

Abstract
Phenotypically distinct cellular (sub)populations are clinically relevant for virulence and antibiotic resistance of a bacterial pathogen, but functionally different cells are usually indistinguishable from each other. Here, we introduce fluorescent activity-based probes as chemical tools for single-cell phenotypic characterization of enzyme activity levels in Staphylococcus aureus. We screened a 1,2,3-triazole urea library to identify selective inhibitors of fluorophosphonate-binding serine hydrolases and lipases in S. aureus and synthesized target-selective activity-based probes. Molecular imaging and activity-based protein profiling studies with these probes revealed a dynamic network within this enzyme family involving compensatory regulation of specific family members and exposed single-cell phenotypic heterogeneity. We propose chemical probe labeling of enzymatic activities as a generalizable method for phenotyping of bacterial cells at the population and single-cell level.
Keywords: Activity-based protein profiling, Molecular imaging, Single-cell analysis, Phenotype, Enzymes
Graphical Abstract
A toolset of clickable and fluorescent triazole urea activity-based probes enables selective manipulation and visualization of different serine hydrolases in live S. aureus cells. We demonstrate that these probes can be used to dissect phenotypic differences among bacterial cells at the population and single-cell level.
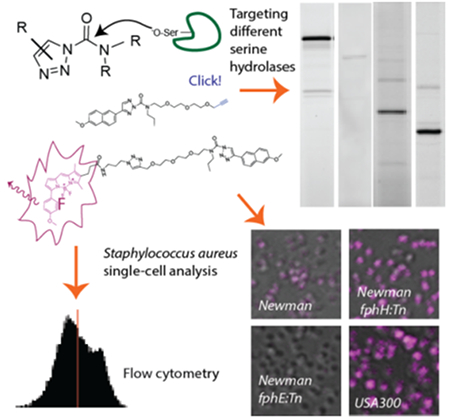
A ‘phenotype’ describes the sum of observable traits of a biological specimen such as a bacterial cell. Although individual cells of an isogenic bacterial population are usually indistinguishable from each other, cells are known to functionally respond to their environment, e.g. through the expression of virulence factors, and functionally distinct cellular subsets have been described. The latter phenomenon, known as phenotypic heterogeneity, becomes apparent and clinically relevant in the formation of surface-associated bacterial communities known as ‘biofilms’[1], the presence of persister cells[2] or in antimicrobial heteroresistance, i.e. the presence of cellular subpopulations with different susceptibilities to antibiotics[3]. Yet, many functional characteristics and responses of these cells remain hidden as non-observable traits unless they involve morphological changes or are visualized by some experimental methodology. Important insights into the phenotypic responses of bacterial pathogens to host factors or other stress or culture conditions have been gained through transcriptomic or proteomic methods.[4] Such studies are commonly performed at the level of entire cell populations, providing a phenotypic snapshot of the ‘averaged’ cell and are thus unable to detect patterns of phenotypic heterogeneity within a population.
Activity-based probes (ABPs) are functionalized active-site directed irreversible enzyme inhibitors that have been used in a range of diverse applications from chemoproteomic profiling and identification of enzymatic targets[5] to non-invasive in vivo imaging in living animals[6]. As molecular imaging with fluorescent ABPs can be used to visualize the localization and distribution of an enzymatic target in its physiological environment with single-cell and subcellular resolution,[7] we reasoned that this class of chemical tools may be exploited for phenotypic characterization of native bacterial populations.
