

Abstract
Head and neck squamous cell carcinoma (HNSCC) represents a model of escape from anti-tumor immunity. The high frequency of p53 tumor suppressor loss in HNSCC leads to genomic instability and immune stimulation through the generation of neoantigens. However, the aggressive nature of HNSCC tumors and significant rates of resistance to conventional therapies highlights the ability of HNSCC to evade this immune response. Advances in understanding the role of co-stimulatory and immune checkpoint receptors in HNSCC-mediated immunosuppression lay the foundation for development of novel therapeutic approaches. This article provides an overview of these co-stimulatory and immune checkpoint pathways, as well as a review of preclinical and clinical evidence supporting the modulation of these pathways in HNSCC. Finally, the synergistic potential of combining these approaches is discussed, along with an update of current clinical trials evaluating combinations of immune-based therapies in HNSCC patients.
Keywords: Head and neck cancer, Immune checkpoints, Costimulatory receptors, CD137, CD40, OX40, PD-1, CTLA-4, Cetuximab, Immunotherapy
Background
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world, affecting over 500,000 people each year [1]. While HPV-associated HNSCC responds well to standard anti-cancer therapies, five-year survival rates of carcinogen-induced HNSCC are 60 % or less [1]. This poor prognosis despite advances in chemotherapy, radiation, and surgical protocols highlights the need for treatments with greater efficacy in the HPV- population, and improved toxicity profiles for HPV+ patients. Advances in understanding the role of the immune system in preventing development and growth of HNSCC has led to renewed focus on immune-targeting therapies as a means of achieving these goals.
In a process termed immune surveillance, recognition of non-self antigens on tumor cells allows for their destruction by the host immune system [2]. The high frequency of p53 tumor suppressor loss in HNSCC leads to significant genomic instability and the generation of neoantigens, which can activate the immune system and attract infiltrates of effector T-lymphocytes and natural killer (NK) cells into the tumor [3–5]. These adaptive anti-tumor immune responses have been correlated with improved outcomes in many cancers, including HNSCC [6, 7]. However, in order for a clinically significant cancer to develop, the tumor must escape from this anti-cancer immunity through a variety of mechanisms [8]. HNSCC represents an ideal model for understanding and targeting these mechanisms of immune escape in order to unleash the full power of the immune response that can be induced by its characteristically high genetic alteration rate. On the other hand, HPV+ HNSCC is an excellent model of viral-induced cancer, in which oncoproteins such as E6 and E7 are by definition antigenic and therefore tumor development is predicated upon evasion of antiviral immunity [9].
Once recruited to the tumor microenvironment, T-cells interact with antigen-presenting cells (APCs) at the “immune synapse,” and require two simultaneous signals from APCs before they can be activated to mediate their anti-tumor effects (Fig. 1). The first, “signal one,” occurs through interaction of the T-cell receptor (TCR) on the surface of the T-cell and a major histocompatibility complex (MHC) molecule presenting tumor antigen on the surface of an APC (Fig. 1, blue). The second, “signal two” is made up of interactions between co-stimulatory molecules on the surface of APCs and T-cells, such as B7 on the APC surface and CD28 on the T-cell (Fig. 1, green) [2]. Both of these signals must also occur in the context of a third signal made up of immune-activating cytokines such as IL-12, type I (IFNα/β) or type II (IFNγ) interferon [10, 11].
Fig. 1.
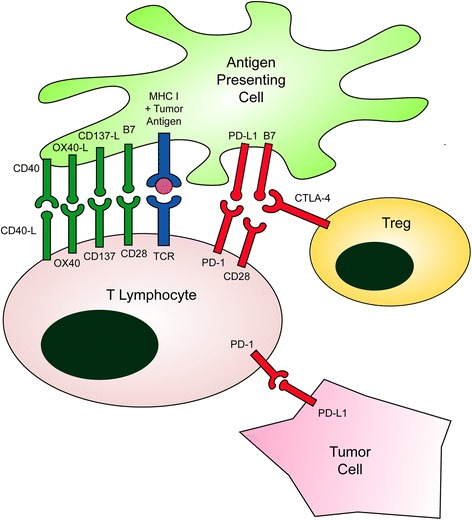
The Immune Synapse. The balance between costimulatory (green) and coinhibitory signals (red) alters the net stimulating effect of TCR signaling mediated through antigen presentation on MHC (blue). Adapted from Ferris RL, J Clin Oncol. 2015;33(29):3293–304
In contrast to these co-stimulatory molecules, the inhibitory “immune checkpoints” prevent T-cell activation. These checkpoints normally function to prevent exaggerated immune responses and subsequent autoimmune disease. However, HNSCC subverts this physiologic function in order to suppress tumor-directed immune activation. The two best known checkpoints include programmed death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), both of which are the targets of FDA-approved inhibitory antibodies [12].
