

Short abstract
Chemotherapy-induced painful peripheral neuropathy (CIPN) is the most common toxicity associated with widely used chemotherapeutics. CIPN is the major cause of dose reduction or discontinuation of otherwise life-saving treatment. Unfortunately, CIPN can persist in cancer survivors, which adversely affects their quality of life. Moreover, available treatments are vastly inadequate, warranting a better understanding of the biochemical and metabolic mechanisms that occur in response to chemotherapeutics which would be critical for the development of novel therapies for CIPN. Using extracellular flux analysis, this study demonstrated that the proteasome inhibitor, bortezomib, enhanced glycolysis while suppressing oxidative phosphorylation in the sensory neurons of mice. This metabolic phenotype is known as aerobic glycolysis. Bortezomib upregulated lactate dehydrogenase A and pyruvate dehydrogenase kinase 1, which consequently enhanced the production of lactate and repressed pyruvate oxidation, respectively. Moreover, lactate dehydrogenase A- and pyruvate dehydrogenase kinase 1-driven aerobic glycolysis was associated with increased extracellular acidification, augmented calcium responses, and pain in bortezomib-induced CIPN. Remarkably, pharmacological blockade and in vivo knockdown of lactate dehydrogenase A or pyruvate dehydrogenase kinase 1 reversed the metabolic phenotype, attenuated calcium responses, and alleviated pain induced by bortezomib. Collectively, these results elucidate the mechanisms by which bortezomib induces aerobic glycolysis. Moreover, these findings establish aerobic glycolysis as a metabolic phenotype that underpins bortezomib-induced CIPN.
Keywords: Neuropathy, CIPN, metabolism, mitochondria, aerobic glycolysis, DRG
Introduction
Chemotherapy-induced painful peripheral neuropathy (CIPN) is the most prevalent toxicity associated with widely used anti-cancer drugs which include taxanes, platinum-based drugs, vinca alkaloids, and proteasome inhibitors. This adverse effect can be severe enough for patients to either reduce the dosage of anti-cancer treatment or stop the treatment altogether. Unfortunately, CIPN can persist in cancer survivors negatively impacting their quality of life.1–3 Currently, treatment options for CIPN are vastly inadequate which warrants a better understanding of the mechanisms that underpin CIPN. Hence, uncovering new mechanisms are critical for the development of novel therapeutic strategies.
Metabolism is defined as the sum of all biochemical reactions in the cell and is divided into two distinct pathways. Biosynthetic pathway, responsible for the production of complex molecules that constitute a living cell, and bioenergetic pathway, linked to energy production.4 Moreover, metabolism is a highly dynamic process where most cells possess the ability to switch energy sources based on their bioenergetic needs. This is especially relevant to neurons which can undergo periods of elevated energy demand following action potentials. Depending on the type of neuron, a single action potential is estimated to consume 107 to 109 ATP molecules.5–9 Neurons are proficient in fully oxidizing glucose and generating over 30 ATP molecules most of which are derived from the Krebs cycle in the mitochondria. Disruption of mitochondrial oxidation would result in reliance on glycolysis which is far less efficient in generating energy. This metabolic phenotype is known as aerobic glycolysis or the Warburg effect. Named after Otto Warburg who almost a century ago observed that the rate of glycolysis in cancer cells was abnormally high and only a small proportion of the resulting pyruvate was catabolized via mitochondrial oxidative phosphorylation.10 Pyruvate that is not oxidized in the mitochondria can get converted to lactate by the enzyme lactate dehydrogenase (LDH). This reaction regenerates the cofactor nicotinamide adenine dinucleotide (NAD+) in the cytosol which is critical for sustaining glycolysis. Finally, lactate is extruded from a cell along with a proton leading to increased extracellular acidification.4,11–13
Mitochondria are the “metabolic hub” of the cell with specialized functions that include energy production, the gamma-aminobutyric acid cycle, amino acid metabolism, iron-sulfur protein synthesis, heme synthesis, fatty acid metabolism and calcium and reactive oxygen species homeostasis.14 Moreover, mitotoxicity has been long-established as a common feature in the pathobiology of CIPN induced by taxanes, platinum-based drugs and the proteasome-inhibitors.15–17 In recent years, the study on the role of metabolism in CIPN has been rapidly progressing where paclitaxel has been demonstrated to enhance glycolysis while reducing oxidative phosphorylation.18 Furthermore, strategies that elevate cellular NAD+ levels either via supplementation of its precursor19 or enhancing its synthesis20 have been demonstrated to alleviate CIPN. However, the mechanisms by which chemotherapeutics alter the metabolism of sensory neurons and how these changes cause pain have remained elusive.
The proteasome inhibitor, bortezomib, is used for the treatment of multiple myeloma and mantle cell lymphoma.21 Upward of 75% of patients treated with bortezomib develop CIPN.22 This study demonstrates that bortezomib alters the metabolism of sensory neurons in a manner consistent with aerobic glycolysis. Moreover, bortezomib treatment enhanced the expression of pyruvate dehydrogenase kinase 1 (PDHK1) and lactate dehydrogenase A (LDHA) which attenuate pyruvate oxidation and enhance the extrusion of metabolites (lactate and protons), respectively. Crucially, inhibition of PDHK1 or LDHA normalized the metabolic phenotype and alleviated bortezomib-induced pain. These findings elucidate the molecular mechanisms through which bortezomib reprograms the metabolism of sensory neurons and uncovers the mechanisms by which aerobic glycolysis causes pain—establishing this metabolic phenotype as a principal contributor to CIPN.
Materials and methods
Experimental animals
Pathogen-free, adult male ICR mice (3–4 weeks old; Envigo) were housed in temperature (23 ± 3°C) and light (12-h light/12-h dark cycle; lights on 07:00–19:00) controlled rooms with standard rodent chow and water available ad libitum. Animals were randomly assigned to treatment or control groups for the behavioral experiments. Animals were initially housed five per cage. All behavioral experiments were performed by experimenters who were blinded to the experimental groups and treatments. The Institutional Animal Care and Use Committee of the University of Maryland approved all experiments. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain.
Bortezomib treatment
In all the experiments, mice were treated with intraperitoneal 0.2 mg/kg of bortezomib (Millipore Sigma, Cat # 5.04314.0001) for five consecutive days for a total dose of 1 mg/kg.16 The vehicle group received intraperitoneal saline for five consecutive days.
Mechanical testing
Male ICR mice were placed in acrylic boxes with wire mesh floors, and baseline mechanical withdrawal thresholds of the left hindpaw were measured after habituation for 1 h using the up-down method.23 After determining the baseline withdrawal thresholds of mice hindpaw using von Frey filaments, the mice were treated with either vehicle or boretezomib.16 Starting on day 7, the tactile withdrawal thresholds were tested. For experiments with intraperitoneal (IP) injections, oxamate (500 mg/kg, Millipore Sigma, Cat # O2751) or dichloroacetate (100 mg/kg, DCA, Millipore Sigma, Cat # 347795) were injected at the indicated time points. For experiments with intrathecal (IT) siRNA treatments on day 7 or later, mice were tested before IT injection and the injections were done between the L4 and L5 vertebrae at the indicated time points under isoflurane anesthesia. Negative control (Millipore Sigma, Cat # SIC001), LDHA (Millipore Sigma, SASI_Mm01_00049543), and PDHK1 (Millipore Sigma, SASI_Mm01_00042115) HPLC-purified siRNAs were injected at a dose of 1 µg in 5 µl of i-Fect (Neuromics, Cat # NI35150).
Conditioned place aversion
The conditioned place aversion (CPA) apparatus consists of three opaque chambers separated by manual doors. A center chamber (60 mm W × 60 mm D × 150 mm H) connects the two end chambers that are identical in size (150 mm W × 150 mm D × 150 mm H) but can be distinguished by the texture of the floor (circular opening vs. diamond pattern mesh), wall color (black walls white ceiling vs. white walls black ceiling), and olfactory cues (vanilla vs. cherry ChapStick™ applied on a cotton swab). Movement of mice and time spent in each chamber were monitored and recorded using custom-built infrared sensors and software. Preconditioning was performed seven days following the initiation of bortezomib treatment for 30 min when mice were exposed to the environment with full access to all chambers. A single-trial conditioning protocol was used in the experiments. On conditioning day (day 2), mice first received vehicle control (IP saline) paired with a randomly chosen chamber in the morning and, 3–4 h later, IP glucose (2 g/kg in saline)24 paired with the other chamber. Food was withheld for 3–4 h prior to the glucose administration. The mouse normal plasma glucose concentration is around 7mM, and fasting for 3–4 hours does not significantly alter these levels. After glucose injection (2 g/kg), the plasma level rapidly reaches to approximately 20 mM for about 30 min, and within 60 min, the glucose levels return back to normal.24 During the conditioning, mice were allowed to stay only in the paired chamber without access to other chambers for 30 min immediately following saline or glucose injection. On the test day, 20 h after the glucose pairing, mice were placed in the middle chamber of the CPA box with all doors open so animals can have free access to all chambers. Movement and duration of each mouse spent in each chamber were recorded for 30 min for analysis of chamber aversion. Difference scores were calculated as (test time – preconditioning time) spent in the glucose chamber. Mice received vehicle or oxamate (500 mg/kg, IP) 2 h prior to the glucose administration. DCA (100 mg/kg, IP) or vehicle was administered 1 h prior to glucose administration.
