All participants shared their SF with immediate family. Thus, an additional theme centered on communication of results to relatives. While most participants described being surprised by their result, several noted that it was not a shock as they understood it was possible to learn a SF. Our interview findings demonstrate that adults who received a SF with a significant health risk reflected on their current circumstances as well in pursuing follow-up actions.
Our study was limited by a relatively small number of SFs per site, limiting site prevalence estimates for ACMG SFs; however, pooling SFs across multiple sites addressed that limitation. The ascertainment of follow-up data on phenotyping and HRU was based on reporting from site personnel; data may have been collected in different ways across sites, such as using EMRs at some sites, or surveys at other sites. Further, data capture may have been incomplete, including: insufficient follow-up time to allow the actions to happen, patients may have received care within another system not captured in the EMR, patients may have left out information reported on surveys, etc. Not all participants were interviewed, therefore, some uncertainty persists in completeness of the data and costs may be underestimated. Lastly, heterogeneity presented in the processes for disclosing SF results across sites (e.g., provider type, report form, etc.), which could have affected the amount of time it took to disclose the SF, but, by utilizing the same case report form we aimed to harmonize capturing some of these differences.
Our findings suggest excessive utilization of limited healthcare resources, overdiagnosis, or anxiety and distress, may not be major consequences to disclosing SFs, despite prior concerns regarding uncertain resource consequences.29 The finding that participants from our study were not anxious or distressed related to their SF is important. These findings should reduce some concerns about adverse psychological reactions perceived as potential harms from SF disclosure.10,29 However, this was a voluntary research population and results may not be fully generalizable to non-research populations. The relative inaction on medical recommendations associated with 1-year follow-up from SF disclosure could indicate several underlying factors including indifference due to competing life priorities and other medical problems requiring care, an underestimation of SF risks by medical providers, poor understanding of evaluations that would be reasonable to undertake, or other factors. Although great concern has been expressed regarding the adverse psychological impact and the inevitable HRU, it may turn out that SFs are like most other medical findings and need to be reported and contextualized to the individual patient - with the provider’s role to encourage the receipt and evaluation of such findings, rather than to avoid and minimize them. These findings argue for development of a rational and practicable approach to establish patient-centered options for disclosing results, with additional support for undergoing the recommended evaluations from SFs.
Supplementary Material
Acknowledgements
The National Human Genome Research Institute (NHGRI) and the National Cancer Institute (NCI) funded the CSER Consortium: CSER Coordinating Center U01HG007307; NEXT Medicine U01HG006507; BASIC3 U01HG006485; MedSeq U01HG006500; NextGen UM1HG007292; PediSeq U01HG006546; HudsonAlpha U01HG007301; DFCI U01 HG006492, NCGENES U01HG006487; ZIAHG200359 09 and ZIAHG200387 04 from the Intramural Research Program of the NHGRI support ClinSeq®. RCG was supported by U01-HG006500, R01-CA154517, R01-AG047866 and funding from the Broad Institute and Department of Defense. JLV is employed by the US Department of Veterans Affairs (VA) and supported by IK2-CX001262 from the VA Office of Research & Development. KDC was supported by K01-HG009173. The views expressed here do not necessarily reflect those of the US Government or VA. We acknowledge Julia Fekecs and Melpi Kasapi for technical and logistical assistance.
Footnotes
Conflict of Interest: LMA, BBB, CLB, JSB, KBB, KLB, KMB, SB, KDC, GMC, LKC, MCD, JNE, KME, CRF, AAG, MJG, KABG, LAH, MRH, GPJ, JJJ, JBK, TLK, WVK, KLL, ALM, CM, JO, EJR, AMS, NBS, ET, MLT, JLV, BSW, none; CSR and HLR are employed by a testing laboratory that offers commercially available sequencing. RCG receives compensation for consultation from AIA, Americord, Helix and Veritas; and is co-founder, advisor and equity holder in Genome Medical, Inc; and is employed by a testing laboratory that offers commercially available sequencing. LGB is an uncompensated advisor to the Illumina Corp, receives royalties from Genentech, Inc, and honoraria from Wiley-Blackwell. SEP is a member of the Scientific Advisory Panel of the Baylor Genetics Laboratory. DLV is a consultant to Roche Sequencing Systems.