Like other cancers, HNSCC tumor cells mediate immunosuppression in the tumor microenvironment (TME) through mechanisms including upregulation of PD-L1 expression and release of immunosuppressive factors [13]. In addition, recruitment or differentiation of immunosuppressive regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) represent key mechanisms of immune escape. This review will focus on co-stimulatory receptors, inhibitory checkpoint receptors, and combination immunotherapies in HNSCC.
Review
Activating co-stimulatory receptors to enhance anti-tumor immune responses
As mentioned previously, the co-stimulatory receptors that make up “signal two” play a central role in the activation of tumor-fighting T-cells. Absence of this co-stimulatory signal can lead to induction of T-cell anergy or apoptosis and decreased strength of the immune response. In addition to the classically described CD28/B7, other key co-stimulatory interactions between T-cells and APCs, respectively, include CD137/CD137-L, OX40/OX40-L, and CD40-L/CD40 (Fig. 1). These costimulatory receptors are members of the tumor necrosis factor receptor superfamily. Decreased expression of CD137, OX40, CD27, and CD28 have been observed on T-cells derived from HNSCC patients compared to those from healthy controls, emphasizing the potential benefits of targeting these co-stimulatory pathways in this population [14–16]. Agonists of co-stimulatory receptors are currently under investigation in multiple trials for HNSCC and other malignancies. Liver toxicity and cytokine storm symptoms have been reported with costimulatory agonists, but these immune-related adverse events (irAEs) may be dose-dependent. Though extensive information on irAEs of these agents is not yet available, they appear to be well tolerated [17].
CD137
CD137 (also known as 4-1BB) is a costimulatory receptor expressed on the surface of activated T-cells, NK cells, and dendritic cells. When bound by its ligand (CD137-L) on the surface of APCs such as macrophages, dendritic cells, and B cells, trimerization of activated CD137 enhances proliferation, cytotoxic capacity, and survival of T-cells [18]. Stimulation of CD137 with an anti-CD137 monoclonal antibody (mAb) has been shown to induce T-cell mediated eradication of established solid tumors in mice [19]. Although anti-CD137 mAb has not been effective as a monotherapy in HNSCC models, it has been shown to synergize with chemoradiation in a model of HPV+ HNSCC to inhibit tumor growth [20, 21].
Two humanized monoclonal antibodies (mAb) against CD137 have been developed including urelumab (IgG4) and PF-05082566 (IgG2) [12]. These antibodies have been evaluated in early phase trials in melanoma, non-small cell lung cancer (NSCLC) and lymphoma, as monotherapy or in combination with rituximab [12]. Studies of CD137 mAb in combination with other immunotherapies are underway in solid cancers including HNSCC, and are discussed further below.
CD40-L
CD40-L expressed on the surface of activated CD4 T-cells binds to CD40 on APCs, playing a key role in the “helper” T-cell function to activate APCs to prime CD8 T-cells [22]. Expression of both CD40 and CD40-L decrease with increasing HNSCC stage, and surgical resection results in increased APC expression of CD40 [23]. These data implicate downregulation of this co-stimulatory pathway in HNSCC immune escape, with subsequent reversal following surgical resection of the tumor bulk. In addition to its expression on immune cells, CD40 has also been identified on HNSCC cell lines and human HNSCC tumors [24, 25]. The precise role of CD40 in this context is controversial, as ligation of CD40 has been shown to inhibit growth of HNSCC cell lines while also inhibiting cancer cell apoptosis and increasing secretion of proangiogenic cytokines [24, 25]. In vitro studies of agonistic CD40 mAb induced APC activation and maturation, and recombinant CD40L increased the ability of APCs to cross-prime naïve T-cells to tumor antigens [26, 27]. These data suggest a role for this approach in augmenting responses to tumor vaccines, which has been demonstrated in a murine solid tumor model [28].
A variety of CD40-targeting therapies have been developed, including agonistic mAbs and recombinant ligands. Although this approach has not been studied specifically in the HNSCC population, one HNSCC patient treated in a phase I trial of recombinant CD40-L experienced a durable and complete response [29]. Phase I trials of agonistic CD40 mAb alone and in combination with standard therapies have shown promise in a variety of solid tumors, encouraging further studies of this approach in combination with other immune-targeting therapies [30–32].
OX40
Like CD137, OX40 is a co-stimulatory molecule expressed on the T-cell surface that promotes T-cell proliferation, cytokine secretion and memory function when bound by its ligand (OX40-L) [12]. The relevance of OX40 to the local immune response in HNSCC has been demonstrated by the observation that close to 30% of T-cells within the tumor and tumor-draining lymph nodes of HNSCC patients expressed OX40, compared to none of the peripheral blood mononuclear cells (PBMCs) [33]. OX40 is also expressed on regulatory T-cells (Tregs), and appears to inhibit Treg-mediated immunosuppression [34]. Although not explicitly evaluated in HNSCC models, OX40 agonism has improved tumor-free survival in a number of solid tumor models through expansion of tumor-specific CD4+ T-cells [34]. An agonistic OX40 mAb was also shown to synergize with cytolytic therapy through apoptosis of Tregs and enhanced CD8 T-cell response [35]. In a sarcoma model, surgical resection followed by adjuvant anti-OX40 treatment resulted in improved survival and increased antigen-specific T-cell proliferation compared to surgical treatment alone [36].