Dorsal root ganglia dissociation
On day 10 following the initiation of vehicle or bortezomib treatment, L4-6 dorsal root ganglia (DRGs) excised aseptically and placed in Hank’s Buffered Salt Solution (Thermo Fisher, Cat # 14170112) on ice. The ganglia were dissociated enzymatically with collagenase A (1 mg/ml, 25 min, Millipore Sigma, Cat # 10103578001) and collagenase D (1 mg/ml, Millipore Sigma, Cat # 11088858001) with papain (30 U/ml, Millipore Sigma, Cat # 10108014001) for 20 min at 37°C. To eliminate debris and large diameter sensory neurons, 70 µm (Thermo Fisher, Cat # 087712) cell strainers were used. The dissociated cells were resuspended in Dulbecco’s Modified Eagle Medium (DMEM)/F12 (Thermo Fisher, Cat # 10565042) containing 1× pen-strep (Thermo Fisher, Cat # 15070063) and 10% fetal bovine serum (Millipore Sigma, Cat # F2442). The cells were plated in either Seahorse XFp Cell Culture Miniplates (Agilent, Cat # 103025–100) or poly-D-lysine-coated, glass-bottomed 35-mm dishes (Mattek, Cat # P35GC-1.5–10). The primary afferent cultures were incubated overnight at 37°C in a humidified 95% air/5%CO2 incubator.
Metabolic assays
The metabolic changes were characterized by analyzing the glycolysis and oxidative phosphorylation rates of sensory neurons using extracellular flux analyzer, Seahorse XFp (Agilent).
Mito Stress Test
On day 10, L4-6 DRGs were dissected from mice treated with vehicle or bortezomib, acutely dissociated, and incubated in the XF analyzer plates overnight which allows for the neurons to adhere to the bottom of the plates. The Mito Stress Test was performed in DMEM medium (Millipore Sigma, Cat # D5030) that contained glucose (10 mM) and pyruvate (1 mM). During the Mito Stress Test, baseline oxygen consumption rate (OCR) measurements were followed by the addition of compounds that target components of the electron transport chain in the mitochondria to reveal key parameters of oxidative phosphorylation. The compounds oligomycin (5 µM, Millipore Sigma, Cat # 75351), FCCP (4 µM, Millipore Sigma, Cat # C2920), and a mix of rotenone (2 µM, Millipore Sigma, Cat # R8875) and antimycin A (2 µM, Millipore Sigma, Cat # A8674) are serially injected to measure ATP-linked respiration, maximal respiration, and non-mitochondrial respiration, respectively. Proton leak and spare respiratory capacity are then calculated using these parameters.12,13
Glycolysis Stress Test
The dissociated L4-6 DRG neurons were incubated in DMEM medium (Millipore Sigma, Cat # D5030) without glucose or pyruvate, and the baseline extracellular acidification rate (ECAR) is measured. The cells were deprived of glucose for about 30–40 min. It should be noted that the DMEM medium contains amino acids that the cells utilize to maintain energetics. In addition to amino acids, the medium contains phosphates where both can serve as mild pH buffers. A saturating concentration of glucose (10 mM, Millipore Sigma, Cat #G8769) is injected to measure the glycolysis rate which is followed by the injection of oligomycin (5 µM) which inhibits mitochondrial ATP production and shifts the energy production to glycolysis, with the subsequent increase in ECAR revealing the cellular maximum glycolytic capacity. The final injection is 2-deoxyglucose (2-DG, 50 mM, Millipore Sigma, Cat # D8375), which inhibits glycolysis by competitively binding to hexokinase, the first enzyme in the glycolytic pathway. The resulting decrease in ECAR confirms that the ECAR produced in the experiment is due to glycolysis. The difference between glycolytic capacity and glycolysis rate defines glycolytic reserve. ECAR, prior to glucose injection, is referred to as non-glycolytic acidification, which is caused by processes in the cell other than glycolysis.12,13
Pyruvate oxidation assay
Dissociated L4-6 primary afferents were incubated in DMEM (Millipore Sigma, Cat # D5030) without glutamine, and baseline OCR measurements were followed by the addition of pyruvate (1 mM, Millipore Sigma, Cat # S8636). The neurons were then treated with the PDHK inhibitor, DCA (20 mM). At the end of the assay, the mitochondrial pyruvate transporter inhibitor, UK5099 (10 µM, Millipore Sigma, Cat # PZ0160), was added. The OCR values obtained following the addition of UK5099 were subtracted from each of the other values to determine the pyruvate oxidation-dependent OCR.
Normalization of metabolic assays
After the completion of the metabolic assays, protein content was determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher, Cat # 23225). The data obtained from the metabolic assays were normalized to the protein content.
Calcium imaging
Dissociated DRG cells were loaded with Fluo4-AM (1 µM, Thermo Fisher, Cat # F14217) for 30 min at 37°C in DMEM (Millipore Sigma, Cat # D5030). The cells were then transferred to a recording chamber placed on an inverted microscope (Olympus IX73, Japan). Images were captures using Micro-Manager 1.4 and analyzed with Fiji/ImageJ 1.52c software (NIH). Neurons measuring between 20 and 35 µm in diameter were analyzed. The Emax and time to half maximum (t1/2) were determined using Graphpad Prism 7. Cells were pretreated with vehicle, oxamate (40 mM), or DCA (20 mM) prior to the addition of glucose (10 mM) at 40 s time point. At the end of the assay, veratridine (30 µM), an inhibitor of voltage-gated sodium channel (VGSC) inactivation, was added. Veratridine is known to increase nerve excitability and intracellular calcium concentrations.25 Cells that did not elicit calcium in transients in response to veratridine were not included in the analysis.
Western blotting
Protein was extracted from the L4-6 DRGs in lysis buffer (50 mM Tris HCl, 1% Triton X-100, 150 mM NaCl, and 1 mM EDTA at pH 7.4) containing protease and phosphatase inhibitor mixtures with an ultrasonicator on ice, and cleared of cellular debris by centrifugation at 14,000 relative centrifugal force for 15 min at 4°C. Fifteen micrograms of protein per well were loaded and separated by standard 7.5% or 10% SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore Sigma, Cat # IPVH00010) and then blocked with 5% dry milk for 3 h at room temperature. The blots were incubated with primary antibody overnight at 4°C and detected the following day with donkey anti-rabbit or goat anti-mouse antibody conjugated to horseradish peroxidase (1:10,000, Jackson Immunoresearch, Cat # 711–036-152, Cat # 115–036-062). Signal was detected by enhanced chemiluminescence on films. For assessment of phospho-proteins membranes were stripped and reprobed for total protein of interest for normalization. Densitometric analyses were done using UN-SCAN-IT 7.1 software (Silk Scientific Corp.). Primary antibodies include phospho-PDH Ser300 (1:1000, Millipore Sigma, Cat # ABS192), PDH, LDHA, PDHK1 (1:1000 Cell Signaling Technology, Cat # 3205, 3582, 3820), and beta-III-tubulin (1:50,000 Promega, Cat # G7121).
Statistical analysis and data presentation
Data are based on the means and the standard error of the means (±SEM). Graph plotting and statistical analysis used GraphPad Prism Version 7 (Graph Pad Software, Inc. San Diego, CA, USA). When analyzing evoked pain behavior data, two-way repeated-measures (RM) analysis of variance (ANOVA) followed by post-hoc pairwise comparisons with Bonferroni correction was used. When analyzing CPA data, two-way ANOVA (pairing vs. treatment) was applied followed by post-hoc pairwise comparisons with Bonferroni correction. Difference scores compared the differences between test times and preconditioning time in each chamber for each mouse. The CPA difference scores, calcium imaging, and XF data were analyzed by one-way ANOVA, followed by Tukey post-hoc test. Western blot data were analyzed by either unpaired t-test or one-way ANOVA, followed by Tukey post-hoc test. A priori level of significance at 95% confidence level was considered at P < 0.05.