We have recently performed a cell-based chemical proteomics study in the Gram-positive opportunistic bacterial pathogen Staphylococcus aureus that identified 12 active serine hydrolase targets, including lipase 1 and 2 (SAL1, SAL2) as well as 10 largely uncharacterized hydrolases that we termed Fluorophosphonate-binding hydrolases (Fph) A-J.[7a] The secreted S. aureus lipases SAL1/2 are encoded as pre-pro-proteins by the gehA and gehB genes.[8] Both have recently been ascribed roles in bacterial virulence by promoting biofilm formation and host cell invasion.[8] Our preliminary functional characterization of Fph enzymes focused on the 34 kDa FphB. We described FphB as a virulence factor whose activity is regulated at the host-pathogen interface.[7a] In a systemic mouse infection model, we also found a mild contribution to liver pathogenicity for the 31 kDa hydrolase FphE.[7a] The catalytic activities and functional relevance of the other 8 Fph enzymes remain unexplored. Competitive activity-based protein profiling of fluorophosphonate(FP)-labeling in S. aureus using our in-house library of ~500 serine-reactive compounds (based on chloroisocoumarin, sulfonyl fluoride or diphenyl phosphonate electrophiles) identified a chloroisocoumarin hit that we developed into an FphB-selective fluorescent ABP. This probe revealed important insight into the localization of FphB in the bacterial cell envelope and its heterogenous distribution among cells of a larger bacterial population[7a]. Inspired by these results, we now aimed to develop further target-selective fluorescent probes directed against these chemically tractable serine hydrolases and to build a chemical toolkit for molecular imaging-based single-cell phenotypic analysis of bacterial populations.
Here, we explore 1,2,3-triazole ureas (TUs) as another class of potent serine-reactive compounds to further develop ABPs for diverse members of the S. aureus Fphs. TUs have been shown by Cravatt and co-workers to be valuable tools for inhibition of serine hydrolase targets and several candidates have emerged as selective inhibitors of human hydrolases with potent and selective in vivo activity.[9] TUs act by carbamoylation of their target enzyme resulting in elimination of the triazole leaving group.[9a] Selective release of the leaving group adds additional versatility to this chemotype allowing for the design of quenched fluorescent ABPs.[10] Initially, we screened a library of ~150 TU compounds for competition with FP-tetramethylrhodamine (FP-TMR) labeling of serine hydrolases active in live S. aureus ATCC35556 grown on Tryptic Soy Agar supplemented with MgCl2 (TSAMg), as used in our previous screen[7a] and reported as biofilm-promoting.[11] Library compounds had high activity against S. aureus serine hydrolases with 53 hit compounds showing >50% competition of FP-TMR labeling of at least 1 hydrolase target at a final concentration of 1 μM (8 compounds exceeding this threshold at 100 nM; Figure S1). To explore the potential of these compounds for development of target-specific inhibitors and probes, we selected four hit scaffolds with diverse selectivity profiles for re-synthesis, validation in dose-response and target identification (Figure 1A). The click-chemistry based synthesis of TUs yields a mixture of 1,4-linked (designated as ‘a’ throughout this work) and 2,4-linked (designated as ‘b’) triazoles. These regioisomers were purified and tested separately. The compounds were tested in dose-response against the screening strain ATCC35556 under biofilm-promoting conditions (Figure S1, S2) and also validated against the clinically relevant Methicillin-resistant (MRSA) strain USA300 and the Methicillin-sensitive strain Newman (Figure 1B, S2, overview of all strains used in Table S1). Transposon mutants with insertions in individual fph genes (designated as ‘:Tn’ strains) are available for both Newman and USA300, allowing rapid assignment of target identities. The screening hit 1b (AA395) preferentially blocked FP-TMR labeling of FphB (IC50 = 128 nM) with a ~ 4-5-fold selectivity over the ~50 kDa FphA as a secondary target (Figure 1B, S2A,B), while its 1,4-regioisomer 1a also targeted a >66.5 kDa and a ~40 kDa hydrolase (IC50 ~ 19 nM), assigned to the pro-protein and matured form of SAL1 or SAL2 (Figure 1B, S2A,B). Validation of the other hit compounds revealed diverse target selectivity profiles, most notably compounds 2b and 3a have a similar labeling profile and preferentially target both FphA and FphH, compounds 4a/b (KT129/KT130) showed selectivity for the 29 kDa hydrolase FphF (Figure 1B, S2, see Supplementary Results in the SI for a detailed description of the selectivity profiles and structure-activity relationship).
Figure 1.
