

Abstract
Autophagy is an evolutionally conserved, highly regulated catabolic process that combines cellular functions required for the regulation of metabolic balance under conditions of stress with those needed for the degradation of damaged cell organelles via the lysosomal machinery. The importance of autophagy for cell homeostasis and survival has long been appreciated. Recent data suggest that autophagy is also involved in non-metabolic functions that impact the immune system. Here, we reflect in two review articles the recent literature pointing to an important role for autophagy in innate immune cells. In this article, we focus on neutrophils, eosinophils, mast cells, and natural killer cells. We mainly discuss the influence of autophagy on functional cellular responses and its importance for overall host defense. In the companion review, we present the role of autophagy in the functions performed by monocytes/macrophages and dendritic cells.
Subject terms: Inflammation, Autophagy
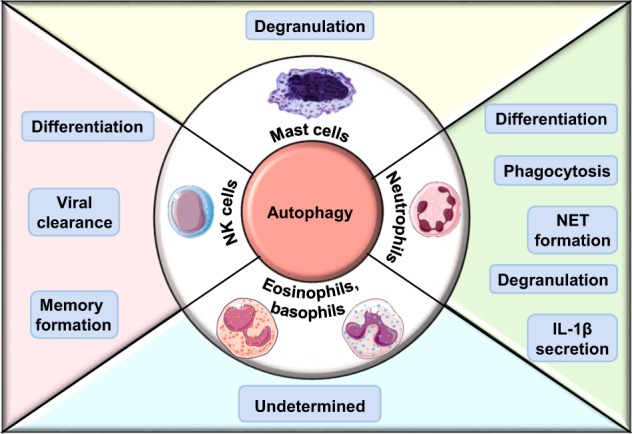
Involvement of autophagy in cellular functions of neutrophils, eosinophils, basophils, NK cells, and mast cells.
Facts
The impact of autophagy on innate immunity extends beyond classical xenophagy, including extensive cross-talk with other host defense mechanisms.
Autophagy in neutrophils is beneficial for fighting various pathogens, preventing their growth and/or chronic parasitism. In addition, autophagy modulates neutrophil functions, including phagocytosis, degranulation, and interleukin (IL)-1β production.
Autophagy plays a crucial role in the degranulation of mast cells (MCs).
In natural killer (NK) cells, autophagy is involved in the processes of their differentiation and transition into memory cells. Autophagy may also modulate NK cell migration into tumor tissues.
Open questions
What are the roles of autophagy in eosinophils, basophils, MCs, and innate lymphocytes? So far, this question has remained poorly investigated.
Does autophagy regulate neutrophil extracellular trap (NET) formation?
What are the mechanisms by which autophagy increases NK cell infiltration into tumor tissues.
Do the autophagy pathways in innate immune cells offer novel therapeutic targets to control immune responses in inflammation and cancer?
Introduction
Autophagy (= self-eating) is a catabolic cellular process that is responsible for a turnover of macromolecules and organelles via the lysosomal degradative pathway. In mammals, it is regulated by the mammalian target of rapamycin (mTOR) and orchestrated by numerous autophagy-related (ATG) proteins. The identification of the ATG genes in yeast and in mammals has provided the impetus for a molecular understanding of autophagy [1, 2]. Autophagy functions have been evolutionarily conserved from Saccharomyces to man. There are now more than 40 recognized ATG genes, some of which bear for historical reasons individual names, such as ATG6, which is usually referred to as Beclin 1 (encoding the BCL-2-interacting coiled-coil protein 1), SQSTM1 (encoding sequestosome 1 and is also known as p62), or FIP200 (which encodes the focal adhesion kinase family-interacting protein of 200 kDa). The proteins encoded by ATG genes are essential for canonical autophagy and p53 and/or p73 activity participates in the regulation of these genes [3, 4]. The canonical autophagic pathways [2, 5–8] and non-canonical pathways that resemble or overlap with autophagy [9–11] have been characterized in excellent reviews. Here, we describe mainly the canonical autophagy pathway that requires ATG proteins.
Currently, the functions of autophagy can be roughly classified into two categories: generation of needed metabolic degradation products and intracellular clearance of defective macromolecules and organelles [1]. Adaptation to starvation is arguably the best conserved function of autophagy among many different organisms. It involves the catabolic production of metabolic intermediates, mainly for sustaining cell survival when nutrients are scarce, or growth requirements increase [12]. Moreover, intracellular quality control of organelles as mediated by autophagy is important for long-lived cells and organisms. For example, neuronal cell- and hepatocyte-specific deletion of ATG genes results in degeneration of the corresponding tissues, with accumulation of abnormal protein aggregates and dysfunctional organelles [13].
An overview of the principal events and players in canonical autophagy is presented here as a guide for further discussion (Fig. 1). The main control gateway to autophagy is via mTOR, a serine/threonine protein kinase that forms two distinct protein complexes known as mTORC1 and mTORC2. Catabolic activity through the process of autophagy is mainly controlled by mTORC1, which integrates signals from several pathways, sensing the levels of nutrients and growth factors [14]. One of the key activators of autophagy is AMP-dependent protein kinase (AMPK), which is the main sensor of cellular energy levels [15–20]. Important regulators of autophagy are also the class I and class III phosphatidylinositol 3-kinases (PI3K) pathways. Class I PI3K, which is activated in response to growth factor ligation to receptors, causes mTORC1 activation and inhibits the initiation of autophagy. On the other hand, activity of class III PI3K vacuolar protein sorting 34 (Vps34) is a direct participant in the induction of autophagy [21].
Fig. 1.