Results
Bortezomib induces aerobic glycolysis in DRG neurons
Glycolysis and oxidative phosphorylation are the two major energy-producing pathways in the cell. Most cells possess the ability to switch between these two pathways, thereby adapting to changes in their environment. Glucose in the cell is catabolized through glycolysis to generate ATP and pyruvate. Pyruvate then enters the mitochondria and is oxidized through the Krebs cycle, ultimately generating ATP, CO2, and H2O while consuming oxygen. Pyruvate which is not oxidized gets converted to lactate and is extruded with a proton to the extracellular medium. The extrusion of protons results in the acidification of the medium surrounding the cell.4,12,13,26
The metabolic changes that bortezomib treatment might exert on sensory neurons were characterized by analyzing the glycolysis and oxidative phosphorylation rates using extracellular flux analyzer.12,13 The XF Analyzer directly and simultaneously measures the ECAR and the OCR, which are measures of glycolysis and respiration rates, respectively.12,13 The effect bortezomib treatment has on oxidative phosphorylation was measured in L4-6 DRGs dissected on day 10 using the Mito Stress Test. During the Mito Stress Test, baseline OCR measurements were followed by the addition of compound oligomycin which measures ATP-linked respiration. The compound FCCP measures maximal respiration which was significantly reduced in DRG neurons dissected from mice treated with bortezomib relative to the vehicle-treated group (Figure 1(a); two-way RM ANOVA revealed a main effect for time (F(10, 110) = 85.6, P < 0.0001) and group (F(1, 110) = 5.801, P = 0.0177)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (*P = 0.0147 and ****P < 0.0001) difference in maximal respiration between the vehicle and bortezomib-treated groups, six mice/group). At the end of the assay, a mix of rotenone and antimycin A was injected to measure non-mitochondrial respiration. This result demonstrates that bortezomib suppresses oxidative phosphorylation rates of DRG neurons.
Figure 1.

(a) Mito Stress Test of lumbar DRGs dissected and dissociated on day 10 following the initiation of the five-day bortezomib treatment. Mito Stress Test profile reveals reduced FCCP (4 µM)-induced maximum respiration in the bortezomib group relative to the vehicle-treated group (*P = 0.0147 and ****P < 0.0001, six mice/group). (B) Dissociated lumbar DRGs demonstrate a Glycolysis Stress Test profile where bortezomib group have significantly elevated oligomycin-evoked maximum ECAR relative to the vehicle group (***P = 0.0003 and ****P < 0.0001, six mice/group). Veh: vehicle; Bor: bortezomib; OCR: oxygen consumption rate; ECAR: extracellular acidification rate; Oligo: oligomycin; Rot/AA: rotenone/antimycin A; Gluc: glucose; 2-DG: 2-deoxyglucose; FCCP: ■.
The reduction in oxidative phosphorylation should result in cells utilizing glycolysis to produce the energy needed. To determine the impact bortezomib has on glycolysis of L4-6 DRGs on day 10, the Glycolysis Stress Test was used. During the Glycolysis Stress Test, DRG neurons are incubated in medium without glucose or pyruvate and the baseline ECAR is measured. Addition of glucose measures the glycolysis rate which is followed by the injection of oligomycin which inhibits mitochondrial ATP production and shifts the energy production to glycolysis. The consequent rise in ECAR measures the cellular maximum glycolytic capacity. Primary afferent neurons from mice treated with bortezomib displayed a significant increase in their glycolytic capacity relative to the vehicle-treated group (Figure 1(b); two-way RM ANOVA revealed a main effect for time (F(11, 120) = 96.98, P < 0.0001) and group (F(1, 120) = 31.3, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (****P = 0.0003 and ****P < 0.0001) difference in the glycolytic capacity between the vehicle and bortezomib-treated groups. Finally, 2-DG is injected which inhibits glycolysis by competitively binding to hexokinase, the first enzyme in the glycolytic pathway. The resulting decrease in ECAR confirms that the ECAR produced in the experiment is due to glycolysis. Noteworthy, DRG neurons have been demonstrated to be the major contributors to the OCR and ECAR measures relative to the non-neuronal cells from DRGs.18 Collectively, these data demonstrate that bortezomib alters the metabolic phenotype of sensory neurons in a manner consistent with aerobic glycolysis.
Bortezomib enhances the expression of LDHA and PDHK1 in DRGs
The maximal rate of respiration is mostly determined by substrate supply and oxidation while the spare respiratory capacity is a function of both basal and maximal respiration rates.12 The Mito Stress Test revealed a reduced maximal respiration and spare respiratory capacities in response to bortezomib treatment (Figure 1(a) and (c)). Pyruvate is one of the main substrates that is oxidized in mitochondria. This oxidation is carried out by the mitochondrial enzyme pyruvate dehydrogenase (PDH) which catabolizes pyruvate into acetyl-CoA. Acetyl-CoA enters the Krebs cycle to generate energy. Phosphorylation of PDH by pyruvate dehydrogenase kinase (PDHK) attenuates the rate of conversion of pyruvate to acetyl-CoA.13 In order to determine the impact bortezomib treatment has on PDH phosphorylation, mice were treated with either vehicle or 0.2 mg/kg of bortezomib once a day for five consecutive days, and on day 10, L4-6 DRGs were dissected. Using Western blot analysis, a threefold upregulation in PDHK1 levels was observed following treatment with bortezomib (Figure 2(a), t = 4.291, df = 6, **P = 0.0051, unpaired t-test, five mice/group). Moreover, twofold increase in the phosphorylation of PDH was observed on serine 300 in DRGs dissected from mice that were treated with bortezomib (Figure 2(b), t = 2.7, df = 6, *P = 0.0356, unpaired t-test, five mice/group). These results elucidate the mechanism by which bortezomib treatment might attenuate the oxidative capacity of sensory neurons.
Figure 2.

(a) Western blot analysis of lumbar DRGs dissected on day 10 showed increased expression of (a) PDHK1 (**P = 0.0051, five mice/group) and (b) enhanced phosphorylation of its substrate PDH on serine 300 in the Bor-treated group (*P = 0.0356, five mice/group). (c) LDHA expression was also enhanced 10 days post bortezomib treatment (*P = 0.0444, five mice/group). Veh: vehicle; Bor: bortezomib; PDHK1: pyruvate dehydrogenase kinase 1; pPDH: phospho-pyruvate dehydrogenase; LDHA: lactate dehydrogenase A.
Pyruvate that does not undergo mitochondrial oxidation can get converted to lactate. This conversion is catalyzed by the enzyme LDH. Hence, Western blot analysis revealed that 10 days post bortezomib treatment, L4-6 DRGs display twofold increase in the expression of LDHA (Figure 2(c), t = 2.534, df = 6, *P = 0.0444, unpaired t-test, five mice/group). This result support the extracellular flux assay where DRG neurons dissected from mice treated with bortezomib exhibit enhanced ECAR.
Blockade of LDHA and PDHK1 reverses bortezomib-induced metabolic phenotype
In an effort to determine if pharmacological inhibition of LDHA can reverse the glycolytic changes observed in sensory neurons in response to bortezomib, the following experiment was performed. L4-6 DRGs were dissected on day 10 following the initiation of the bortezomib treatment. This was followed by the Glycolysis Stress Test. After establishing baseline ECAR, glucose and oligomycin were injected to measure glycolysis and glycolytic capacity, respectively. Although glycolysis rates were not affected, DRGs dissected from mice treated with bortezomib displayed a significant increase in the glycolytic capacity. Next, oxamate was injected which caused a profound decline in ECAR. Oxamate is a compound which selectively inhibits LDH.27–29 ECAR was further decreased following the injection of 2-DG, which inhibits glycolysis by targeting hexokinase (Figure 3(a); two-way RM ANOVA revealed a main effect for time (F(14, 150) = 47.35, P < 0.0001) and group (F(1, 150) = 23.95, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (*P = 0.0101, **P = 0.0035) difference in glycolytic capacity (post oligomycin addition, six mice/group) between the control and the bortezomib-treated group). These results show that inhibition of LDHA by oxamate severely abrogates extracellular acidification.
Figure 3.

