
Fig. 2 |. Annotation strategies for lncRNAs.
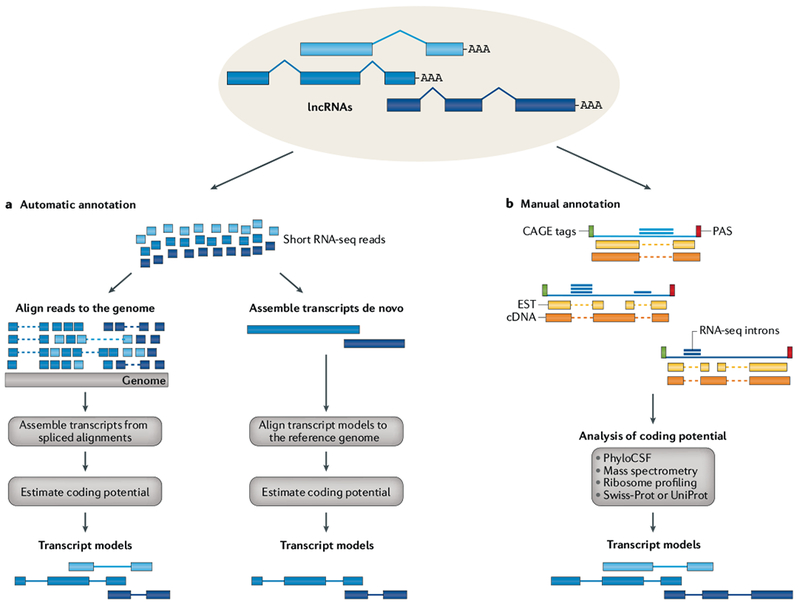
a | Automatic annotation based on RNA sequencing (RNA-seq) may follow two distinct strategies that differ in how the genome reference is used. The align-then-assemble strategy (left) aligns reads to the reference genome to reveal possible splicing events and then assembles reads into transcript models. The assemble-then-align strategy (right) builds transcript models de novo, directly from the RNA-seq reads, and then aligns them to the reference genome to determine their exon–intron structure. De novo transcriptome assembly has more explorative potential than alignment-based assembly but tends to have worse performance68. b | In manual annotation, human annotators employ various sources of data to build transcript models. Expressed sequence tags (ESTs) and cDNA form the primary evidence for transcript models and are often supplemented with RNA-seq reads to validate introns, cap analysis of gene expression (CAGE) clusters to identify 5′ ends45 and poly(A)-position profiling by sequencing (3P-seq) to identify polyadenylation sites (PASs)47. A key step in the annotation process is to assess the protein-coding potential of transcripts, usually on the basis of a combination of methods. lncRNA, long non-coding RNA.