Mechanism of autophagy and its regulation in mammalian cells. The autophagy machinery is regulated by different functional groups of ATG proteins. By integrating several upstream signals, autophagy is initiated by the activation of ULK kinase complex, consisting of proteins ULK1/2, ATG13, ATG101, and FIP200. The autophagic process is inhibited in the presence of growth factors through the action of the class I PI3K-mTORC1 axis, whereas autophagy is induced by amino acid starvation or lack of energy through the AMPK pathway. The pharmacological inhibitors rapamycin, torin 1, and resveratrol act as mTORC1 inhibitors and therefore induce autophagy. Nucleation of the autophagosomal membrane is also regulated by the class III PI3K complex, consisting of proteins Beclin 1, ATG14, ATG9, Vps34, and Vps15. Wortmannin, LY294002, and 3-MA are commonly used autophagy inhibitors, acting as blockers of class III PI3K (known as Vps34) enzymatic activity. ATG proteins that are involved in the elongation process belong to two conjugation systems: ATG12 covalently binds to ATG5 with the assistance of ATG7 and ATG10 enzymes, forming a complex through interaction with ATG16. A second conjugation system leads to conjugation of LC3-I with PE. The lipidated LC3-II exists as a part of the autophagosomal membrane, and allows p62 to target ubiquitinated cargo to the expanding autophagosomal membrane by binding to LC3-II. The completed autophagosome fuses with the lysosome, and the autophagolysosomal contents are degraded by lysosomal hydrolytic enzymes. Chemical agents, such as CQ, bafilomycin A1, and ammonium chloride, can inhibit degradation of engulfed cytoplasmic material as well as the fusion between autophagosomes and lysosomes
The autophagy process consists of five principal steps, referred to as initiation, elongation, autophagosome completion, fusion with the lysosome, and degradation [22]. When mTORC1 is inactivated following a lack of nutrients and energy in cells, it can no longer inhibit the autophagy initiation complex involving the protein kinases unc-51-like kinases 1 and 2 (ULK1/2), ATG13, ATG101, and FIP200, allowing formation of a newly formed phagophore membrane carrying the endoplasmic reticulum-associated protein ATG14. The nascent phagophore membrane recruits Beclin 1, Vps34, and Vps15, leading to the formation of an activated class III PI3K complex that produces phosphatidylinositol 3-phosphate (PI3P) locally and promotes ATG9-dependent membrane acquisition from endosome vesicles [23].
The elongation step involves the enlargement and eventual closure of an expanding membrane, resulting in a completed autophagosome. Formation of a double-membrane organelle requires two protein conjugation systems, essential for the elongation phase. One involves a covalently linked ATG12-ATG5 conjugate, which dissociates from the outer autophagosomal membrane after vesicle formation is complete. The other conjugation system includes microtubule-associated proteins 1A/1B light chains (LC3), which are cleaved by ATG4 upon autophagy induction, resulting in cytoplasmic LC3-I. Further lipidation with phospholipid phosphatidylethanolamine (PE) produces the LC3-II conjugation complex, which is then integrated into both inner and outer autophagosomal membranes. Since LC3-II is present in the autophagosome, it is a commonly used marker for detection of double-membrane autophagic organelles. p62 associates with polyubiquitinated protein aggregates and binds to LC3-II to allow the phagophore to engulf cytosolic elements, to grow, and finally to close the autophagosome. Since protein p62 is generally degraded by autophagy and accumulates when autophagy is inhibited, the reduction of p62 indicates successful vesicle degradation and autophagic flux [24–26]. At the final steps, the autophagosome fuses with the lysosome and auto(phago)lysosomal content is degraded by hydrolytic enzymes [27–30] (Fig. 1).
Several well-characterized inhibitors can block the process of autophagy at different steps. For example, inhibitors of the class I and III PI3Ks such as wortmannin, LY294002, and 3-methyladenine (3-MA), have been widely used as autophagy inhibitors based on their inhibitory effect on class III PI3K activity, which is known to be essential for induction of autophagy. The same inhibitors, however, are reported also to block the class I PI3K that is needed to maintain mTORC1 activity [31]. In a later step of the autophagic process, inhibitors that prevent lysosomal acidification such as chloroquine (CQ), bafilomycin A1, and ammonium chloride (NH4Cl) essentially block the autophagic degradation. Chemicals, such as CQ and ammonium ions are weak bases, raising the pH value in lysosomes, deactivating enzymes, and consequently disabling degradation of vesicle cargo. Bafilomycin A1 regulates intracellular pH by acting as a strong and specific inhibitor of the vacuolar H+ adenosine triphosphate (ATP)ase, known as a proton pump. The change in pH can also inhibit fusion of autophagosomes with lysosomes, resulting in accumulation of autophagosomes [32] (Fig. 1).
Several molecules are able to increase autophagy activity, such as rapamycin, torin 1, and resveratrol. Rapamycin allosterically inhibits the kinase activity of mTORC1 and mimics cellular starvation by blocking signals required for cell growth and proliferation [33]. Torin 1 is a potent and selective ATP-competitive inhibitor of mTOR kinase, the catalytic subunit of two functionally distinct complexes (mTORC1 and mTORC2) that coordinately promote cell growth, proliferation, and survival. Unlike classical mTOR inhibitors, such as rapamycin, torin 1 is able to effectively block phosphorylation of mTORC1 and mTORC2 [33]. Resveratrol (3,4′,5-trihydroxy-trans-stilbene) is a polyphenol compound found in plants known to possess anti-inflammatory and chemopreventive properties. Resveratrol induces autophagy through its capability to both inhibit mTOR and activate AMPK [34] (Fig. 1).
Innate immunity provides an early line of defense against microbes. It consists of cellular and biochemical defense mechanisms that are in place even before infection takes place and are poised to respond rapidly to infections. These mechanisms react to products of microbes and injured cells, and they respond in essentially the same way to repeated exposures. The mechanisms of innate immunity are specific for structures that are common to groups of related microbes, but may not distinguish fine differences between microbes. The principal components of innate immunity are (1) physical and chemical barriers (such as epithelia and the antimicrobial chemicals produced at their surfaces), (2) granulocytes (neutrophils, eosinophils, basophils), mast cells, monocytes/macrophages, dendritic cells, natural killer (NK) cells, and (3) blood proteins, including members of the complement system and other mediators of inflammation [35].
As mentioned above, autophagy contributes to cellular homeostasis and thus, it is not surprising that autophagy regulates functions within the immune system. Consequently, the role of autophagy in regulating innate immune cells is important and under intense investigation. In this article, we summarize the currently available knowledge about the role of autophagy in the functions performed by innate immune cells, such as neutrophils, eosinophils, mast cells, and NK cells. In an accompanying article published in this issue, we focus on functions of monocytes/macrophages and dendritic cells which are regulated or at least modulated by autophagy [36].
Role of autophagy in neutrophils
Neutrophils form the first line of defense against pathogens. About 55–60% of the bone marrow is dedicated to the production of one cell type, the neutrophil, a very important component of the innate immune system, and the main source of reactive oxygen species (ROS) production. Approximately 100 billion neutrophils are generated in normal adults daily, clearly an indication of their important role in defense against microorganisms. On the other hand, in chronic inflammatory responses neutrophils often cause tissue damage associated with dysfunction of the affected tissue/organ. Therefore, it is important to understand the molecular mechanisms leading to overall granulocyte activation. Our present concept of inflammation-associated autophagy in neutrophils is quite unclear, largely due to the fact that this aspect has not yet been extensively investigated [37]. Several lines of evidence, both in the human system and in mouse models, suggest a critical role for autophagy in neutrophil-driven inflammation and defense against pathogens. Autophagy appears to be important for major neutrophil functions, including differentiation, phagocytosis, cytokine production, degranulation, cell death, and, perhaps, neutrophil extracellular trap (NET) formation.