(a) Glycolysis Stress Test profile on day 10 of dissociated L4-6 DRGs of mice treated with either vehicle or bortezomib. Glucose (10 mM) injection did not cause a significant difference in ECAR in either group. Oligomycin (5 µM) induces significantly higher ECAR in the bortezomib group which was dramatically reduced following the application of LDH inhibitor, oxamate (40 mM), in both groups (*P = 0.0101, **P = 0.0035, six mice/group). (b) Treatment of DRG cultures with DCA (20 mM) for 10 min reduces the phosphorylation of PDH on S300 (****P < 0.0001, six wells/group). (c) Pyruvate oxidation assay of dissociated L4-6 DRGs on day 10 post bortezomib treatment. DRGs dissected from mice treated with bortezomib display a reduced pyruvate oxidation. Additional pyruvate administration does not alter OCR of both groups. Treatment with the PDHK inhibitor, DCA (20 mM), normalizes the OCR of the sensory neurons dissected from mice treated with bortezomib. The mitochondrial pyruvate transporter inhibitor, UK5099 (10 µM), was added at the end of the assay to measure pyruvate-dependent OCR (vehicle vs. bortezomib *P = 0.0161, **P = 0.004, DCA vs. bortezomib ###P < 0.0001, 9–12 mice/group). (d) Glycolysis Stress Test profile of L4-6 DRGs dissected on day 10 post vehicle or bortezomib treatment. Following baseline measurement, either vehicle (Veh) or DCA (20 mM) was added to the cells. DCA treatment normalized the glycolytic capacity in cells dissected from mice treated with bortezomib (Bor→Veh vs. other groups ****P < 0.0001, 6–10 mice/group). Veh: vehicle; Bor: bortezomib; Oligo: oligomycin; Gluc: glucose; Oxa: oxamate; OCR: oxygen consumption rate; ECAR: extracellular acidification rate; DCA: dichloroacetate; UK: UK5099; 2-DG: 2-deoxyglucose; pPDH: phospho-pyruvate dehydrogenase.
The pharmacological inhibition of PDHK1 should normalize pyruvate oxidation and glycolytic capacity, thereby increasing respiration rates, while limiting extracellular acidification by diverting pyruvate away from LDHA-mediated lactate formation.30–32 DCA is a selective inhibitor of PDHK.30 Western blot analysis revealed that the treatment of DRG cultures with DCA for 10 min caused a profound reduction in the phosphorylation of PDH (Figure 3(b), t = 10.24, df = 10, ****P < 0.0001, unpaired t-test, six wells/group). Moreover, pyruvate oxidation assay on L4-6 DRGs dissected from mice treated with either vehicle of bortezomib showed that treatment with bortezomib caused a significant reduction of baseline pyruvate oxidation relative to the vehicle-treated group (Figure 3(c)). Addition of pyruvate did not alter OCR confirming that pyruvate production is not affected in both groups. However, the addition of DCA rapidly increased pyruvate-dependent OCR demonstrating that inhibition of PDHK can normalize pyruvate oxidation (Figure 3(c); two-way RM ANOVA revealed a main effect for time (F(7, 152) = 6.558, P < 0.0001) and group (F(1, 152) = 61.03, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (*P = 0.0161, **P = 0.004) difference in pyruvate oxidation between the mice treated with vehicle or bortezomib. Post-hoc pairwise comparisons with Bonferroni correction also showed that the treatment with DCA significantly (###P < 0.0001) elevated OCR of the sensory neurons dissected from bortezomib-treated mice relative to the baseline, 9–12 mice/group). Finally, pyruvate-dependent OCR was determined by the addition of the mitochondrial pyruvate transporter inhibitor, UK5099.
The effect of PDHK inhibition on glycolysis was determined by performing the Glycolysis Stress Test on L4-6 DRGs dissected on day 10 post bortezomib treatment. After establishing baseline ECAR, DRG neurons were treated with either vehicle or DCA. This was followed by the addition of glucose which caused a significant reduction in ECAR in DCA-treated neurons dissected from the bortezomib-pretreated mice (Figure 3(d)). Crucially, the addition of oligomycin revealed that DCA normalizes the glycolytic capacity of DRG neurons dissected from bortezomib-pretreated mice (Figure 3(d)); two-way RM ANOVA revealed a main effect for time (F(11, 240) = 297.1, P < 0.0001) and group (F(33, 240) = 1.687, P = 0.0144)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (****P < 0.0001) difference in glycolytic capacity (post oligomycin addition) between the Bor → Veh and the other groups, 6–10 mice/group). At the end of the assay, 2-DG was added to determine non-glycolytic ECAR. Collectively, these results show that pharmacological inhibition of PDHK profoundly reduces the phosphorylation of PDH, normalizing glycolytic capacity and pyruvate oxidation in DRG neurons excised from mice pretreated with bortezomib.
Bortezomib enhances calcium responses in DRG neurons and inhibition of LDH and PDHK attenuate them
Since bortezomib treatment enhances the glycolytic capacity of sensory neurons evidenced by the enhanced extrusion of protons, calcium imaging experiments were performed to determine the impact of the extrusion of metabolites on the induction of calcium responses. Lumbar 4–6 DRGs from mice that were treated with either vehicle or bortezomib were dissected on day 10. The DRGs were loaded with calcium probe Fluo4-AM. Imaging of DRG neurons was performed in DMEM without glucose, pyruvate, or glutamine. The cells were pretreated with vehicle, oxamate, or DCA and baseline measurements were made for 40 s. At the 40-smark, glucose was added and imaging of calcium responses continued for another 400 s. Addition of glucose led to a significant increase in the calcium responses of neurons excised from bortezomib-treated mice relative to the vehicle group (Figure 4(a)). Neurons with diameters measuring 20–35 µm were analyzed. Glucose addition revealed a significantly higher maximum response, Emax, in DRG neurons dissected from mice pretreated with bortezomib relative to the vehicle-pretreated groups. Treatment with DCA caused a significant reduction in the Emax of calcium responses in neurons pretreated with bortezomib (Figure 4(b), One-way ANOVA revealed a significant difference between the groups (F(5, 148) = 65.58, P < 0.0001). Tukey post-hoc analysis revealed significant (**P < 0.0092, ****P < 0.0001) difference between the Bor + Veh and the other groups (20–30 neurons from five animals/group). Treatment of DRG neurons with oxamate or DCA significantly extended the time to half-maximum, t1/2, in response to glucose addition in both the vehicle and bortezomib pretreated groups (Figure 4(c); F(5, 148) = 128.3, P < 0.0001). Tukey post-hoc analysis revealed significant (****P < 0.0001) difference between the vehicle and the drug-treated groups (20–30 neurons from five animals/group). Oxamate is a noncompetitive inhibitor of LDH, known to reduce the rate of conversion of pyruvate to lactate.33 Analysis of calcium responses captures this aspect of LDH enzymology where oxamate extends the time to half-maximum without significantly influencing the Emax. In contrast, DCA enhances the mitochondrial oxidation of pyruvate diverting it away from the LDH mediated conversion to lactate. This reaction robs LDH of its substrate thus causing a reduction in the Emax as well as extending the time to half-maximum. Collectively, these results show that DRG neurons dissected from mice pretreated with bortezomib display a significantly higher extracellular acidification and calcium responses relative to the vehicle pretreated group. Crucially, the blockade of LDHA or PDHK attenuates both parameters.
Figure 4.