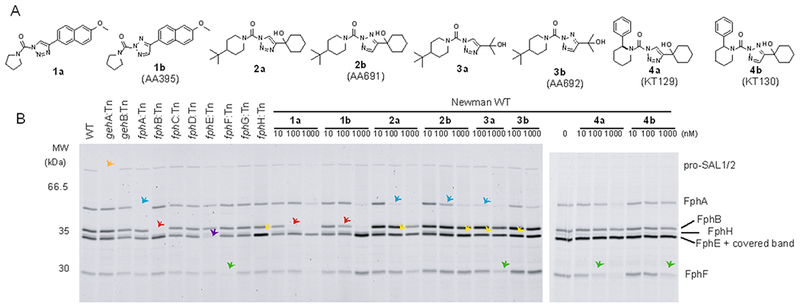
Inhibitory profile of TU screening hits and labeling profile of clickable probes. A) Chemical structures of hit compounds and their regioisomers 1-4 a/b. B) Competitive FP-TMR labelling profiles of S. aureus Newman WT or indicated transposon mutant (:Tn) strains after preincubation with compounds 1-4 a/b. Cells were harvested from TSAMg plates, preincubated with inhibitors for 60 min, labelled with FP-TMR (1 μM), lysed and analysed by SDS-PAGE/fluorescence scan. Preferred targets of each probe (according to transposon mutant analysis) are highlighted by colored arrows: Orange: SAL1, blue: FphA, red: FphB, green: FphF, yellow: FphH, purple: FphE.
To convert these inhibitors into fluorescent ABPs, we focused on analogs of 1a and 1b as their reactivity profile suggested they might yield potent fluorescent ABPs for FphB and open a pathway to generate quenched fluorescent probes for this target. Furthermore, structural modifications may switch the selectivity profile and generate specificity for secondary targets of the parent inhibitor. We synthesized a series of clickable analogs (5-7a/b) where an alkyne handle was installed after a C2 (5,6) or PEG3 linker (7) replacing the pyrrolidinyl substituent of the parent compound. In order to generate a secondary amine as present in the pyrrolidinyl substituent of 1, compounds 6 and 7 feature an additional propyl substituent (Figure 2A). Alkyne probes 6 and 7 could easily be converted into fluorescent ABPs through copper-catalyzed azide-alkyne cycloaddition with a bodipyTMR dye leading to probes 8a/b and 9a/b, respectively. In contrast, our attempts to synthesize fluorescent analogs of probes 5a/b by this method failed due to the decomposition of the primary amine triazole urea group.
Figure 2.
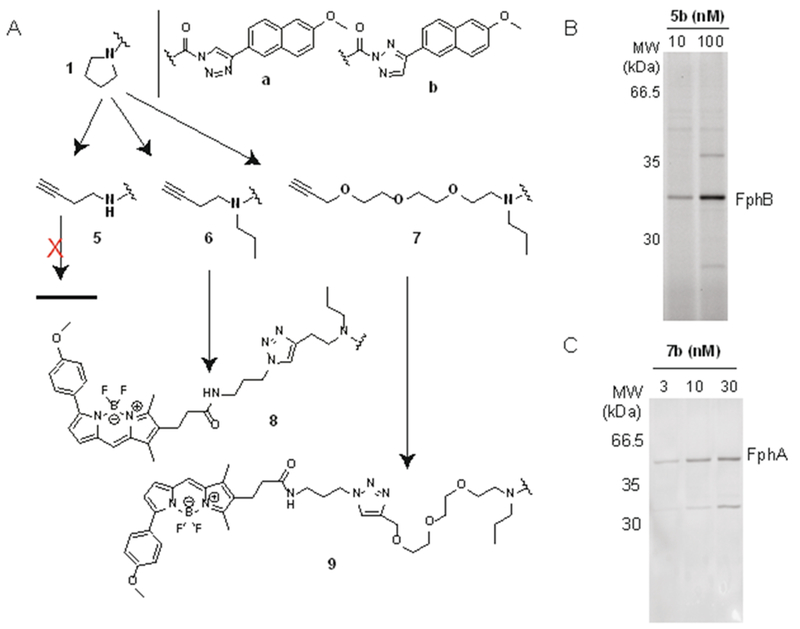
Strategy for the design of fluorescent TU activity-based probes and labeling profiles of select clickable intermediates. A) Schematic overview of ABP design with chemical structures of the clickable probes 5-7a/b and the corresponding fluorescent probe 8a/b and 9a/b. B) Direct cellular labeling profiles of S. aureus Newman cultures with probe 5b and C) 7b. For direct visualization of labeling with clickable probes in B,C, labeled proteins were fluorescently tagged by attachment of N3-TAMRA by click-chemistry in situ after bacterial lysis and analysed by SDS-PAGE. Data show whole cell extracts excluding secreted proteins in the culture supernatant.