References
- 1.Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370(25):2418–2425. [DOI] [PubMed] [Google Scholar]
- 2.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in Medicine. 2013;15(7):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 updated (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine. 2017;19(2):249–255. [DOI] [PubMed] [Google Scholar]
- 4.Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91(1):97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Natarajan P, Gold NB, Bick AG, et al. Aggregate penetrance of genomic variants for actionable disorders in European and African Americans. Science Translational Medicine. 2016;8(364ra151). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25(3):305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorschner MO, Amendola LM, Turner EH, et al. Actionable, pathogenic incidental findings in 1,000 participants’exomes. Am J Hum Genet. 2013;93(4):631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole- exome sequencing. JAMA. 2014;312(18):1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olfson E, Cottrell CE, Davidson NO, et al. Identification of Medically Actionable Secondary Findings in the 1000 Genomes. PLoS One. 2015;10(9):e0135193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burke W, Antommaria AH, Bennett R, et al. Recommendations for returning genomic incidental findings? We need to talk! Genet Med. 2013;15(11):854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evaluation of Genomic Applications in P, Prevention Working G. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009; 11(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knowles JW, Rader DJ, Khoury MJ. Cascade Screening for Familial Hypercholesterolemia and the Use of Genetic Testing. JAMA Insights. 2017;318(4):381–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Douglas MP, Ladabaum U, Pletcher MJ, Marshall DA, Phillips KA. Economic evidence on identifying clinically actionable findings with whole-genome sequencing: a scoping review. Genet Med. 2016;18(2):111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennette CS, Gallego CJ, Burke W, Jarvik GP, Veenstra DL. The cost-effectiveness of returning incidental findings from next-generation genomic sequencing. Genet Med. 2015;17(7):587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen KD, Vassy JL, Phillips KA, et al. Short Term Costs of Integrating Whole Genome Sequencing into Primary Cary and Cardiology Settings: A Pilot Randomized Trial. Genet Med. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green RC, Goddard KAB, Jarvik GP, et al. Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine. Am J Hum Genet. 2016;98(6): 1051–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen KD, Bernhardt BA, Jarvik GP, et al. Anticipated responses of early adopter genetic specialists and nongenetic specialists to unsolicited genomic secondary findings. Genet Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berg JS, Amendola LM, Eng C, et al. Processes and preliminary outputs for identification of actionable genes as incidental findings in genomic sequence data in the Clinical Sequencing Exploratory Research Consortium. Genet Med. 2013; 15(11):860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsons DW, Roy A, Yang Y, et al. Diagnostic Yield of Clinical Tumor and Germline Whole- Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vassy JL, Lautenbach DM, McLaughlin HM, et al. The MedSeq Project: a randomized trial of integrating whole genome sequencing into clinical medicine. Trials. 2014;15(85). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kauffman TL, Wilfond BS, Jarvik GP, et al. Design of a randomized controlled trial for genomic carrier screening in healthy patients seeking preconception genetic testing. Contemp Clin Trials. 2017;53:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bowling KM, Thompson ML, Amaral MD, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biesecker LG, Mullikin JC, Facio FM, et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19(9):1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foreman AK, Lee K, Evans JP. The NCGENES Project: Exploring the New World of Genome Sequencing. North Carolina Medical Journal. 2013;74(6):500–504. [PubMed] [Google Scholar]
- 26.Gallego CJ, Bennette CS, Heagerty P, et al. Comparative effectiveness of next generation genomic sequencing for disease diagnosis: design of a randomized controlled trial in patients with colorectal cancer/polyposis syndromes. Contemp Clin Trials. 2014;39(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354(6319). [DOI] [PubMed] [Google Scholar]
- 28.Thompson ML, Finnila CR, Bowling KM et al. Genomic sequencing identifies secondary findings in a cohort of parent study participants. Genetics in Medicine. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright CF, Middleton A, Burton H, et al. Policy challenges of clinical genome sequencing. BMJ. 2013;347:f6845. [DOI] [PubMed] [Google Scholar]
- 30.Vassy JL, Christensen KD, Schonman EF, et al. The Impact of Whole-Genome Sequencing on the Primary Care and Outcomes of Healthy Adult Patients: A Pilot Randomized Trial. Ann Intern Med. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohane IS, Masys DR, Altman RB. The Incidentalome, A Threat to Genomic Medicine. JAMA. 2006;296(2):212–215. [DOI] [PubMed] [Google Scholar]
- 32.Ding A, Eisenberg JD, Pandharipande PV. The economic burden of incidentally detected findings. Radiol Clin North Am. 2011;49(2):257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.