A phase I trial of the murine agonistic anti-human OX40 mAb 9B12 in patients with refractory solid tumors demonstrated a mild toxicity profile and promising immunologic correlates (NCT01644968) [37]. Although no objective responses were observed, 12 out of the 30 patients experienced regression of at least one metastatic tumor deposit [37]. Additional therapies targeting OX40, including the anti-OX40 mAbs MEDI6469, MEDI0562, PF-04518600, and the OX40L fusion protein MEDI6383, are currently in phase I trials in patients with advanced solid tumors (NCT02205333, NCT02318394, NCT02315066, NCT02221960). In addition, based on the promising preclinical data combining anti-OX40 treatment with surgery [36], a phase Ib trial of OX40 agonistic mAb MEDI6469 prior to definitive surgical resection is currently recruiting patients with locoregionally advanced HNSCC (NCT02274155).
Inhibiting immune checkpoint receptors to enhance anti-tumor immune responses
A reciprocal approach to agonism of co-stimulatory receptors is the inhibition of the immunosuppressive checkpoint receptors. These molecules are upregulated by immune activation, and serve a physiologic role by preventing excessive inflammation and autoimmune disease. However, when overexpressed in the TME, these checkpoints contribute to tumor-promoting immunosuppression, and therefore represent a promising target for disinhibiting immune responses against tumor cells and improving HNSCC patient outcomes. Immune-related adverse events (irAEs) may occur in patients treated with these drugs, including rash, gastrointestinal symptoms, thyroid disorders or autoimmune pneumonitis; however these irAEs are now easily recognized and treated with steroids and/or cessation of the drug in most cases [38].
PD-1/PD-L1
PD-1 is expressed by activated CD8 T-cells, NK cells, B cells, monocytes, and dendritic cells, and normally serves to prevent overactivation of the immune response [12]. However, chronic antigen exposure can lead to chronic upregulation of PD-1 and subsequent T-cell fatigue [39]. Ligation by PD-L1 inhibits activation signaling through the TCR. PD-L1 is expressed by the majority of HNSCC tumors, and blockade of PD-L1 has been shown to synergize with T-cell immunotherapy in an animal model of HNSCC [40]. CD8 T-cells derived from HPV+ HNSCC samples expressed high levels of PD-1, and HPV+ HNSCC cells were observed to express greater levels of PD-L1 compared to HPV- samples [41, 42]. In addition, infiltration of T-cells expressing PD-1 has been associated with a better prognosis in HPV+ disease, emphasizing the role of prior immune activation in patient prognosis [42].
A variety of antibodies have been developed against both PD-1 and PD-L1, including pembrolizumab (anti-PD1 mAb; FDA approved for HNSCC, melanoma and NSCLC) and nivolumab (anti-PD1; FDA approved for melanoma, NSCLC, and renal cell carcinoma). Numerous clinical trials targeting the PD-1/PD-L1 pathway have been extensively discussed in recent reviews [2, 13, 43]. Therefore we will only briefly highlight a few current late-phase trials in HNSCC, then further discuss checkpoint inhibitor combination trials below. Of note, the Keynote 40 and Keynote 48 phase III trials comparing pembrolizumab to standard of care are currently recruiting patients with recurrent or metastatic HNSCC who have failed platinum-based treatment (NCT02252042), or as first-line therapy (NCT02358031). The Keynote 55 phase II trial is evaluating pembrolizumab in HNSCC patients who have failed both platinum and cetuximab therapy (NCT02255097). Pembrolizumab was recently FDA approved based on long-term data from the KEYNOTE-012 trial including HNSCC patients with recurrent or metastatic HNSCC on or following platinum-based chemotherapy. KEYNOTE-012 showed an overall response rate of 17.7 %, median overall survival of 8.5 months, and 6-month progression-free survival (PFS) rate of 25 %; 12 % of patients had grade 3–4 adverse events [44]. The Checkmate 141 phase III trial comparing nivolumab to investigator’s choice in patients with recurrent or metastatic, platinum-refractory HNSCC (NCT02105636) was stopped early due to significant survival benefit including a 30 % reduction in risk of death and doubling of one-year survival from 17 to 36 % [45]. Median overall survival in Checkmate 141 was 7.5 months with nivolumab vs. 5.1 months for investigator’s choice therapy; overall response rate ranged from 18–33 %, with higher response rates noted in patients whose tumors expressed higher levels of PD-L1 [46]. Similar to KEYNOTE-012, in Checkmate 141 the rate of grade 3–4 adverse events was 13 % [45, 46].