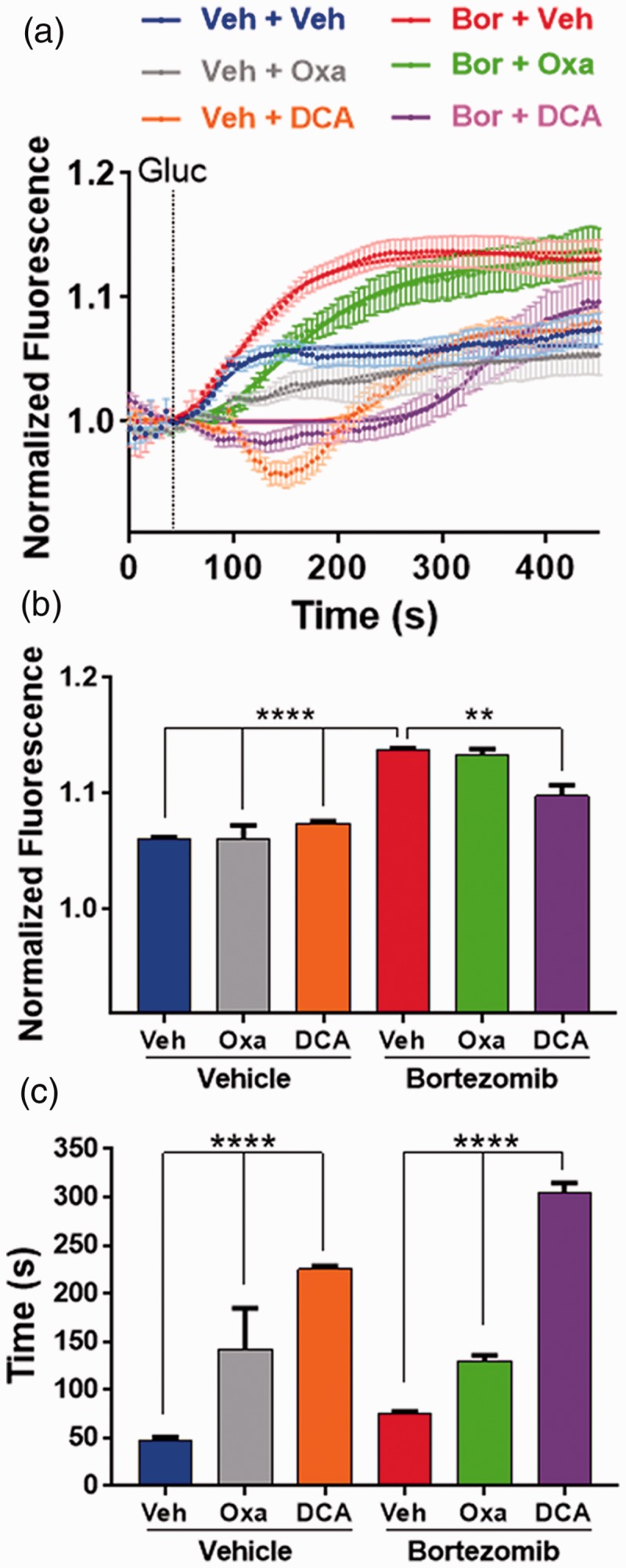
(a) Cumulative average of calcium imaging traces showing the changes in Fluo4 fluorescence in dissociated DRG neurons dissected on day 10, from mice treated with vehicle or bortezomib. Cells were treated with vehicle, oxamate (40 mM), or DCA (20 mM). After baseline measurement was established for 40 s, glucose (10 mM) was added and the Fluo4 fluorescence was recorded for 400 s. (b) The maximum response, Emax, to glucose revealed that DRG neurons dissected form mice treated with bortezomib display higher Emax of calcium transients which was attenuated by the treatment of cells with DCA (**P < 0.0092, ****P < 0.0001, 20–30 neurons/group). (c) Time to half maximum (t1/2) response revealed that oxamate and DCA treatment of DRG cells extended the t1/2 of calcium transients (****P < 0.0001, 20–30 neurons/group). Veh: vehicle; Bor: bortezomib; Gluc: glucose; Oxa: oxamate; DCA: dichloroacetate.
Inhibition of LDHA and PDHK1 alleviates bortezomib-induced pain
The role of increased extracellular acidification in promoting allodynia was measured in mice treated with either vehicle or bortezomib. On day 10, bortezomib-treated mice display a profound tactile hypersensitivity measured by von Frey filaments. Both vehicle and bortezomib groups received IP treatment with either vehicle or oxamate. Oxamate reversed the tactile hypersensitivity for several hours in the bortezomib treatment group without affecting the tactile thresholds of the vehicle group (Figure 5(a); two-way RM ANOVA revealed a main effect for time (F(7, 28) = 59.76, P < 0.0001) and group (F(3, 12) = 266.8, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (###P < 0.0001) difference between the bortezomib and the vehicle groups treated with either vehicle or oxamate. Post-hoc pairwise comparisons with Bonferroni correction also revealed a significant (****P < 0.0001) difference between the bortezomib → vehicle and bortezomib → oxamate groups, five mice/group). These results demonstrate that increased extracellular acidification due to enhanced glycolysis causes allodynia.
Figure 5.

(a) Bortezomib-induced allodynia on day 10 was reversed for several hours by inhibiting LDH with oxamate (IP 500 mg/kg) (Bortezomib vs. vehicle ###P < 0.0001, bortezomib →vehicle vs. bortezomib→oxamate ****P < 0.0001, five mice/group). (b) Intrathecal administration of siRNA that targets LDHA reversed bortezomib-induced CIPN. Control siRNA did not affect the tactile thresholds (Bor vs. Veh ###P < 0.0001, Bor → IT LDHA siRNA and Bor → IT Cont siRNA ****P < 0.0001, five mice/group). (c) L4-6 DRGs was dissected to confirm knockdown of LDHA (*P = 0.0363, ***P = 0.0002, five mice/group). (d) Intrathecal siRNA administration did not significantly alter the expression of LDHA in the L4-6 spinal cord. Veh: vehicle; Bor: bortezomib; IT: intrathecal; LDHA: lactate dehydrogenase A; Cont: control.
Metabolism is crucial for cellular development and healthy function. Hence, to avert adverse events that a complete loss of metabolic gene can cause, a knockdown approach was used to attenuate the expression of LDHA. Intrathecal delivery of 2–5 µg of siRNA for three to four days into the general lumbar region has been shown to knockdown a gene of interest both in lumbar DRGs and spinal cord.34–36 Crucially, knockdown of LDHA will not completely eliminate the target gene and it is reversible. Hence, IT administration between L4 and L5 vertebrae of only 1 µg of siRNA (using i-Fect transfection reagent for two consecutive days) that targets LDHA gene reversed the allodynia associated with bortezomib-induced pain (Figure 5(b); two-way RM ANOVA revealed a main effect for time (F(3, 62) = 53.23, P < 0.0001) and group (F(3, 62) = 135.4, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (###P < 0.0001) difference between the bortezomib and the vehicle groups. Post-hoc pairwise comparisons with Bonferroni correction also revealed a significant (****P < 0.0001) difference between the Bor → IT LDHA siRNA and Bor → IT control siRNA, five mice/group). Since the distance between L4-6 DRGs and the spinal cord section that is innervated by L4-6 DRGs is around 17 mm,37 this injection paradigm limited the reduction of LDHA levels to L4-6 DRGs, without affecting L4-6 spinal cord (Figure 5(c) and (d); one-way ANOVA revealed a significant difference between the groups (F(3, 16) = 10.61, P = 0.0004)). Tukey post-hoc analysis revealed significant (*P = 0.0363, ***P = 0.0002) difference between bortezomib + control siRNA and the other groups, five mice/group). These data demonstrate that normalizing LDHA levels was sufficient in alleviating bortezomib-induced allodynia. The control groups received siRNA in i-Fect that does not target any mouse genes.
The role of pyruvate oxidation in bortezomib-induced pain was explored next. Similar to the aforementioned experiment, 10 days following bortezomib treatment mice were injected with IP DCA. DCA normalized tactile hypersensitivity for several hours in the bortezomib group without adversely affecting the tactile thresholds of the control group (Figure 6(a); two-way RM ANOVA revealed a main effect for time (F(7, 128) = 49.69, P < 0.0001) and group (F(3, 128) = 554.4, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (###P < 0.0001) difference between the bortezomib and the vehicle groups treated with either vehicle or DCA. Post-hoc pairwise comparisons with Bonferroni correction also revealed a significant (****P < 0.0001) difference between the bortezomib → vehicle and bortezomib → DCA groups, five mice/group). IT administration of siRNA (1 µg) that targets PDHK1 reversed the bortezomib-induced allodynia (Figure 6(b); two-way RM ANOVA revealed a main effect for time (F(3, 62) = 75.01, P < 0.0001) and group (F(3, 62) = 181.3, P < 0.0001)). Post-hoc pairwise comparisons with Bonferroni correction revealed a significant (####P < 0.0001) difference between the bortezomib and the vehicle groups. Post-hoc pairwise comparisons with Bonferroni correction also revealed a significant (****P < 0.0001) difference between the Bor → IT PDHK1 siRNA and Bor → IT control siRNA, five mice/group). Western blot analysis of L4-6 DRGs confirmed knockdown of PDHK1 in the DRGs but not in the spinal cord (Figure 6(c) and (d); one-way ANOVA revealed a significant difference between the groups (F(3, 16) = 16.19, P < 0.0001). Tukey post-hoc analysis revealed significant (*P = 0.02, ***P = 0.0002) difference between bortezomib + control siRNA and the other groups, five mice/group) and L4-6 spinal cord).
Figure 6.