Differentiation
ATG proteins are key players in the neutrophil differentiation pathway. ATG5 is indispensable in both canonical and non-canonical autophagy. ATG5’s role in the autophagy process may shed light on the link between autophagy and the immune response and could lead to the development of new therapies for autoimmune and autoinflammatory diseases. Moreover, ATG5, being essential for the autophagy process, has been suggested as a “guardian of immune integrity” [38]. For instance, ATG5 has been demonstrated to have a role in neutrophil differentiation [39]. Atg5f/fLyz2Cre/Cre mice, in which Atg5 is deleted within the myeloid cell lineage, exhibit an increased proliferation rate in the neutrophil precursor cells of the bone marrow as well as an accelerated process of neutrophil differentiation. As a result, mature neutrophils accumulate in the bone marrow, blood, spleen, and lymph nodes [39]. As discussed above, mTORC1 has a central role in the regulation of autophagy [23], and pharmacological inhibition of p38 mitogen-activated protein kinase (MAPK)- or mTORC1-induced autophagy in neutrophilic precursor cells blocks their differentiation. Thus, autophagy and differentiation exhibit a reciprocal regulation by the p38–mTORC1 axis [39].
On the other hand, it has been shown that metabolism and autophagy are developmentally programmed and are essential for neutrophil differentiation in vivo. Atg7-deficient neutrophil precursors were unable to carry out a shift toward mitochondrial respiration, displaying instead excessive glycolysis, but impaired mitochondrial respiration, decreased ATP production, and lipid droplet accumulation. Just inhibiting autophagy-mediated lipid degradation or fatty acid oxidation within the mitochondria was sufficient to cause defective differentiation, while administration of free fatty acids or pyruvate for mitochondrial respiration rescued differentiation in autophagy-deficient neutrophil precursors and restored normal glucose metabolism [40] (Fig. 2). A recent study reported the differential expression of 22 autophagy-related genes between monocytic and granulocytic differentiation, proposing a role for autophagy in the late stages of differentiation of myeloid precursors toward granulocytes and monocytes, indicating the products of these autophagy genes could be important factors involved in the differentiation of myeloid progenitors into monocytes and granulocytes [41].
Fig. 2.

Involvement of autophagy in neutrophil differentiation and function. Neutrophils are generated in the bone marrow from hematopoietic stem cells (HSC), giving rise to mature neutrophils. Autophagy is involved in the differentiation of neutrophils, and it is requiered for the control of their functions, such as phagocytosis, degranulation, ROS production, NET formation, and cytokine release. Numbers in brackets designate references: + , enhanced function; −, without effect; red indicates an inhibition
Reduced ATG gene expression was observed in primary acute myeloid leukemia (AML) samples and human AML cell lines. In these systems, treatment with all-trans retinoic acid (ATRA) induced neutrophil differentiation and autophagic activity, while inhibition of ATG3, ATG4D, and ATG5 impaired both. ATRA-induced autophagy was Beclin 1-independent, suggesting a non-canonical autophagy pathway. Taken together, these data suggest that restoring autophagic activity might be beneficial for differentiation therapies [42].
Bacterial clearance
The first observation of autophagy as a defense mechanism in neutrophils came with in vitro rickettsia-infected guinea pig peritoneal neutrophils. These neutrophils contained vacuoles which were interpreted as autophagosomes within which phagocytized extracellular material could be observed [43]. Autophagy could be induced in human neutrophils either in a phagocytosis-independent (rapamycin, Toll-like receptor (TLR)-agonists, phorbol-12-myristate 13-acetate (PMA)) or a phagocytosis (Escherichia coli)-dependent manner. ROS activation is a positive mechanism for autophagy induction in the case of PMA, TLR activation, and phagocytosis [44]. In bone marrow-derived mouse neutrophils, the engagement of either TLRs or the fragment crystallizable region (Fc)γ receptors during phagocytosis induced recruitment of the autophagy protein LC3 to phagosomes. Both receptors are known to activate the NADPH oxidase 2 (NOX2), which plays a central role in microbial killing by phagocytes through the generation of ROS [45].
Human neutrophils and the human neutrophil-like cell lines (PLB-985) differentiated into mature neutrophils were found to be able to activate the autophagic machinery as an immediate response to fight pathogenic, adherent-invasive Escherichia coli (AIEC) infections, and to limit the production of inflammatory cytokines. AIEC were found within endosomes and autophagic vesicles characterized by a double-membrane, showing that AIEC bacteria entered cells via the endosomal compartment and were further captured in double-membrane autophagosomes, indicating that AIEC bacteria are subjected to canonical autophagy instead of LC3-associated phagocytosis (LAP) [46]. Moreover, intravenous immunoglobulin (IVIG) preparations were also shown to increase the autophagic activity of human neutrophils in vitro. Inhibiting the late phase of autophagy with bafilomycin A1 reduced neutrophil-mediated bactericidal activity [47]. The bactericidal capacity of human neutrophils was also reduced by addition of the 3-MA [48]. These findings indicate that autophagy plays a critical role in the bactericidal activity mediated by human neutrophils (Fig. 2).
A role of autophagy for bacterial killing in neutrophils was also demonstrated by genetic approaches. For instance, Streptococcus pneumoniae (S. pneumoniae) induced autophagy in neutrophils in a class III PI3K-dependent fashion that also required Atg5. Phagocytosis was enhanced by autophagy, while killing was inhibited by autophagy [49]. Another study proposed an in vivo role of ATG5 in protection against experimental Mycobacterium tuberculosis (M. tuberculosis) by preventing neutrophil-mediated immunopathology in lungs. Loss of Atg5 in neutrophils sensitized mice to M. tuberculosis [50].