For validation of these probes we first assessed the cellular selectivity profile of the alkyne probes, labeling live cells with the probe and clicking on a fluorescent-tag to probe-labeled targets after bacterial lysis in situ followed by SDS-PAGE analysis. We found that the structural modifications required for installing an alkyne handle had dramatic effects on the selectivity profile of the resulting compounds (Supplementary Results in the SI, Figure S3) yielding probes that preferentially labeled FphB (5a/b, Figure 2B), had multiple targets (6a/b, 7a, Figure S3) or were selective for FphA (7b, Figure 2C). Introduction of the bodipyTMR fluorophore again invoked major changes in the selectivity profile. We then continued with analysis of the fluorescent probes and found that probe 8a selectively labeled the secreted lipases SAL2 and more weakly SAL1 but did not react with any of the Fph enzymes (Figure 3A, S4) labeled by 6a (Figure S3C). Reactivity towards SAL1/2 is a shared feature with the parent probe 6a (Fig. S3G). It is conceivable that this selectivity is due to a decreased accessibility of the cell-associated Fph enzymes to the fluorescent probe. While SAL1/2 levels were low when bacteria were harvested from biofilm-like growth on agar, late stationary phase cultures of both strains Newman and USA300 show elevated levels of lipase activity. However, while strain Newman only produced detectable levels of SAL1 (Figure S4), USA300 cultures were characterized by high levels of SAL2 activity (present both as the pro-protein and in its matured form; Figure 3A). While probe 8a is promising as a selective inhibitor and research tool to study the function of SAL1/2 biology, this probe is not suited to provide phenotypic information at the cellular level as its targets are secreted.
Figure 3.
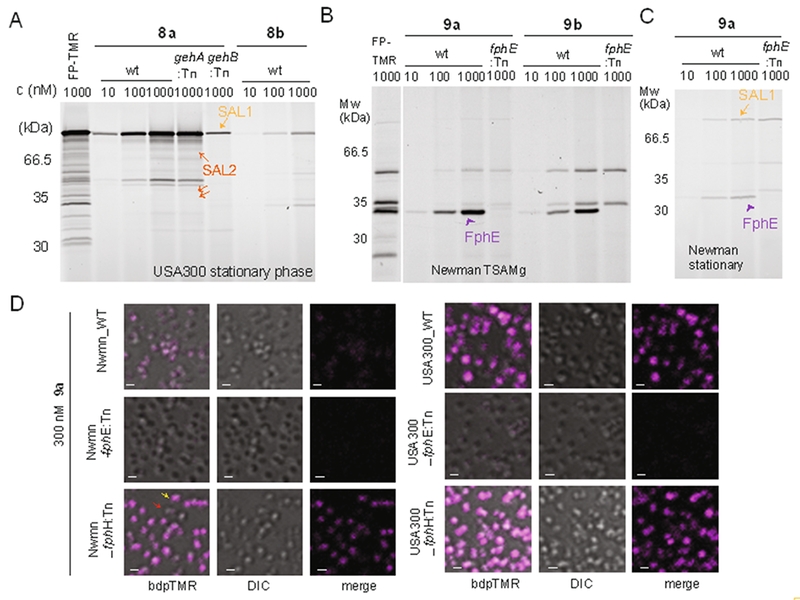
Labeling profiles of fluorescent triazole urea probes show selectivity for SAL1/2 or FphE. A) Labeling profile of S. aureus USA300 and its transposon mutant (:Tn) cultures in late stationary phase with fluorescent probes 8a, 8b or FP-TMR. Arrows indicate pro-form and matured forms of SAL1 (light orange) and SAL2 (dark orange). B, C) Labeling profile of S. aureus Newman and its fphE transposon mutant cultures harvested from TSAMg plates (B) or cultures in late stationary phase (C) labelled with fluorescent probes 9a, 9b or FP-TMR. All samples include full cultures with whole cell extracts and secreted culture supernatants. Arrows indicate FphE (purple), SAL1 (orange). D) Confocal micrographs of indicated S. aureus wt or transposon mutant cells. Cells were harvested from TSAMg and labelled with 300 nM 9a in TSB for 30 min, before washing and fixation. Bodipy-TMR fluorescence is shown in magenta. The red and yellow arrows highlight cells exemplifying the heterogeneity of cellular labeling (red: no or weak labeling, yellow: strong labeling). Scale bar: 1 μm.