(a) PDHK inhibitor, DCA (IP 100 mg/kg), reversed bortezomib-induced allodynia on day 10 (bortezomib vs. vehicle ###P < 0.0001, bortezomib →vehicle vs. bortezomib→DCA ****P < 0.0001, five mice/group). (b) Intrathecal (IT) administration of siRNA that targets PDHK1 reversed bortezomib-induced CIPN (Bor vs. Veh ###P < 0.0001, Bor → IT PDHK1 siRNA and Bor → IT Cont siRNA ****P < 0.0001, five mice/group). (c) Knockdown of PDHK1 in L4-6 DRGs was confirmed (*P = 0.02, ***P = 0.0002, five mice/group). (d) Intrathecal siRNA administration did not alter the expression of PDHK1 in the L4-6 spinal cord. Veh: vehicle; Bor: bortezomib; DCA: dichloroacetate; BL: baseline IT: intrathecal; Cont: control; PDHK1: pyruvate dehydrogenase kinase 1.
Paresthesia and dysesthesia are common clinical symptoms of CIPN which cannot be measured using reflexive behavioral assays (von Frey). Hence, CPA assay was developed to determine if targeting LDH or PDHK alleviate pain/dysesthesia. This study uncovered increased extracellular acidification due to enhanced glycolysis as a mechanism that contributes to bortezomib-mediated pain. This suggests that promoting glycolytic flux should exacerbate bortezomib-induced pain. Using a single-trial conditioning protocol in the CPA experiments,38,39 baseline measurements were performed for 30 min when mice were exposed to the environment with full access to all chambers. The next day, in the morning the mice received IP saline paired with a randomly chosen chamber for 30 min and 3–4 h later, IP glucose, paired with the other chamber for 30 min. It should be noted that food was withheld between the saline and glucose treatments (3–4 h) which allows for a rapid spike in the levels of plasma glucose peaking at around 15–30 min and normalizes by 60 min.24 On the test day, 20 h posttraining, mice had free access to all chambers. Movement and duration of each mouse spent in each chamber were recorded for 30 min for analysis of chamber aversion (Figure 7(a); two-way RM ANOVA revealed a main effect for group (F(6, 144) = 4.072, P = 0.0008)). Post-hoc pairwise comparisons with Bonferroni correction also revealed a significant (**P = 0.001) difference between the saline and glucose-paired groups in mice pretreated with bortezomib, 10 mice/group). Difference scores were calculated as test time minus baseline time spent in the drug chamber (Figure 7(b); one-way ANOVA revealed a significant difference between the groups (F(5, 51) = 2.432, P = 0.0472)). Tukey post-hoc analysis revealed significant (*P = 0.04) difference between vehicle and bortezomib groups, 10 mice/group). Glucose administration induced CPA in mice pretreated with bortezomib while the vehicle-pretreated mice did not show aversion to either chamber. The peak anti-allodynic effect of oxamate and DCA was determined to be between 2–3 h and 1–2 h, respectively (Figures 5(a) and 6(a)). Hence, IP oxamate and DCA were administered 2 h and 1 h prior to the glucose treatment, respectively. Both oxamate and DCA blocked the CPA produced by glucose administration in the bortezomib pretreated mice (Figure 7(a) and 7(b)). Neither oxamate nor DCA affected the chamber preference of mice in the vehicle group. Collectively, these results demonstrate that the enhancement of glycolytic flux due to aerobic glycolysis promotes pain/dysesthesia following bortezomib treatment. Moreover, targeted inhibition of the enzymes that maintain this metabolic phenotype might serve as a valid therapeutic strategy for treating bortezomib-mediated pain.
Figure 7.

(a) Intraperitoneal glucose administration (IP 2 g/kg) induced CPA in bortezomib-pretreated mice on day 10 post chemotherapy. BL measurements ensured that absence of chamber bias prior to conditioning. After a single-trial conditioning protocol, bortezomib-pretreated mice spent significantly shorter time in glucose-paired chamber, whereas vehicle-pretreated mice showed no chamber preference. Treatment with oxamate (IP 500 mg/kg) or DCA (IP 500 mg/kg) blocked the CPA produced by glucose administration (**P = 0.001, 10 mice/group). (b) Difference scores (test time – baseline time spent in glucose-paired chamber) demonstrate that glucose produced CPA in bortezomib—but not vehicle-pretreated mice. Treatment with oxamate or DCA blocked the glucose-induced CPA (*P = 0.0488, 10 mice/group). Veh: vehicle; Bor: bortezomib; Oxa: oxamate; BL: baseline; DCA: dichloroacetate.
Discussion
The adverse effect bortezomib has on mitochondria of sensory neurons has been long-established.16 Moreover, paclitaxel has been demonstrated to enhance glycolysis while reducing oxidative phosphorylation in sensory neurons.18 However, the molecular mechanisms that sustain changes in the metabolism of primary afferents and how these changes cause pain have remained elusive. This study demonstrates that bortezomib alters the metabolic phenotype of sensory neurons in a manner consistent with aerobic glycolysis. Bortezomib enhanced the expression of PDHK1 which phosphorylates PDHA attenuating pyruvate oxidation. Moreover, bortezomib treatment enhanced the expression of LDHA which catalyzes the conversion of unoxidized pyruvate to lactate (Figure 8). Crucially, inhibition of PDHK1 and LDHA (1) reversed the biochemical changes, (2) normalized the metabolic phenotype which abrogated extracellular acidification, (3) normalized or delayed glucose-induced calcium responses, and (4) alleviated bortezomib-induced pain. These results establish aerobic glycolysis as a mechanism that drives bortezomib-mediated CIPN.
Figure 8.

Schematic diagram summarizing finding of this study. (1) Glucose is imported into cells by the glucose transporter (Glut) and converted to glucose-6p by hexokinase 1 (HK1). (2) Glycolysis eventually generates 2 pyruvate, 2 NADH (NAD+ + H) and 2 ATP molecules. (3) Pyruvate is imported into the mitochondria and converted to acetyl-CoA by pyruvate dehydrogenase (PDH). Acetyl-CoA enters the Krebs cycle where further oxidation can generate around 30 ATP molecules. The rate of PDH enzymatic reaction is attenuated via phosphorylation of PDH by pyruvate dehydrogenase kinase (PDHK). (4) Pyruvate that is not oxidized is converted to lactate by lactate dehydrogenase (LDH), regenerating NAD+. (5) Lactate and a proton are extruded to the extracellular space via monocarboxylate transporter (MCT), which can lead to the activation of a variety of ion channels and receptors that are expressed in different combinations on sensory neurons. Crucially, these targets are known to sensitize primary afferents. In this study, bortezomib treatment increased the expression of PDHK1 in DRGs, resulting in increased phosphorylation of PDH which attenuates pyruvate oxidation. Moreover, bortezomib enhanced the expression of LDHA. Blockade of LDHA or PDHK1 alleviate pain, normalize pyruvate oxidation, and abrogate extracellular acidification.
The key feature of aerobic glycolysis is the reliance on glycolysis for energy production which leads to enhanced extrusion of metabolites (protons and lactate) which can sensitize primary afferents through well-established mechanisms (Figure 8). Sensory neurons express distinct combinations of several types of proton-gated channels which include transient receptor potential vanilloid receptor-1 (TRPV1),40,41 acid-sensing ion channels (ASICs),42,43 certain two-pore domain potassium channels (TWIK and TASK),44 and purinergic P2X receptors.45 Moreover, lactate enhances the ASIC response to protons46 and potentiates the electrophysiological properties of VGSCs.47 Lactate is also known to potentiate toll-like receptor (TLR) signaling.48 This is particularly relevant to CIPN where chemotherapeutics have been shown to activate and recruit the innate immune system to the DRGs via TLRs.49–51 Hence, chemotherapy-induced aerobic glycolysis might activate and recruit immune cells into the DRGs. Activated immune cells can release a multitude of proinflammatory mediators that further sensitize DRG neurons52 leading to increased generation of action potentials where glycolysis provides the majority of the energy. This leads to increased release of metabolites which would exacerbate the sensitization of DRG neurons and extend the activation of immune cells. This bi-directional regulatory mechanism between the immune system and sensory neurons might underpin the maintenance of CIPN that can outlast the chemotherapy administration.