CTLA-4
CTLA-4 is transiently expressed by activated T-cells upon binding of an antigen-bearing MHC molecule to the TCR, thereby limiting exaggerated immune responses [47]. CTLA-4 is also constitutively expressed on the surface of Tregs in the HNSCC microenvironment [48]. In animal models, CTLA-4 expression was necessary to the immunosuppressive function of Tregs, and conditional knockout of CTLA-4 in Tregs protected from tumor development [49]. CTLA-4 binds B7 ligands CD80 and CD86 with higher affinity than CD28, thereby competitively inhibiting “signal two” in the T-cell activation cascade [47]. Preclinical studies in solid tumor models demonstrated regression of established tumors and the rejection of further tumor challenge following anti-CTLA-4 mAb treatment [50].
Since that time, two humanized anti-CTLA-4 mAbs, ipilimumab (IgG1) and tremelimumab (IgG2), have been developed and evaluated in phase III trials in advanced melanoma [47]. Based on results of two phase III trials of ipilimumab demonstrating enhanced survival and improved tumor responses, ipilimumab became the first FDA-approved checkpoint inhibitor in 2011 [51, 52]. For patients with platinum-refractory, recurrent or metastatic, PD-L1-negative HNSCC, an ongoing phase II/III study includes tremelimumab and durvalumab (anti-PD-L1) as separate monotherapies or in combination (NCT02319044). Trials combining multiple checkpoint inhibitors are further discussed below.
LAG-3
LAG-3 is another inhibitory checkpoint that is expressed on the surface of Tregs in HNSCC patients [53]. LAG-3 has been identified as a key regulatory molecule involved in prevention of autoimmune disease, as well as the development of tumor tolerance [54, 55]. Knockout of LAG-3 in murine models has been shown to reduce the immunosuppressive activity of Tregs, and conversely ectopic expression of LAG-3 has been shown to confer immunosuppressive capacity upon CD4 T-cells [56]. In addition to playing a role in the immunosuppressive functions of Tregs, LAG-3 expression has also been observed on effector CD8 T-cells at the immunologic synapse [57]. Anti-LAG-3 mAb treatment in solid tumor models has shown success in inhibiting primary tumor growth through activation of antigen-specific T-cells in the TME [55]. In murine solid tumor models, LAG-3 and PD-1 co-expression has been identified on the surface of TILs, and combination anti-LAG-3 and anti-PD-1 antibody treatment cured the majority of established tumors in mice [58]. Early phase clinical trials evaluating anti-LAG-3 mAb in combination with other checkpoint inhibitors are reviewed in Table 1.
Table 1.
Current Combination Immunotherapy Trials Including HNSCC Patients
| Targets | Treatments | Phase | Clinical Trial ID | Patient Eligibility | Status |
|---|---|---|---|---|---|
| Costimulatory/Checkpoint Combinations | |||||
| CD137 (4-1BB) PD-L1 |
PF-05082566 + Avelumab | Ib/II | NCT02554812 | Advanced/metastatic solid tumors | Recruiting |
| CD137 (4-1BB) PD-1 |
PF-05082566 + Pembrolizumab | I | NCT02179918 | Advanced/metastatic solid tumors | Recruiting |
| OX40 PD-L1 |
MEDI6383 +/−Durvalumab | I | NCT02221960 | Recurrent or metastatic solid tumors | Recruiting |
| OX40 CTLA-4 PD-L1 |
MEDI6469 Alone, + Tremelimumab, or + Durvalumab | Ib/II | NCT02205333 | Advanced solid tumors | Ongoing, not recruiting |
| CD27 PD-L1 |
Varlilumab + Atezolizumab | I/II | NCT02543645 | Advanced cancers including HNSCC | Recruiting |
| CD27 PD-1 |
Varlilumab + Nivolumab | I/II | NCT02335918 | Advanced solid tumors | Recruiting |
| Checkpoint/Checkpoint Combinations | |||||
| CTLA-4 B7-H3 |
Ipilimumab + MGA271 | I | NCT02381314 | Advanced/metastatic B7-H3+ HNSCC, melanoma, or NSCLC | Recruiting |
| CTLA-4 PD-L1 |
Tremelimumab + Durvalumab | III | NCT02551159 | HNSCC with no prior chemotherapy | Recruiting |
| CTLA-4 PD-L1 |
Tremelimumab + Durvalumab | I | NCT02262741 | Recurrent or metastatic HNSCC | Recruiting |
| CTLA-4 PD-L1 |
Tremelimumab + Durvalumab (monotherapy or combination) | II | NCT02319044 | Recurrent or metastatic HNSCC | Ongoing, not recruiting |
| CTLA-4 PD-L1 Vaccine |
Tremelimumab + Durvalumab + PolyICLC | I/II | NCT02643303 | Advanced solid tumors including HPV- HNSCC or HPV+ HNSCC after prior treatment failure | Not yet recruiting |
| PD-L1 CTLA-4 |