Finally, functionalization of the alkyne 7b with bodipyTMR leading to probe 9b increased the activity for the secondary target FphE resulting in a loss of the FphA-selectivity seen for 7b. In contrast, its regioisomer 9a displayed selectivity for FphE among the cell-associated Fph targets (Figure 3B). Compared to the alkyne 7a (Fig. S3D), for which FphE is also a major target, the fluorescent probe 9a lost activity against FphA, FphH and FphF. Probe 9a also labeled SAL1/2 under stationary phase conditions where lipase activities were more abundant (Figures 3C, S4). As SAL1 and SAL2 are secreted, we reasoned that any cellular labeling of 9a is dependent on FphE. Indeed, confocal fluorescence microscopy experiments with WT and FphE mutant (fphE:Tn) cells (harvested from TSAMg plates) revealed that cell-associated labeling of 9a is specific for FphE (Figure 3D) suggesting FphE activity labeling may be harnessed as a functional parameter for phenotypic characterization of S. aureus cells at the single-cell level. Collectively, the data from our probe design efforts indicate that the path from a small inhibitory hit scaffold to the design of a selective ABP is not straightforward and that each functionalization (e.g. with an alkyne or fluorophore) invoked major changes to the selectivity profile with often unanticipated results. Whether these changes are the result of different cellular permeability and target accessibility or due to altered binding affinity to their molecular target remains to be determined.
As a proof-of-principle demonstration of the utility of activity-based probes for phenotypic characterization of bacterial populations, we used probe 9a to visualize and quantify FphE activity levels and distribution of different S. aureus strains under varying culture conditions. Confocal microscopy revealed that Newman cells were only weakly (and heterogeneously) labeled by this probe (Figure 3D). In contrast, USA300 cells gave a much brighter signal. Given the increased FphE-band intensity in the fphH-deficient Newman strain (Figure S3D,E), we tested if the FphE-probe 9a was able to report any corresponding differences in cellular FphE levels by fluorescence microscopy. Indeed, compared to WT cells, probe labeling in Newman fphH:Tn cells was highly increased and reached levels comparable to those observed for strain USA300 (Figure 3D). Interestingly, this potential compensatory or phenotypic response to the lack of FphH was not shared by all cells in the population, as individual cells still failed to label with the probe (Figure 3D).
Given these striking differences, we aimed to use probe 9a to achieve a quantitative read-out of cellular FphE activities under diverse bacterial growth conditions (exponential growth, late stationary phase and after biofilm-promoting growth on TSAMg) by flow cytometry (Figure 4A,B, Figure S5). In both USA300 and Newman strains, FphE-specific labeling was highest in cells harvested from TSAMg, reaching a mean fluorescence intensity signal of 14.5 or 9.8-fold, respectively, over that observed for the corresponding fphE:Tn strain. In USA300, FphE-specific labeling of probe 9a under these conditions was ~4-fold increased compared to exponential phase. Throughout all culture stages, USA300 showed higher levels of probe-labeling than strain WT Newman. Of note, in stationary phase cultures of USA300 a unique subpopulation of ~25% of 9a-positive cells was identified even in the fphE:Tn mutant strain (Figure S5). It is tempting to speculate that cells of this subpopulation interact with secreted SAL2, which is highly abundant under this condition and is efficiently labeled by 9a (Figure S4B). Activity-dependent labeling of other off-targets (e.g. FphA) or non-specific probe binding would be other conceivable explanations for this cellular phenotype.
Figure 4.