In a recent study, the role of the nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome in neutrophil-dependent bacterial clearance in polymicrobial sepsis was investigated. NLRP3-deficient (Nlrp3−/−) and NLRP3 inhibitor-treated wild-type mice showed protection from death and displayed a lower bacterial load, but no impairment in neutrophil recruitment to the peritoneum. Intriguingly, following induction of polymicrobial sepsis, Nlrp3−/− peritoneal cells, primarily neutrophils, demonstrated decreased autophagy, augmented phagocytosis, and enhanced scavenger receptor and mannose-binding leptin expression. Taken together, deletion of Nlrp3 augments the survival of mice during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis, indicating that NLRP3 plays a critical role in modulating autophagy and phagocytosis in neutrophils [51]. On the other hand, interleukin (IL)-1β secretion in human neutrophils is accomplished by an autophagy-mediated secretory pathway. Inhibition of autophagy with 3-MA or wortmannin markedly reduced IL-1β secretion induced by lipopolysaccharide (LPS) and ATP, as did the disruption of autophagic flux with bafilomycin A1 and E64d, inhibitors of autolysosomal digestion. Moreover, siRNA-mediated knockdown of ATG5 markedly reduced IL-1β secretion in neutrophil-differentiated PLB-985 cells [52] (Fig. 2).
Degranulation and cell death
Atg5-knockout neutrophils showed no evidence of abnormalities in morphology, granule protein content, apoptosis regulation, migration, or effector functions [39]. In contrast, neutrophils from myeloid-specific mice with deletions of Atg7 or Atg5 exhibited diminished NADPH oxidase-mediated production of ROS and reduced degranulation [53] (Fig. 2). Moreover, these mice demonstrated a reduced severity with several neutrophil-mediated inflammatory and autoimmune disease models, including PMA-induced ear inflammation, LPS-induced breakdown of the blood–brain barrier, and experimental autoimmune encephalomyelitis [53]. Further work is required to understand the consequences of a deficiency in autophagy for neutrophil degranulation events.
In human neutrophils, a caspase-independent form of cell death has been observed. This form of neutrophil death was induced by sialic acid-binding immunoglobulin-like lectin-9 (Siglec-9) ligation, but required concurrent stimulation with survival cytokines that blocked caspases. This non-apoptotic cell death was characterized by cytoplasmic vacuolization, mitochondrial swelling, nuclear condensation, and normal plasma membrane integrity before cell death actually occurs [54, 55]. Furthermore, in agreement with these studies, neutrophils exposed to granulocyte macrophage-colony stimulating factor (GM-CSF) and other inflammatory cytokines followed by CD44 ligation undergo an autophagy-associated, caspase-independent death characterized by large cytoplasmic vacuoles, apparently generated by fusion events between several organelles, including endosomes, autophagosomes, and secondary granules [56]. These data point to the possibility that adhesion molecules have the capacity to trigger a caspase-independent cell death associated with autophagy induction in neutrophils that is largely necrotic in its nature. Vacuolized neutrophils were observed in septic shock, cystic fibrosis, rheumatoid arthritis, and several neutrophilic skin diseases [56], suggesting that induction of autophagy in these cells is a general phenomenon of neutrophilic inflammatory responses independent of the trigger [57].
It has recently been reported that the adhesion receptor-triggered death of neutrophils in the presence of GM-CSF also exhibits features of necroptosis, as the presence of a receptor interacting protein kinase-3 (RIPK3)–mixed lineage kinase-like (MLKL) signaling pathway has been demonstrated, which in conjunction with p38 MAPK and PI3K, activates the NADPH oxidase, resulting in the production of high levels of ROS [58]. In conclusion, the non-apoptotic death of adherent and cytokine-primed neutrophils shows both autophagic and necroptotic features. Moreover, ATG5 has been shown to trigger apoptosis upon cleavage by calpain [59].
Neutrophil extracellular traps
In recent years, it has been recognized that microbial killing occurs not only after phagocytosis, but also in the extracellular space owing to the formation of extracellular traps (ETs), designated neutrophil extracellular traps (NETs) for neutrophils [60]. NETs consist of extracellular fibers, primarily composed of deoxyribonucleic acid (DNA) and granule proteins released from activated, viable neutrophils [61]. The formation of NETs is an important function of the innate immune system acting against infections. NETs bind and kill both Gram-positive and -negative bacteria, as well as fungi and have been identified in appendicitis, sepsis, and pre-eclampsia, suggesting that these structures are of general in vivo importance. There is a dispute as to whether cell death is required for the release of NETs, and the question, to what extent autophagy is involved in NET formation, is still controversial [62, 63].
Interestingly, Atg5-knockout neutrophils do not exhibit abnormalities in formation of NETs upon physiological activation or exposure to low concentrations of PMA [64]. Investigation has confirmed that human and mouse neutrophils, after pre-treatment with inhibitors of class III PI3K, showed a block both in ROS production and in NET formation. The so-called late autophagy inhibitors bafilomycin A1 and CQ, on the other hand, were without effect. These data indicate that NET formation occurs independently of autophagy and that the inhibition of both ROS production and NET formation in the presence of 3-MA and wortmannin is probably owing to their additional ability to block the class I PI3Ks, which are involved in the signaling cascades initiated by the triggers of NET formation [64]. Non-specific inhibition of ROS activity by PI3K inhibitors such as wortmannin, and their additional ability to block the class I PI3Ks, has led to several contradictory reports about the role of autophagy in NET formation [65–67] (Fig. 2).
Similarly, in whole-blood cultures ex vivo, or in vitro in the presence of platelets, LPS of different bacterial sources induced “vital” NET formation independent of autophagy. This platelet-dependent release of NETs occurred rapidly without neutrophil cell death and required platelet TLR4 and CD62P (P-selectin)-dependent platelet–neutrophil interactions [68].
On the other hand, it has recently been reported that autophagy primes neutrophils for increased NET formation, which is important for proper neutrophil effector functions during sepsis (Fig. 2). Neutrophils isolated from patients who survived sepsis showed an increased autophagy induction and were primed for NET formation in response to subsequent PMA stimulation. In contrast, neutrophils isolated from patients who did not survive sepsis showed dysregulated autophagy and a decreased response to PMA stimulation, indicating that induction of autophagy primed healthy neutrophils for NET formation and vice versa. In a mouse model of sepsis, the augmentation of autophagy improved survival via a NET-dependent mechanism [69]. Similarly, neutrophils from familial Mediterranean fever (FMF) patients during remission were resistant to inflammatory stimuli that induce NET release. Lower basal autophagy levels were identified in neutrophils during remission, while induction of autophagy facilitated NET formation [70, 71].