Durvalumab +/−Tremelimumab | III | NCT02369874 | Recurrent or metastatic HNSCC | Recruiting |
| PD-1 B7-H3 |
Pembrolizumab + MGA271 | I | NCT02475213 | B7-H3+ advanced HNSCC | Recruiting |
| PD-L1 HPV E7 |
Durvalumab + ADXS 11–001 |
I/II | NCT02291055 | Recurrent or metastatic HPV-associated HNSCC | Ongoing, not recruiting |
| LAG-3 PD-1 |
BMS-986016 +/− Nivolumab | I | NCT01968109 | Advanced solid tumors | Recruiting |
| LAG-3 PD-1 |
LAG525 +/− PDR001 | I/II | NCT02460224 | Advanced solid tumors | Recruiting |
| TIM-3 PD-1 |
MBG453 +/−PDR001 | I/II | NCT02608268 | Advanced solid malignancies | Recruiting |
| Cetuximab Combinations | |||||
| CD137 (4-1BB) | Urelumab + Cetuximab | Ib | NCT02110082 | Advanced/metastatic HNSCC or CRC | Ongoing, not recruiting |
| CTLA-4 | Iplimumab + Cetuximab + IMRT | Ib | NCT01860430, NCT01935921 | Stage III-IVB HNSCC p16- or intermediate-risk p16+ | Recruiting |
| TLR8 | Cetuximab + SOC Chemo (CDDP + 5-FU) +/− VTX-2337 | II | NCT01836029 | Recurrent or metastatic HNSCC | Ongoing, not recruiting |
| TLR8 | Cetuximab + VTX-2337 window of opportunity before surgery | Ib | NCT02124850 | Stage II-IVA resectable HNSCC | Recruiting |
TIM-3
TIM-3 represents an additional inhibitory checkpoint that has been implicated both in the immunosuppressive function of Tregs and in the exhaustion of effector T cells in the TME. Elevated expression of TIM-3 has been observed on intratumoral Tregs derived from patients with HNSCC and non-small cell lung cancer, and has been observed to correlate with worse clinical outcomes [53, 59]. TIM-3 has also been implicated in the exhaustion of effector T cells through upregulation of TCR signaling [60]. Anti-TIM-3 mAbs have been shown to modestly inhibit solid tumor growth in murine models, and have induced more impressive control of tumor growth in combination with CTLA-4 and PD-1 targeting therapies [61, 62].
B7-H3
Initially identified as a co-stimulatory receptor of T cell function [63], B7-H3 has since been described as a co-inhibitory checkpoint expressed in a variety of tumor types [64, 65]. Although the specific immunological role of B7-H3 in cancer remains controversial, B7-H3 expression has been correlated with poor prognosis in multiple cancer types, including HNSCC [66]. Antibodies targeting the B7-H3 molecule have been shown to exhibit antitumor activity in solid tumor models with surface expression of B7-H3 [67]. Early phase trials combining these agents with other checkpoint inhibitors are currently underway (Table 1).
Combination immunotherapies for maximal enhancement of anti-tumor immune responses
Although many of the above mentioned immunotherapy approaches have shown significant efficacy in certain patients, there is room for improvement with regards to expanding response rates. Current efforts focus on the rational combination of immunotherapy approaches in order to increase the breadth and depth of patient responses (Table 1). It is important to note, however, that combination immunotherapies may increase the frequency and/or severity of immune-related adverse events [38].
Cetuximab
Cetuximab is a human-mouse chimeric IgG1 antibody against epidermal growth factor receptor (EGFR) that is FDA approved as a monotherapy for recurrent/metastatic HNSCC, in combination with radiation therapy for advanced HNSCC, and in combination with chemoradiation for recurrent/metastatic HNSCC [13]. Although more than 80 % of HNSCC tumors overexpress EGFR, only 10-20 % of patients respond to cetuximab treatment [68]. In patients who respond, cetuximab is thought to mediate part of its effect through inhibition of EGFR signaling and downstream proliferation signals. However, evidence suggests that much of the therapeutic effect of cetuximab is derived from activation of NK cells and antibody-dependent cell-mediated cytotoxicity (ADCC) [43]. Extracellular binding of cetuximab to EGFR exposes the constant region (Fc) of cetuximab to binding by the activating Fc receptor (CD16/FcγRIII) expressed on NK cells. This activation signal induces ADCC mediated by NK cells resulting in tumor cell lysis (Fig. 2). The variability of patient response to cetuximab is thought to be in part due to polymorphisms in NK cell FcγRIII, which lead to variation in its affinity for the Fc region of cetuximab [69].
Fig. 2.