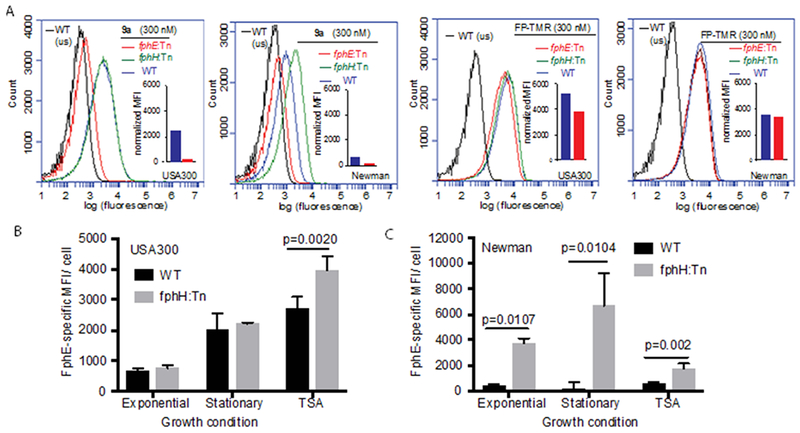
Single-cell analysis of S. aureus cells labeled with probe 9a by flow cytometry. A) Representative plot of cellular fluorescence levels for indicated S. aureus strains harvested from TSAMg and labelled with 9a or FP-TMR. Cells were analyzed by flow cytometry (552 nm laser excitation, 586 nm emission filter) after fixation. Insets show corresponding normalized mean fluorescence intensity (MFI) values for wt and transposon mutant strain fphE:Tn after subtracting MFI from unstained control samples. B, C) Plots of FphE-specific MFI values of PFA-fixed USA300 (B) or Newman (C) wt or fphH:Tn cells labeled with 300 nM of 9a at indicated growth conditions and analyzed by flow cytometry. Primary MFI values were normalized by subtracting the average MFI value of fphE:Tn control samples. Graphs shows means ± S.D. of three independent culture replicates per condition. Statistical significance was tested by unpaired two-tailed Student’s t-test.
In continuation of our phenotypic characterization we found that Newman fphH:Tn showed a statistically significant increase in FphE-specific labeling by 9a over WT under all culture conditions. Surprisingly, the highest level of labeling in this strain was observed in late stationary phase (>10-fold increase; Figure 4C). In contrast, in strain USA300 FphH-deficiency only induced a mild upregulation of FphE activity (~42%) when grown on TSAMg and had no effect on 9a-labeling under exponential and stationary phase condition. This large increase in FphE activity in the Newman fphH:Tn mutant suggests a particular relevance of FphH activity in late stationary phase, which merits further investigation. Overall, the quantitative assessment of single cell-labeling by 9a has revealed that FphE levels are dynamically regulated and are subject to the bacterial growth state and environment in a strain-specific manner. A growth environment-dependent relevance of FphE activity for bacterial physiology would be concurrent with our previous observation that, in a systemic mouse infection model, Newman fphE:Tn bacteria showed a mild but statistically significant reduction in only some organs.[7a] Importantly, FphE-specific labeling of cells by probe 9a was also obtained in the context of live imaging (Fig. S6). Thus activity-based probe labeling can enable the separation of cells within bacterial populations based on probe-labeling status (e.g. corresponding to single target or more global enzymatic activities depending on the probe used) by FACS-sorting and downstream functional analysis of purified subpopulations.
In conclusion, we have used the electrophilic TU scaffold to design multiple activity-based probes with selectivity towards distinct bacterial serine hydrolases and enable their selective manipulation and visualization for a variety of potential applications. The use of these probes in molecular imaging studies has provided insight into the regulatory networks underlying these largely uncharacterized enzymes. This study demonstrates that target-selective ABPs are useful tools for single-cell phenotypic characterization of bacteria according to their enzymatic activities and are able to expose a previously hidden level of functional cellular diversity among different bacterial strains and within isogenic populations. Future studies will address the correlation of such cellular phenotypes with clinically relevant parameters such as virulence, metabolic status or antibiotic resistance parameters.