Mincle is a C-type lectin receptor that functions as an activating receptor for host- and pathogen-associated molecular patterns. Attenuated NET formation in Mincle-deficient neutrophils correlates with impaired autophagy activation in vitro and in vivo, whereas ROS formation in these neutrophils remains intact. The requirement of autophagy for Mincle-mediated NET formation was further supported by exogenous treatment with the autophagy inducer tamoxifen, which rescued the NET formation defect in Mincle−/− neutrophils. These findings identify a previously unrecognized role of Mincle as a regulator of autophagy, which mediates NET formation without affecting ROS generation [72].
Deficient NET formation predisposes humans to severe infection, but, paradoxically, dysregulated NET formation contributes to inflammatory vascular injury and tissue damage. It was found that mTOR regulates NET formation by posttranscriptional control of expression of hypoxia-inducible factor 1 α (HIF-1α), a critical modulator of antimicrobial defenses. mTOR controls NET formation and translation of HIF-1α mRNA in response to LPS. Inhibition of mTOR and HIF-1α expression and activity prevented NET-mediated extracellular bacterial killing [73]. In contrast, another study reported that rapamycin accelerated the rate of NET release following neutrophil stimulation with the bacteria-derived peptide formyl-Met-Leu-Phe (fMLP), while autophagosome formation was enhanced by mTOR inhibitors. Increased mTOR-dependent NET release was sensitive to inhibition of respiratory burst or blockade of cytoskeletal dynamics [74].
Taken together, there is evidence that activated neutrophils are associated with both increased autophagic activity and NET formation. However, it remains unclear whether autophagy is indeed required for NET formation.
Role of autophagy in eosinophils
Eosinophils, like neutrophils and basophils, are bone marrow-derived granulocytes. Although present in all species of vertebrates, eosinophils are still without a firm, exclusive function. There are relatively few mature eosinophils to be found in the peripheral blood of a healthy human (less than 400 per mm3). In response to inflammatory stimuli, eosinophils mature in the bone marrow in increasing numbers and are recruited to and activated in the inflamed tissue(s). In contrast to neutrophils, which are absolutely required for antibacterial defense, pharmacological, or genetic ablation of eosinophils does not result in any obvious functional consequences [75]. Eosinophils are rare cell types and less well studied than neutrophils; hence, the importance of autophagy for regulating eosinophil hematopoiesis and functions remains largely unknown.
Eosinophils in an ovalbumin (OVA)-specific mouse model of allergic asthma showed prominent formation of autophagosomes and increased LC3 expression compared with other inflammatory cells in bronchoalveolar lavage fluid (BALF) and lung tissue. Inhibition of autophagy by 3-MA or Atg5 short-hairpin RNA (shRNA) treatment greatly improved the airway hyper-responsiveness, eosinophilia, and IL-5 levels in BALF, and histological analysis of lung tissue sections revealed decreased numbers of inflammatory cells and mucus-containing goblet cells. Finally, treatment with an anti-IL-5 monoclonal antibody decreased autophagic flux and the number of eosinophils. These findings suggested that IL-5 stimulates autophagy in eosinophils that somehow regulates the severity of eosinophilic inflammation [76].
Under inflammatory conditions, eosinophils can undergo a caspase-independent cell death that is associated with the release of intact granules in the extracellular space. This non-apoptotic cell death is called eosinophil cytolysis. It has recently been reported that eosinophil cytolysis is regulated by components of the necroptotic pathway, which are counter-regulated by rapamycin-induced autophagy [77]. Taken together, although very limited information is available on the regulation of autophagy in eosinophils, the currently available knowledge suggests that autophagy levels in eosinophils can regulate the severity of inflammation.
Differentiation
No information is available regarding the effect of autophagy in the regulation of eosinophil differentiation. However, it has been demonstrated that inhibition of mTORC1 with rapamycin effectively blocks eosinophil differentiation [78]. The authors of this study also observed that rapamycin significantly attenuated OVA-induced allergic airway inflammation and markedly decreased the number of eosinophils in local airways, peripheral blood, and bone marrow, independent of IL-5 levels. In vitro eosinophil colony-forming unit (CFU) assays and liquid cultures demonstrated that rapamycin directly inhibited IL-5-induced eosinophil differentiation. Rapamycin was also capable of reducing the eosinophil levels in IL-5 transgenic mice (line NJ.1638) in vivo, again regardless of constitutive high levels of IL-5. Interestingly, rapamycin inhibition of eosinophil differentiation in turn resulted in an accumulation of eosinophil lineage-committed progenitors in the bone marrow. These results demonstrated a direct inhibitory role for rapamycin in eosinophil differentiation and re-emphasized the importance of rapamycin and possibly, mTOR, in allergic airway disease [78].
The same group studied the role of mTORC1 and mTORC2 in eosinophil differentiation by establishing a genetically modified mouse model with specific deletion of mTOR in myeloid cells [79]. Intriguingly, inhibition of mTOR activity, either by genetic deletion or with the pharmacological inhibitor torin 1, which blocks both mTORC1 and mTORC2 activity, resulted in accelerated eosinophil differentiation, causing elevated levels of eosinophil infiltration in an experimental mouse model of allergy. However, this was not observed to have any considerable effect on eosinophil apoptosis. Ablation of mTOR in myeloid cells also resulted in increased numbers of eosinophil lineage-committed progenitors in allergic mice. The results of the above two studies suggest that mTOR has differential effects on the regulation of eosinophil differentiation, perhaps owing to the distinct functions of mTORC1 and mTORC2 [79]. However, a note of caution is warranted since the latter report is based on a questionable mouse model. The authors used the LysM-cre (Lyz2-Cre) strain to delete mTOR in the eosinophil lineage, which is usually used to ascertain the role of neutrophils, macrophages, and dendritic cell populations in mouse models, but not eosinophils [80].
Eosinophil extracellular traps
Eosinophil extracellular traps (EETs) are part of the innate immune response and seen in multiple infectious, allergic, and autoimmune eosinophilic diseases [81–84]. Interestingly, the DNA within the EETs appears to have its origin in the release of mitochondrial DNA (mtDNA), whereby the cells remain still viable, exhibiting no evidence of a reduced life span. Multiple eosinophil activation mechanisms are represented; TLR as well as cytokine, chemokine, and adhesion receptors can all initiate transmembrane signal transduction processes leading to the formation of EETs capable of killing bacteria. One of the key signaling events required for DNA release is the activation of the NADPH oxidase [85, 86]. So far, there is only one report which studied the requirement of autophagy for EET formation: Neither Atg5-knockout mouse eosinophils nor autophagy-deficient human eosinophils exhibited defects in EET formation [64].