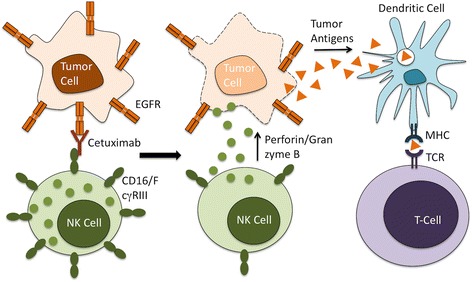
Cetuximab-Mediated ADCC. The Fab portion of Cetuximab binds to EGFR on the surface of tumor cells, while its Fc region binds to the Fc receptor CD16/FcγRIII on the NK cell surface. This leads to NK cell activation and release of cytolytic granules containing perforin and granzyme B that result in tumor cell lysis and release of tumor antigen. This tumor antigen is subsequently presented on APCs to activate antigen-specific T-cells
In addition to mediating ADCC, cetuximab-activated NK cells have been shown to promote maturation of APCs and the development of an adaptive immune response [68]. Recent evidence also suggests that cetuximab treatment decreases the function of immunosuppressive myeloid cells [70]. Increased numbers of monocytic MDSCs were observed in patients who did not respond to cetuximab therapy, suggesting potential for improving responses through combining cetuximab with MDSC-targeting treatments [70]. Given the low response rate to cetuximab as a monotherapy or in combination with standard therapies, recent efforts have focused on combining cetuximab with additional immunotherapies to enhance ADCC.
Human NK cells have been shown to upregulate surface expression of CD137 following exposure to cetuximab and EGFR-expressing cell lines [71]. In preclinical studies, sequential treatment with cetuximab followed by anti-CD137 mAb eradicated established tumors in an NK-cell dependent manner [71]. In addition to this enhancement of ADCC, preclinical evidence also supports a mechanistic role for the adaptive immune “vaccinal effect.” Mice previously cured with this combination therapy rejected rechallenge with both EGFR-positive and negative cell lines, which supports immunologic memory and epitope spreading [72]. Based on this promising preclinical data, a phase Ib trial combining cetuximab with the anti-CD137 mAb urelumab is currently underway (NCT02110082).
In addition to FcγRIII polymorphisms, proposed mechanisms of resistance to cetuximab-mediated ADCC include an increase in the number of Tregs within the HNSCC TME following cetuximab treatment [73]. These CTLA-4+ Tregs were shown to suppress cetuximab-mediated ADCC, and their increased numbers correlated with poor patient prognosis [73]. Ex vivo treatment of HNSCC tumor-infiltrating lymphocytes with the anti-CTLA-4 mAb ipilimumab depleted Tregs and restored NK cell-mediated ADCC [73]. Based on this promising preclinical data, two phase Ib studies combining ipilimumab with intensity modulated radiation therapy (IMRT) and cetuximab are currently recruiting patients with untreated advanced HNSCC (NCT01860430, NCT01935921).
Toll like receptor agonists
Toll-like receptors (TLRs) are transmembrane receptors that recognize microbial invasion and respond through activation of the innate immune system [74]. TLR7 and TLR8 have been particular targets for improving anti-cancer immunity. An early topical TLR7/8 agonist, imiquimod, is FDA approved for actinic keratosis and basal cell carcinoma. In addition, novel stabilized immune-modulatory RNA (SIMRA) compounds are also under study for their dual TLR7/8 agonism [75]. However, recent development of a potent and selective TLR8 agonist has focused attention on this endosomal TLR that is naturally activated by viral single-stranded RNA. Stimulation of TLR8 results in activation of dendritic cells and macrophages and subsequent secretion of immune-activating cytokines. TLR8 signaling has also been implicated in reversal of Treg function [76]. VTX-2337, a TLR8 agonist, has been shown to induce TNFα and IL-12 secretion by monocytes and myeloid dendritic cells, in addition to increasing NK cell cytotoxicity and secretion of IFNγ [77]. VTX-2337 has also been reported to enhance rituximab and trastuzumab-induced ADCC in lymphoma and breast cancer cells lines, respectively [77]. Subsequent preclinical studies using PBMCs from healthy individuals and HNSCC patients demonstrated the ability of VTX-2337 to enhance cetuximab-mediated ADCC against HNSCC cells [78].
Based on these promising preclinical data regarding selective TLR8 agonism, VTX-2337 was studied in a phase I trial in advanced solid tumors [79]. This trial demonstrated clinical tolerability in addition to increases in plasma levels of immune-activating cytokines G-CSF, monocyte chemoattractant protein-1, macrophage inflammatory protein-1β, and TNFα when administered at higher doses. Based on this information, phase II placebo-controlled trials of combination therapy with VTX-2337 have been initiated, including a comparison of chemotherapy + cetuximab + VTX-2337 to chemotherapy + cetuximab alone in recurrent or metastatic HNSCC (NCT01836029). In addition, a phase Ib study of neoadjuvant cetuximab + VTX-2337 vs. cetuximab + VTX-2337 + nivolumab is currently recruiting patients with stage II-IVA surgically resectable HNSCC (NCT02124850).
Checkpoint inhibitors
Although the degree and durability of response to checkpoint inhibitor monotherapy has been impressive, objective response rates remain low. For example, preliminary data from the KEYNOTE-012 expansion cohort showed an objective response in 18.2 % of recurrent/metastatic HNSCC patients treated with pembrolizumab monotherapy [80]. For this reason, much attention is currently directed towards combining checkpoint inhibitors with a variety of immune-based therapies to achieve higher response rates in both preclinical and clinical studies (Table 1).