Supplementary Material
Acknowledgements
This work was supported by a National Institutes of Health grant R01 EB026332 (to M.B.), a China Scholarship Council fellowship No. 201604910274 (to L.C.), a Youth Innovation Promotion Association of CAS grant No.2017329 (to L.C.) and a German Research Foundation (DFG) postdoctoral research fellowship (LE3289/1-1 to C.S.L.). L.J.K received support from Stanford ChEM-H Chemistry/Biology Interface Predoctoral Training Program, Stanford Molecular Pharmacology Training Grant and a Stanford Graduate Fellowship. We thank Benjamin Cravatt (The Scripps Research Institute) for providing the triazole urea screening library. We are grateful to Jessica Sheldon and Eric Skaar (Vanderbilt University) for S. aureus transposon mutant strains.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Moormeier DE, Bayles KW, Mol Microbiol 2017, 104, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lewis K, Annu Rev Microbiol 2010, 64, 357–372. [DOI] [PubMed] [Google Scholar]
- [3].El-Halfawy OM, Valvano MA, Clin Microbiol Rev 2015, 28, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hecker M, Becher D, Fuchs S, Engelmann S, Int J Med Microbiol 2010, 300, 76–87 [DOI] [PubMed] [Google Scholar]; b) Thanert R, Goldmann O, Beineke A, Medina E, Nat Commun 2017, 8, 14268. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tan X, Qin N, Wu C, Sheng J, Yang R, Zheng B, Ma Z, Liu L, Peng X, Jia A, Sci Rep 2015, 5, 11997. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Attia AS, Cassat JE, Aranmolate SO, Zimmerman LJ, Boyd KL, Skaar EP, Pathog Dis 2013, 69, 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Liu Y, Patricelli MP, Cravatt BF, Proc Natl Acad Sci U S A 1999, 96, 14694–14699 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van Rooden EJ, Florea BI, Deng H, Baggelaar MP, van Esbroeck ACM, Zhou J, Overkleeft HS, van der Stelt M, Nat Protoc 2018, 13, 752–767. [DOI] [PubMed] [Google Scholar]
- [6].a) Blum G, Mullins SR, Keren K, Fonovic M, Jedeszko C, Rice MJ, Sloane BF, Bogyo M, Nat Chem Biol 2005, 1, 203–209 [DOI] [PubMed] [Google Scholar]; b) Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M, Nat Chem Biol 2007, 3, 668–677. [DOI] [PubMed] [Google Scholar]
- [7].a) Lentz CS, Sheldon JR, Crawford LA, Cooper R, Garland M, Amieva MR, Weerapana E, Skaar EP, Bogyo M, Nat Chem Biol 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sanman LE, van der Linden WA, Verdoes M, Bogyo M, Cell Chem Biol 2016, 23, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kasperkiewicz P, Altman Y, D’Angelo M, Salvesen GS, Drag M, J Am Chem Soc 2017, 139, 10115–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Poreba M, Rut W, Vizovisek M, Groborz K, Kasperkiewicz P, Finlay D, Vuori K, Turk D, Turk B, Salvesen GS, Drag M, Chem Sci 2018, 9, 2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Rollof J, Hedstrom SA, Nilsson-Ehle P, Biochim Biophys Acta 1987, 921, 364–369 [DOI] [PubMed] [Google Scholar]; b) Cadieux B, Vijayakumaran V, Bernards MA, McGavin MJ, Heinrichs DE, J Bacteriol 2014, 196, 4044–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF, Nat Chem Biol 2011, 7, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hsu KL, Tsuboi K, Chang JW, Whitby LR, Speers AE, Pugh H, Cravatt BF, J Med Chem 2013, 56, 8270–8279 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hsu KL, Tsuboi K, Whitby LR, Speers AE, Pugh H, Inloes J, Cravatt BF, J Med Chem 2013, 56, 8257–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Rooden EJ, Kohsiek M, Kreekel R, van Esbroeck ACM, van den Nieuwendijk A, Janssen APA, van den Berg R, Overkleeft HS, van der Stelt M, Chem Asian J 2018. [DOI] [PubMed] [Google Scholar]
- [11].Koch G, Yepes A, Forstner KU, Wermser C, Stengel ST, Modamio J, Ohlsen K, Foster KR, Lopez D, Cell 2014, 158, 1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.