Role of autophagy in mast cells
Mast cells (MCs) are hematopoietic cells that reside in virtually all vascularized tissues and represent potential sources for a wide variety of biologically active secreted products, including diverse cytokines and growth factors. MCs have been proposed to influence many other biological processes, including responses to bacteria and viruses [87]. Since their discovery in 1863, MCs physiological roles have remained unknown, and their most prominent feature has been linked to promoting various chronic inflammatory disorders, such as asthma, allergic rhinitis urticaria, and rheumatic disease, due to their excessive recruitment into inflamed sites. It is now well established that MCs play a crucial role in generating protective innate host responses after infection. Additionally, activated MCs influence the lymph node composition to regulate the induction of adaptive immune responses [88]. Furthermore, MCs as a granule-containing immune cells that are widely distributed in tissues can interact with the external environment. It is well-known that MCs are significantly involved in IgE-mediated allergic reactions, but because of their location, it has also long been hypothesized that MCs can act as sentinel cells that sense pathogens and initiate protective immune responses [89]. However, little is known about the role of autophagy in MCs funtions.
Degranulation
It has been shown that autophagic flux is constitutive in bone marrow-derived mast cells (BMMCs) under fully sufficient nutrient conditions and that LC3-II is located in secretory granules of mast cells [90]. Deletion of Atg7 in BMMCs resulted in a severe impairment in degranulation, but did not impair their differentiation or cytokine production upon FcεRI cross-linking. Intriguingly, LC3-II but not LC3-I was co-localized with CD63, a secretory lysosomal marker, and was released extracellularly along with degranulation in Atg7+/+ but not Atg7−/− BMMCs. Moreover, passive cutaneous anaphylaxis reactions were severely impaired in MC-deficient mice reconstituted with Atg7−/− BMMCs compared with Atg7+/+ BMMCs. These results suggest that autophagy is not essential for their differentiation, but plays a crucial role in the degranulation of mast cells [90] (Fig. 3).
Fig. 3.

The role of autophagy in mast cells function. Autophagy is essential for the degranulation of bone marrow-derived MCs, but has no role in MC differentiation, generation of cysteinyl leukotriens, or cytokine release. Numbers in brackets designate references: + , enhanced function; −, without effect
Role of autophagy in natural killer cells
NK cells originally were defined as large granular lymphocytes with natural cytotoxic ability against tumor cells. They were later recognized as a separate lymphocyte lineage having both cytotoxicity and cytokine-producing effector functions. Having cytotoxic ability makes it important for these cells to be able to discriminate target cells from healthy “self” cells. NK cells are equipped with a variety of cell surface activating and inhibitory receptors that regulate NK cell activities [91]. NK cells are well studied for their anti-tumor and anti-inflammatory functions. Here, we describe some recent reports about the role of autophagy in NK cell differentiation, and tumor infiltration.
Differentiation
Interleukin 15 (IL-15) controls both the homeostasis and the peripheral activation of NK cells. The metabolic checkpoint kinase mTOR is activated and boosts bioenergetic metabolism after exposure of NK cells to high concentrations of IL-15, whereas low doses of IL-15 trigger only phosphorylation of the transcription factor STAT5. mTOR stimulated the growth and nutrient uptake of NK cells and showed a positive feedback on the receptor for IL-15. This process is essential for sustaining NK cell proliferation during differentiation and for the acquisition of a cytolytic potential during inflammation or viral infection [92]. These data suggest that autophagy might be involved in the process of NK cell differentiation.
Indeed, robust autophagy appears in immature NK (iNK) cells and is required for NK cell development. Autophagy protects NK cell viability during development through removal of the damaged mitochondria and consequent reduction in the intracellular ROS levels. Phosphorylated FoxO1 is located in the cytoplasm of iNKs and interacts with ATG7, which promotes autophagy induction. FoxO1 deficiency or an inactive FoxO1AAA mutant abolishes the initiation of autophagy in iNKs and impairs NK cell development and viral clearance [93]. In agreement with this study, NK cell-specific deletion of Atg5 revealed a pronounced reduction in the frequency and absolute numbers of mature NK cells, while mice quickly succumbed to mouse cytomegalovirus (MCMV) infection [94]. Viral infection induces dynamic changes in NK cell mitochondrial quality. As proliferating NK cells accumulate dysfunctional mitochondria during viral infection, a protective mitophagy is induced to promote NK cell survival through removal of the depolarized mitochondria. The autophagic machinery is essential during the transition from effector cells to long-lived memory cells and a pharmacologic induction of autophagy enhances NK cell memory formation through an ATG3-dependent mechanism [95] (Fig. 4).
Fig. 4.

The role of autophagy in differentiation and viral clearance of NK cells. Autophagy is required for the differentiation of mature NK cells from bone marrow-derived HSC. In addition, autophagy is essential for NK cell viral clearance and for enhancement of NK cell memory formation. Numbers in brackets designate references: + , enhanced function
Tumor infiltration
While blocking tumor growth by inhibiting autophagy is a well-established strategy, its role in the infiltration of NK cells into tumors remains unknown. Thus, the impact of targeting Beclin 1 protein expression in tumor cells on the infiltration of NK cells into melanomas was investigated. It was found that, in addition to inhibiting tumor growth, Beclin 1-deficient melanoma cells increased the infiltration of functional NK cells into melanoma tumors. Driving NK cells into the tumor bed relied on the ability of autophagy-defective tumors to transcriptionally overexpress the chemokine (C-C motif) ligand 5 (CCL5) gene. Such infiltration and tumor regression were abrogated by silencing CCL5 in Beclin 1-deficient tumors. Similar to Beclin 1, targeting other autophagy genes such as ATG5 and p62, or inhibiting autophagy pharmacologically with CQ also induced the expression of CCL5 in melanoma cells. Clinically, a positive correlation between CCL5 and the NK cell marker NKp46 expression was found in melanoma patients, and a high expression level of CCL5 correlated with a significant improvement of melanoma patients’ survival. In conclusion, targeting autophagy results in increased NK infiltration into melanoma tissues [96].
Concluding remarks
Autophagy is a key pathway required to fulfill important functions of cells of the innate immune system. As each cell undergoes dramatic changes during differentiation, it is not surprising that autophagy is also involved in this process. On the other hand, autophagy also modifies cell-specific functions of neutrophils, such as phagocytosis, cytokine release, and degranulation. Autophagy is essential for NK cell viral clearance and for enhancement of NK cell memory function, and is required for bone marrow-derived mast cell degranulation, but not for differentiation. The role of autophagy in the pathophysiology of inflammatory diseases represents an exciting new field of investigation. A better understanding of autophagy functions will doubtless suggest novel therapy options.