Glycolysis is less efficient in producing ATP than oxidative phosphorylation. Moreover, reduced levels of ATP have been demonstrated to correlate with the pain phenotype following chemotherapy treatment. This has led to the proposal of a hypothesis that links deficits in ATP production to pain due to CIPN.15,18,52 However, several lines of evidence refute this hypothesis. (1) Energy deficits would result in the activation of AMP-activated protein kinase (AMPK).53–56 The pharmacological activation of AMPK has been demonstrated to prevent the development of CIPN.57 (2) CIPN is associated with increased frequency of action potentials in sensory nerves.17,58 Given that a single action potential can consume up to a billion ATP molecules,5–9 the reduction of ATP levels seen in many CIPN studies are very likely due to the increased consumption of ATP rather than its production. (3) Despite being less efficient than oxidative phosphorylation in producing ATP, glycolysis can maintain cellular energetics during energy intensive processes in many cell types. For instance, activated and proliferating immune cells acquire the aerobic glycolysis phenotype.59 Moreover, aerobic glycolysis is the most prevalent metabolic phenotype of cancer cells.60 (4) This study tests this hypothesis by administering glucose which would augment glycolytic and mitochondrial ATP production. To support the hypothesis, glucose administration must alleviate pain in bortezomib-treated mice. However, we demonstrated that the enhancement of glycolytic flux increased calcium responses and exacerbated pain, suggesting that the pain in response to bortezomib treatment is not related to ATP levels. Moreover, limiting the production of lactate and protons blocks pain in bortezomib-treated mice. Collectively, these results suggest that the pain associated with CIPN is not due to deficits in ATP levels but related to the augmented production of metabolites due to aerobic glycolysis.
During glycolysis, the oxidation of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate by glyceraldehyde-3-phosphate dehydrogenase reduces cytosolic pool of NAD+ to NADH. The conversion of pyruvate to lactate by LDH is a crucial reaction for the regeneration of cytosolic pool of NAD+, which is indispensable for sustaining glycolysis. This suggests that strategies that enhance NAD+ levels would replenish the cytosolic pool, counteracting LDH-mediated regeneration of NAD+ and production of lactate. Such a strategy has been shown to alleviate paclitaxel-induced pain where cellular NAD+ levels were augmented either via supplementation of its precursor19 or enhancement of its synthesis.20 These studies further support aerobic glycolysis as the principle mechanism that sustains CIPN.
Aerobic glycolysis in primary afferents might also elucidate the distinct clinical characteristics of CIPN. Chemotherapeutics are known to predominately affect sensory neurons with long axons. Long axons significantly add to the bioenergetic requirements of neurons to reestablish resting membrane potential following action potentials.61 Chemotherapy-induced aerobic glycolysis in sensory neurons could potentially lead to increased reliance on glycolytic pathways in neurons with long fibers. This might be a mechanism by which length-dependent distal sensory neuropathy develops. Moreover, the augmented bioenergetic requirements would divert nutrients away from biosynthetic pathways that are crucial for maintaining axonal structure.62 This can potentially result in the loss of intraepidermal innervation that leads to distal sensory loss and reduced reflexes which are observed with chemotherapy use in patients.22,63
Perhaps the most compelling evidence that affirms the role of aerobic glycolysis in the pathobiology of CIPN originates in failed clinical trials. These trials include lipoic acid which proved to be ineffective in treating CIPN while acetyl-L-carnitine exacerbated pain.64 Lipoic acid is a cofactor critical for PDH function.4 However, this study demonstrates that bortezomib enhances the expression of PDHK1 in sensory neurons which in turn phosphorylates PDH suppressing its activity. Hence, supplementing a cofactor (Lipoic acid) for an inactive enzyme (PDH) would not be effective in treating CIPN. Furthermore, bortezomib (Figure 1) and paclitaxel18 treatment enhance glycolysis, which can sensitize primary afferents due to increased extracellular acidification. It is well-established that acetyl-L-carnitine potentiates glycolysis,65–68 which might have been the underlying cause of increased paclitaxel-induced pain severity in a clinical trial.64
A crucial consideration in the development of novel therapies for CIPN is that they must not interfere with anticancer treatments. Otto Warburg postulated that aerobic glycolysis is the fundamental cause of cancer.10,69 The Warburg effect is estimated to occur in up to 80% of cancers, and positron emission tomography has demonstrated that >90% of human cancers exhibit increased glucose uptake indicating aerobic glycolysis is a pervasive property of the malignant phenotype.60 Crucially, targeting LDH or PDHK using oxamate or DCA, respectively, have been shown to potentiate anti-cancer therapy.70–87 Hence, therapeutic strategies that prevent the conversion of pyruvate to lactate (inhibition of LDH) or promote its mitochondrial oxidation (inhibition of PDHK), might serve as a valid therapeutic strategy for not only treating CIPN but potentially enhancing anti-cancer therapy.
In conclusion, these results elucidate the mechanisms by which bortezomib-induced CIPN reprogram the metabolism of sensory neurons in a manner consistent with aerobic glycolysis. Moreover, this study uncovers the mechanisms by which this altered metabolic phenotype causes pain. Crucially, the data reveal that targeted inhibition of the enzymes that maintain aerobic glycolysis can serve as a valid strategy for treating CIPN—a highly significant clinical challenge.
Author Contributions
TL designed and performed research. OKM designed research, performed research, analyzed data, and wrote the paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Department of Neural and Pain Sciences, School of Dentistry, University of Maryland Baltimore (to OKM) and Future Leaders in Pain Research, American Pain Society (to OKM).
References
- 1.Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol 2014; 10: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol 2014; 10: 694–707. [DOI] [PubMed] [Google Scholar]
- 3.Boyette-Davis JA, Walters ET, Dougherty PM. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manage 2015; 5: 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehninger AL, Nelson DL, Cox MM. Lehninger principles of biochemistry. 6th ed New York: W.H. Freeman, 2013. [Google Scholar]
- 5.Lewis JE, Gilmour KM, Moorhead MJ, Perry SF, Markham MR. Action potential energetics at the organismal level reveal a trade-off in efficiency at high firing rates. J Neurosci 2014; 34: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 2001; 21: 1133–1145. [DOI] [PubMed] [Google Scholar]
- 7.Lennie P. The cost of cortical computation. Curr Biol 2003; 13: 493–497. [DOI] [PubMed] [Google Scholar]
- 8.Sengupta B, Stemmler M, Laughlin SB, Niven JE. Action potential energy efficiency varies among neuron types in vertebrates and invertebrates. PLoS Comput Biol 2010; 6: e1000840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alle H, Roth A, Geiger JR. Energy-efficient action potentials in hippocampal mossy fibers. Science 2009; 325: 1405–1408. [DOI] [PubMed] [Google Scholar]
- 10.Warburg O. The metabolism of carcinoma cells. J Cancer Res 1925; 9: 148–163. [Google Scholar]
- 11.Bender DA. Amino acid metabolism. 3rd ed Chichester, West Sussex: Wiley-Blackwell, 2012, p. xxi, 456 p. [Google Scholar]
- 12.Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Meth Enzymol 2014; 547: 309–354. [DOI] [PubMed] [Google Scholar]
- 13.Yang C, Jiang L, Zhang H, Shimoda LA, DeBerardinis RJ, Semenza GL. Analysis of hypoxia-induced metabolic reprogramming. Meth Enzymol 2014; 542: 425–455. [DOI] [PubMed] [Google Scholar]
- 14.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 2011; 435: 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng H, Xiao WH, Bennett GJ. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol 2011; 232: 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng H, Xiao WH, Bennett GJ. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol 2012; 238: 225–234. [DOI] [PubMed] [Google Scholar]
- 17.Xiao WH, Zheng H, Bennett GJ. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 2012; 203: 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duggett NA, Griffiths LA, Flatters SJL. Paclitaxel-induced painful neuropathy is associated with changes in mitochondrial bioenergetics, glycolysis, and an energy deficit in dorsal root ganglia neurons. Pain 2017; 158: 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats. Pain 2017; 158: 962–972. [DOI] [PubMed] [Google Scholar]
- 20.LoCoco PM, Risinger AL, Smith HR, Chavera TS, Berg KA, Clarke WP. Pharmacological augmentation of nicotinamide phosphoribosyltransferase (NAMPT) protects against paclitaxel-induced peripheral neuropathy. eLife 2017; 6: pii: e29626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pazdur R, Coia LR, Hoskins WJ, Wagman LD. Cancer management: a multidisciplinary approach: medical, surgical, & radiation oncology. 13 ed. Oncology Group, 2011. USA: UBM Medica.