Signaling through various immune checkpoints and downregulation of costimulatory receptors each represent a distinct mechanism of tumor-mediated immunosuppression. Combining inhibitors that target different checkpoints is a logical strategy to generate synergy and target potential mechanisms of resistance to therapy. For example, melanoma patients with high PD-L1 expression did not respond to anti-CTLA4 mAb and radiation, implicating PD-1/PD-L1 signaling in this resistance (NCT01497808) [81]. However, in preclinical studies of melanoma, combined targeting of CTLA-4 and PD-1 more than doubled the rate of tumor rejection and increased tumor-infiltrating T-cells while reducing Tregs and MDSCs in the TME [82]. Synergism has also been described between antibodies targeting PD-1 and TIM-3 [61], and PD-1 and LAG-3 [58] in solid tumor models, leading to current clinical trials evaluating these combinations in patients with advanced solid tumors (NCT02608268, NCT01968109, NCT02460224).
A phase I trial of combined nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) in advanced melanoma showed an overall response of 40 % and objective responses in 53 % of patients treated with the maximal tolerated dose [83]. These outcomes exceed responses seen with either drug as a monotherapy. Preliminary results from another phase I trial combining durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) in NSCLC patients showed an overall response rate of 25 %, and the interesting finding that this efficacy did not depend on PD-L1 expression in the tumor [84]. Many further studies combining checkpoint inhibitors with one another and with co-stimulatory molecules are currently underway and summarized in Table 1.
In addition, simultaneous agonism of co-stimulatory pathways and antagonism of inhibitory checkpoints allows one to “step on the gas while taking the foot off the brakes.” In solid tumor models, combined treatment with agonistic anti-OX40 mAb and anti-CTLA-4 mAb improved survival and induced tumor regression through expansion of effector CD8 T-cells [85]. Combined targeting of OX40 and PD-L1 or OX40 and CTLA-4 is currently under study in early phase trials in advanced solid tumors (NCT02221960 & NCT02205333). Studies in solid tumor models also demonstrated synergy between mAbs targeting CD137, PD-1, and CTLA-4 [86]. Based on these data, two current phase I/II trials are evaluating the combination of anti-CD137 and anti-PD-1/PD-L1 mAb in advanced solid tumors (NCT02554812 & NCT02179918).
Combination of immunotherapies with standard or targeted therapies
In addition to combination immunotherapies for HNSCC, other promising strategies under investigation include the combination of these agents with standard-of-care or targeted therapies. Studies in HNSCC and other cancer types suggest that radiotherapy, cisplatin chemotherapy, and other cytotoxic drugs may enhance anti-tumor adaptive immunity in the TME [87–90]. As a result, radiation and cisplatin may also increase immune checkpoint expression [89–92]. These findings suggest a strong rationale for combining radiation and/or chemotherapy with checkpoint inhibitors and other immune therapies, and such combinations are currently under study in multiple clinical trials.
The Cancer Genome Atlas and other studies have revealed specific genomic alterations in HNSCC that may be targeted by specific therapies [93]. Although cetuximab is so far the only targeted agent that is FDA-approved for HNSCC, a myriad of targeted agents are under investigation in preclinical and clinical studies of HNSCC. Future treatment strategies are likely to utilize combinations of targeted agents with immune and standard therapies.
HPV-specific immunotherapies
As mentioned above, HPV-associated HNSCC represents distinct mechanisms of antiviral immune escape, and this disease entity also may benefit from specific antiviral immunotherapies. Since patients with HPV-associated disease generally have an excellent prognosis and high cure rates, newer therapeutic strategies have focused on improving upon long-term toxicities seen with current therapies. Therapeutic vaccines and adoptive transfer of immune cells have been studied in HPV-associated HNSCC [2] and will be combined with surgery, chemoradiation, targeted therapies or other immune therapies in ongoing and future trials. Trials of combination immunotherapies specific to patients with HPV-associated disease are detailed in Table 1.
Conclusions
Recent advances in understanding the balance between costimulatory and inhibitory immune pathways at the immune synapse have encouraged interest in re-directing these signals from tumor-promoting immunosuppression towards tumor-fighting immunity. With the development of an array of costimulatory agonists and checkpoint inhibitors, these immune-based strategies have become a focal point for research in many cancers, including HNSCC. In addition to focusing on clinical application of these novel immunotherapies, much work is underway to investigate mechanisms of resistance in those patients who do not achieve durable responses. Rational design of combination strategies represents a promising approach to target resistance, while care must be taken to avoid immune over-activation and serious autoimmune consequences.
Acknowledgements
Not applicable.
Funding
This work was supported by NIH, National Institute on Deafness and Other Communication Disorders intramural project number ZIA-DC-DC000090. The NIH had no direct role in the writing of this manuscript.
Availability of data and materials
Not applicable.