Acknowledgements
The authors apologize to investigators whose contributions may not have been cited because of space limitations. The original work of the authors described here was supported by the Swiss National Science Foundation (31003A_173215 to S.Y. and 310030_166473 to H.U.S.) and the European Union Horizon 2020 Research and Innovation Program (Marie Sklodowska-Curie grant no. 642295; MEL-PLEX). N.G. and Z.F. are PhD students of the Graduate School of Cellular and Biomedical Sciences of the University of Bern.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Melino
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP, et al. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ. 2013;20:1415–24. doi: 10.1038/cdd.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu D, Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25:169–79. doi: 10.1038/cdd.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16:1014–24. doi: 10.1038/ni.3273. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 10.Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–76. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niso-Santano M, Malik SA, Pietrocola F, Bravo-San Pedro JM, Mariño G, Cianfanelli V, et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015;34:1025–41. doi: 10.15252/embj.201489363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 14.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–71. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranieri R, Ciaglia E, Amodio G, Picardi P, Proto MC, Gazzerro P, et al. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018;25:353–67. doi: 10.1038/cdd.2017.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Lee Y, Kang Y, Dai B, Perez MR, Pratt M, et al. Hypoxia-induced autophagy of stellate cells inhibits expression and secretion of lumican into microenvironment of pancreatic ductal adenocarcinoma. Cell Death Differ. 2019;26:382–93. doi: 10.1038/s41418-018-0207-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Lambelet M, Terra LF, Fukaya M, Meyerovich K, Labriola L, Cardozo AK, et al. Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic β-cell apoptosis. Cell Death Dis. 2018;9:96. doi: 10.1038/s41419-017-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun J, Mu Y, Jiang Y, Song R, Yi J, Zhou J, et al. Inhibition of p70 S6 kinase activity by A77 1726 induces autophagy and enhances the degradation of superoxide dismutase 1 (SOD1) protein aggregates. Cell Death Dis. 2018;9:407. doi: 10.1038/s41419-018-0441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Jiang Y, Sun J, Geng S, Pan Z, Prinz RA, et al. Activation of TGF-β-activated kinase 1 (TAK1) restricts Salmonella Typhimurium growth by inducing AMPK activation and autophagy. Cell Death Dis. 2018;9:570. doi: 10.1038/s41419-018-0612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sridharan S, Jain K, Basu A. Regulation of autophagy by kinases. Cancers. 2011;3:2630–54. doi: 10.3390/cancers3022630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signalling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon HU, Friis R, Tait SWG, Ryan KM. Retrograde signaling from autophagy modulates stress responses. Sci Signal. 2017;10:eaag2791. doi: 10.1126/scisignal.aag2791. [DOI] [PubMed] [Google Scholar]
- 24.Bitto A, Lerner CA, Nacarelli T, Crowe E, Torres C, Sell C. p62/SQSTM1 at the interface of aging, autophagy, and disease. Age. 2014;36:9626. doi: 10.1007/s11357-014-9626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32:431–6. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 26.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, He Z, Simon HU. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol. 2013;23:352–60. doi: 10.1016/j.semcancer.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol. 2011;11:294–300. doi: 10.1016/j.coph.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12:1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–61. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–40. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, et al. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep. 2016;6:21772. doi: 10.1038/srep21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yousefi S, Simon HU. Autophagy in cells of the blood. Biochim Biophys Acta. 2009;1793:1461–4. doi: 10.1016/j.bbamcr.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 36.Germic N, Frangez Z, Yousefi S, Simon HU Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. (in press). [DOI] [PMC free article] [PubMed]
- 37.Mihalache CC, Simon HU. Autophagy regulation in macrophages and neutrophils. Exp Cell Res. 2012;318:1187–92. doi: 10.1016/j.yexcr.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 38.Ye X, Zhou XJ, Zhang H. Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front Immunol. 2018;9:2334. doi: 10.3389/fimmu.2018.02334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rozman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ. 2015;22:445–56. doi: 10.1038/cdd.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity. 2017;47:466–80. doi: 10.1016/j.immuni.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y, Tan P, Wang X, Yi Y, Hu Y, Wang D, et al. Transcriptomic insights into temporal expression pattern of autophagy genes during monocytic and granulocytic differentiation. Autophagy. 2018;14:558–9. doi: 10.1080/15548627.2018.1425060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin J, Britschgi A, Schläfli AM, Humbert M, Shan-Krauer D, Batliner J, et al. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid Med Cell Longev. 2018;2018:1482795. doi: 10.1155/2018/1482795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rikihisa Y. Glycogen autophagosomes in polymorphonuclear leukocytes induced by rickettsiae. Anat Rec. 1984;208:319–27. doi: 10.1002/ar.1092080302. [DOI] [PubMed] [Google Scholar]
- 44.Mitroulis I, Kourtzelis I, Kambas K, Rafail S, Chrysanthopoulou A, Speletas M, et al. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol. 2010;40:1461–72. doi: 10.1002/eji.200940025. [DOI] [PubMed] [Google Scholar]
- 45.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci. 2009;106:6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One. 2012;7:e51727. doi: 10.1371/journal.pone.0051727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itoh H, Matsuo H, Kitamura N, Yamamoto S, Higuchi T, Takematsu H, et al. Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. J Leukoc Biol. 2015;98:107–17. doi: 10.1189/jlb.4A0813-422RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramachandran G, Gade P, Tsai P, Lu W, Kalvakolanu DV, Rosen GM, et al. Potential role of autophagy in the bactericidal activity of human PMNs for Bacillus anthracis. Pathog Dis. 2015;73:ftv080. doi: 10.1093/femspd/ftv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ullah I, Ritchie ND, Evans TJ. The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun. 2017;23:413–23. doi: 10.1177/1753425917704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–9. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol. 2017;198:1253–62. doi: 10.4049/jimmunol.1601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lula L, Keitelman IA, Sabbione F, Fuentes F, Guzman M, Galletti JG, et al. Autophagy mediates interleukin-1β secretion in human neutrophils. Front Immunol. 2018;9:269. doi: 10.3389/fimmu.2018.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. 2015;12:1731–9. doi: 10.1016/j.celrep.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 54.von Gunten S, Yousefi S, Seitz M, Jakob SM, Schaffner T, Seger R, et al. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the pro-inflammatory cytokine environment. Blood. 2005;106:1423–31. doi: 10.1182/blood-2004-10-4112. [DOI] [PubMed] [Google Scholar]