- 22.Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 2008; 112: 1593–1599. [DOI] [PubMed] [Google Scholar]
- 23.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth 1994; 53: 55–63. [DOI] [PubMed] [Google Scholar]
- 24.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metabol 2008; 295: E1323–E1332. [DOI] [PubMed] [Google Scholar]
- 25.Mohammed ZA, Doran C, Grundy D, Nassar MA. Veratridine produces distinct calcium response profiles in mouse dorsal root ganglia neurons. Scientific Rep 2017; 7: article 45221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3: 721–732. [DOI] [PubMed] [Google Scholar]
- 27.Jones NP, Schulze A. Targeting cancer metabolism – aiming at a tumour’s sweet-spot. Drug Discov Today 2012; 17: 232. [DOI] [PubMed] [Google Scholar]
- 28.TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Meth Enzymol 2014; 542: 91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winer LSP, Wu M. Rapid analysis of glycolytic and oxidative substrate flux of cancer cells in a microplate. PLoS One 2014; 9: e109916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papandreou I, Goliasova T, Denko NC. Anticancer drugs that target metabolism: is dichloroacetate the new paradigm? Int J Cancer 2011; 128: 1001. [DOI] [PubMed] [Google Scholar]
- 31.Schulze A, Downward J. Flicking the Warburg switch—tyrosine phosphorylation of pyruvate dehydrogenase kinase regulates mitochondrial activity in cancer cells. Mol Cell 2011; 44: 846. [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov 2013; 12: 829. [DOI] [PubMed] [Google Scholar]
- 33.Powers JL, Kiesman NE, Tran CM, Brown JH, Bevilacqua VL. Lactate dehydrogenase kinetics and inhibition using a microplate reader. Biochem Mol Biol Educ 2007; 35: 287–292. [DOI] [PubMed] [Google Scholar]
- 34.Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, Dong Y, Jelinek J, Cesaroni M, Issa JP, Pan HL. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci 2015; 18: 1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai YQ, Chen SR, Han HD, Sood AK, Lopez-Berestein G, Pan HL. Role of M2, M3, and M4 muscarinic receptor subtypes in the spinal cholinergic control of nociception revealed using siRNA in rats. J Neurochem 2009; 111: 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo MC, Zhang DQ, Ma SW, Huang YY, Shuster SJ, Porreca F, Lai J. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Mol Pain 2005; 1: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakla FB. Quantitative studies on the postnatal growth of the spinal cord and the vertebral column of the albino mouse. J Comp Neurol 1969; 136: 237–251. [DOI] [PubMed] [Google Scholar]
- 38.He Y, Tian X, Hu X, Porreca F, Wang ZJ. Negative reinforcement reveals non-evoked ongoing pain in mice with tissue or nerve injury. J Pain 2012; 13: 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 2009; 12: 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998; 21: 531–543. [DOI] [PubMed] [Google Scholar]
- 41.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997; 389: 816–824. [DOI] [PubMed] [Google Scholar]
- 42.Li WG, Xu TL. ASIC3 channels in multimodal sensory perception. ACS Chem Neurosci 2011; 2: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dussor G. ASICs as therapeutic targets for migraine. Neuropharmacology 2015; 94: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morenilla-Palao C, Luis E, Fernandez-Pena C, Quintero E, Weaver JL, Bayliss DA, Viana F. Ion channel profile of TRPM8 cold receptors reveals a role of TASK-3 potassium channels in thermosensation. Cell Rep 2014; 8: 1571–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Browne LE, Nunes JP, Sim JA, Chudasama V, Bragg L, Caddick S, North RA. Optical control of trimeric P2X receptors and acid-sensing ion channels. Proc Natl Acad Sci USA 2014; 111: 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci 2001; 4: 869–870. [DOI] [PubMed] [Google Scholar]
- 47.Rannou F, Leschiera R, Giroux-Metges MA, Pennec JP. Effects of lactate on the voltage-gated sodium channels of rat skeletal muscle: modulating current opinion. J Appl Physiol 2012; 112: 1454–1465. [DOI] [PubMed] [Google Scholar]
- 48.Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol 2009; 182: 2476–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain 2014; 15: 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, Dougherty PM. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain 2016; 17: 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu XJ, Zhang Y, Liu T, Xu ZZ, Park CK, Berta T, Jiang D, Ji RR. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res 2014; 24: 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flatters SJL, Dougherty PM, Colvin LA. Clinical and preclinical perspectives on chemotherapy-induced peripheral neuropathy (CIPN): a narrative review. Br J Anaesth 2017; 119: 737–749. [DOI] [PubMed] [Google Scholar]
- 53.Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, Price TJ. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 2011; 7: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melemedjian OK, Khoutorsky A. Translational control of chronic pain. Progr Mol Biol Transl Sci 2015; 131: 185–213. [DOI] [PubMed] [Google Scholar]
- 55.Melemedjian OK, Mejia GL, Lepow TS, Zoph OK, Price TJ. Bidirectional regulation of P body formation mediated by eIF4F complex formation in sensory neurons. Neurosci Lett 2014; 563: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ, Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain 2012; 8: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, Cleeland C, Heijnen CJ. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS One 2014; 9: e100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao WH, Bennett GJ. Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. Pain 2012; 153: 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol 2014; 32: 609–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Epstein T, Gatenby RA, Brown JS. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS One 2017; 12: e0185085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hille B. Ion channels of excitable membranes. 3rd ed Sunderland, MA: Sinauer, 2001, p. xviii, 814 p. [Google Scholar]
- 62.Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci 2009; 29: 5525–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst 2008; 13: 27–46. [DOI] [PubMed] [Google Scholar]
- 64.Majithia N, Temkin SM, Ruddy KJ, Beutler AS, Hershman DL, Loprinzi CL. National Cancer Institute-supported chemotherapy-induced peripheral neuropathy trials: outcomes and lessons. Support Care Cancer 2016; 24: 1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerlinger M, Santos CR, Spencer-Dene B, Martinez P, Endesfelder D, Burrell RA, Vetter M, Jiang M, Saunders RE, Kelly G, Dykema K, Rioux-Leclercq N, Stamp G, Patard JJ, Larkin J, Howell M, Swanton C. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J Pathol 2012; 227: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mouradian M, Kikawa KD, Dranka BP, Komas SM, Kalyanaraman B, Pardini RS. Docosahexaenoic acid attenuates breast cancer cell metabolism and the Warburg phenotype by targeting bioenergetic function. Mol Carcinog 2015; 54: 810–820. [DOI] [PubMed] [Google Scholar]
- 67.Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, Uchikado Y, Natsugoe S, Nakagawa H, Avadhani NG. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016; 35: 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fodor T, Szanto M, Abdul-Rahman O, Nagy L, Der A, Kiss B, Bai P. Combined treatment of MCF-7 cells with AICAR and methotrexate, Arrests cell cycle and reverses Warburg metabolism through AMP-activated protein kinase (AMPK) and FOXO1. PLoS One 2016; 11: e0150232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Warburg O. On the origin of cancer cells. Science 1956; 123: 309–314. [DOI] [PubMed] [Google Scholar]
- 70.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 2010; 107: 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP, Tan M. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer 2010; 9: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013; 4: e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecologic Oncol 2008; 109: 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishiguro T, Ishiguro R, Ishiguro M, Iwai S. Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepato-Gastroenterol 2012; 59: 994–996. [DOI] [PubMed] [Google Scholar]
- 75.Tong J, Xie G, He J, Li J, Pan F, Liang H. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol 2011; 2011: 740564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H. Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PLoS One 2012; 7: e39949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010; 120: 253–260. [DOI] [PubMed] [Google Scholar]
- 78.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99: 989–994. DOI: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008; 68: 1223–1231. [DOI] [PubMed] [Google Scholar]
- 80.Dhar S, Lippard SJ. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc Natl Acad Sci 2009; 106: 22199–22204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao H, Yan L, Zhang Y, Qi R, Li W, Wang R, Liu S, Huang Y, Li Y, Jing X. A dual-targeting hybrid platinum(IV) prodrug for enhancing efficacy. Chem Commun 2012; 48: 10730–10732. [DOI] [PubMed] [Google Scholar]
- 82.Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, de Lassalle EM, Maboudou P, Formstecher P, Polakowska R, Mortier L, Marchetti P. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res 2012; 72: 5035–5047. [DOI] [PubMed] [Google Scholar]
- 83.Fiebiger W, Olszewski U, Ulsperger E, Geissler K, Hamilton G. In vitro cytotoxicity of novel platinum-based drugs and dichloroacetate against lung carcinoid cell lines. Clin Transl Oncol 2011; 13: 43–49. [DOI] [PubMed] [Google Scholar]
- 84.Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, Liu SH, Cheng AL. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer 2013; 108: 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishiguro T, Ishiguro M, Ishiguro R, Iwai S. Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncol Lett 2012; 3: 726–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, Liu B, Fodstad O, Chen J, Wu Y, Price JE, Tan M. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res 2011; 71: 4585–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 2011; 3: 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]