- 55.von Gunten S, Schaub A, Vogel M, Stadler BM, Miescher S, Simon HU. Immunological and functional evidence for anti-Siglec-9 autoantibodies in intravenous immunoglobulin (IVIg) preparations. Blood. 2006;108:4255–9. doi: 10.1182/blood-2006-05-021568. [DOI] [PubMed] [Google Scholar]
- 56.Mihalache CC, Yousefi S, Conus S, Villiger PM, Schneider EM, Simon HU. Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J Immunol. 2011;186:6532–42. doi: 10.4049/jimmunol.1004055. [DOI] [PubMed] [Google Scholar]
- 57.Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011;18:1457–69. doi: 10.1038/cdd.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J Immunol. 2016;197:4090–4100. doi: 10.4049/jimmunol.1600051. [DOI] [PubMed] [Google Scholar]
- 59.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 60.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 61.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 62.Yousefi S, Simon HU. NETosis - does it really represent nature’s “suicide bomber”? Front Immunol. 2016;7:328. doi: 10.3389/fimmu.2016.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, et al. Untangling “NETosis” from NETs. Eur J Immunol. (in press). [DOI] [PubMed]
- 64.Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology. 2017;152:517–25. doi: 10.1111/imm.12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Remijsen Q, van den Berghe T, Wirawan E, Asselbergh B, Parthoens E, de Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One. 2011;6:e29318. doi: 10.1371/journal.pone.0029318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol. 2016;7:879. doi: 10.3389/fmicb.2016.00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol. 2016;7:484. doi: 10.3389/fimmu.2016.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med. 2017;196:577–89. doi: 10.1164/rccm.201603-0596OC. [DOI] [PubMed] [Google Scholar]
- 70.Apostolidou E, Skendros P, Kambas K, Mitroulis I, Konstantinidis T, Chrysanthopoulou A, et al. Neutrophil extracellular traps regulate IL-1β-mediated inflammation in familial Mediterranean fever. Ann Rheum Dis. 2016;75:269–77. doi: 10.1136/annrheumdis-2014-205958. [DOI] [PubMed] [Google Scholar]
- 71.Skendros P, Chrysanthopoulou A, Rousset F, Kambas K, Arampatzioglou A, Mitsios A, et al. Regulated in development and DNA damage responses 1 (REDD1) links stress with IL-1β-mediated familial Mediterranean fever attack through autophagy-driven neutrophil extracellular traps. J Allergy Clin Immunol. 2017;140:1378–1387. doi: 10.1016/j.jaci.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 72.Sharma A, Simonson TJ, Jondle CN, Mishra BB, Sharma J. Mincle-mediated neutrophil extracellular trap formation by regulation of autophagy. J Infect Dis. 2017;215:1040–8. doi: 10.1093/infdis/jix072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood. 2012;120:3118–25. doi: 10.1182/blood-2012-01-405993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Itakura A, McCarty OJ. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. 2013;305:C348–C354. doi: 10.1152/ajpcell.00108.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Radonjic-Hoesli S, Valent P, Klion AD, Wechsler ME, Simon HU. Novel targeted therapies for eosinophil-associated diseases and allergy. Annu Rev Pharm Toxicol. 2015;55:633–56. doi: 10.1146/annurev-pharmtox-010814-124407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016;48:e243. doi: 10.1038/emm.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radonjic-Hoesli S, Wang X, de Graauw E, Stoeckle C, Styp-Rekowska B, Hlushchuk R, et al. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J Allergy Clin Immunol. 2017;140:1632–42. doi: 10.1016/j.jaci.2017.01.044. [DOI] [PubMed] [Google Scholar]
- 78.Hua W, Liu H, Xia LX, Tian BP, Huang HQ, Chen ZY, et al. Rapamycin inhibition of eosinophil differentiation attenuates allergic airway inflammation in mice. Respirology. 2015;20:1055–65. doi: 10.1111/resp.12554. [DOI] [PubMed] [Google Scholar]
- 79.Zhu C, Xia L, Li F, Zhou L, Weng Q, Li Z, et al. mTOR complexes differentially orchestrates eosinophil development in allergy. Sci Rep. 2018;8:6883. doi: 10.1038/s41598-018-25358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. 2011;127:1260–6. doi: 10.1016/j.jaci.2010.12.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon HU. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol. 2011;127:194–9. doi: 10.1016/j.jaci.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 83.Yousefi S, Simon D, Simon HU. Eosinophil extracellular DNA traps: molecular mechanisms and potential roles in disease. Curr Opin Immunol. 2012;24:736–9. doi: 10.1016/j.coi.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 84.Simon D, Radonjic-Hösli S, Straumann A, Yousefi S, Simon HU. Active eosinophilic esophagitis is characterized by epithelial barrier defects and eosinophil extracellular trap formation. Allergy. 2015;70:443–52. doi: 10.1111/all.12570. [DOI] [PubMed] [Google Scholar]
- 85.Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- 86.Morshed M, Yousefi S, Stöckle C, Simon HU, Simon D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy. 2012;67:1127–37. doi: 10.1111/j.1398-9995.2012.02868.x. [DOI] [PubMed] [Google Scholar]
- 87.Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. 2018;282:121–50. doi: 10.1111/imr.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson-Weaver B, Choi HW, Abraham SN, Staats HF. Mast cell activators as novel immune regulators. Curr Opin Pharmacol. 2018;41:89–95. doi: 10.1016/j.coph.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piliponsky AM, Romani L. The contribution of mast cells to bacterial and fungal infection immunity. Immunol Rev. 2018;282:188–97. doi: 10.1111/imr.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127:1267–76. doi: 10.1016/j.jaci.2010.12.1078. [DOI] [PubMed] [Google Scholar]
- 91.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 92.Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749–57. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang S, Xia P, Huang G, Zhu P, Liu J, Ye B, et al. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun. 2016;7:11023. doi: 10.1038/ncomms11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Sullivan TE, Geary CD, Weizman OE, Geiger TL, Rapp M, Dorn GW, 2nd, et al. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep. 2016;15:1910–9. doi: 10.1016/j.celrep.2016.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015;43:331–42. doi: 10.1016/j.immuni.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci. 2017;114:E9271–E9279. doi: 10.1073/pnas.1703921114. [DOI] [PMC free article] [PubMed] [Google Scholar]