Authors’ contributions
RJD drafted the manuscript and carried out a literature review, NCS carried out the primary literature review and helped to draft the manuscript, RLF conceived of the review and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
RLF serves as a paid member of committees, panels or boards for Merck, Celgene, Bristol Myers Squibb, and AstraZeneca/Medimmune, and receives research funding from VentiRx, Bristol Myers Squibb, and AstraZeneca/Medimmune.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- APC
Antigen-presenting cell
- CTLA-4
Cytotoxic T lymphocyte-associated protein 4
- EGFR
Epidermal growth factor receptor
- HNSCC
Head and neck squamous cell carcinoma
- HPV
Human papillomavirus
- IMRT
Intensity modulated radiation therapy
- LAG-3
Lymphocyte-activation gene 3
- mAb
Monoclonal antibody
- MDSC
Myeloid-derived suppressor cell
- MHC
Major histocompatibility complex
- NSCLC
Non small-cell lung cancer
- PBMC
Peripheral blood mononuclear cell
- PD-1
Programmed death-1
- PD-L1
programmed death-1 ligand
- TCR
T-cell receptor
- TIM-3
T-cell immunoglobulin mucin 3
- TLR
Toll-like receptor
- TLR
Toll-like receptor
- TME
Tumor microenvironment
- Treg
Regulatory T-cell
Contributor Information
Ruth J. Davis, Email: ruth.davis2@nih.gov
Robert L. Ferris, Phone: 412-623-0327, Email: Ferrrl@upmc.edu
Nicole C. Schmitt, Email: nicole.schmitt@nih.gov
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol. 2015;33:3293–304. doi: 10.1200/JCO.2015.61.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Champiat S, Ferte C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology. 2014;3:e27817. doi: 10.4161/onci.27817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keck MK, Zuo Z, Khattri A, Stricker TP, Brown CD, Imanguli M, et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin Cancer Res. 2015;21:870–81. doi: 10.1158/1078-0432.CCR-14-2481. [DOI] [PubMed] [Google Scholar]
- 5.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balermpas P, Michel Y, Wagenblast J, Seitz O, Weiss C, Rodel F, et al. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br J Cancer. 2014;110:501–9. doi: 10.1038/bjc.2013.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schantz SP, Shillitoe EJ, Brown B, Campbell B. Natural killer cell activity and head and neck cancer: a clinical assessment. J Natl Cancer Inst. 1986;77:869–75. [PubMed] [Google Scholar]
- 8.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ressing ME, Sette A, Brandt RM, Ruppert J, Wentworth PA, Hartman M, et al. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154:5934–43. [PubMed] [Google Scholar]
- 10.Sckisel GD, Bouchlaka MN, Monjazeb AM, Crittenden M, Curti BD, Wilkins DE, et al. Out-of-Sequence Signal 3 Paralyzes Primary CD4(+) T-Cell-Dependent Immunity. Immunity. 2015;43:240–50. doi: 10.1016/j.immuni.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22:333–40. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauman JE, Ferris RL. Integrating novel therapeutic monoclonal antibodies into the management of head and neck cancer. Cancer. 2014;120:624–32. doi: 10.1002/cncr.28380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen CT, Clavijo PE, Van Waes C, Chen Z. Anti-Tumor Immunity in Head and Neck Cancer: Understanding the Evidence, How Tumors Escape and Immunotherapeutic Approaches. Cancers (Basel) 2015;7:2397–414. doi: 10.3390/cancers7040900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baruah P, Lee M, Odutoye T, Williamson P, Hyde N, Kaski JC, et al. Decreased levels of alternative co-stimulatory receptors OX40 and 4-1BB characterise T cells from head and neck cancer patients. Immunobiology. 2012;217:669–75. doi: 10.1016/j.imbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Kuss I, Donnenberg AD, Gooding W, Whiteside TL. Effector CD8 + CD45RO-CD27-T cells have signalling defects in patients with squamous cell carcinoma of the head and neck. Br J Cancer. 2003;88:223–30. doi: 10.1038/sj.bjc.6600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukishiro T, Donnenberg AD, Whiteside TL. Rapid turnover of the CD8(+)CD28(−) T-cell subset of effector cells in the circulation of patients with head and neck cancer. Cancer Immunol Immunother. 2003;52:599–607. doi: 10.1007/s00262-003-0395-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan JM, Wasser JS, Adler AJ, Vella AT. Enhancing the safety of antibody-based immunomodulatory cancer therapy without compromising therapeutic benefit: Can we have our cake and eat it too? Expert Opin Biol Ther. 2016;16:655–74. doi: 10.1517/14712598.2016.1152256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynch DH. The promise of 4-1BB (CD137)-mediated immunomodulation and the immunotherapy of cancer. Immunol Rev. 2008;222:277–86. doi: 10.1111/j.1600-065X.2008.00621.x. [DOI] [PubMed] [Google Scholar]
- 19.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–5. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 20.Vahle AK, Hermann S, Schafers M, Wildner M, Kerem A, Ozturk E, et al. Multimodal imaging analysis of an orthotopic head and neck cancer mouse model and application of anti-CD137 tumor immune therapy. Head Neck. 2016;38:542–9. doi: 10.1002/hed.23929. [DOI] [PubMed] [Google Scholar]
- 21.Lucido CT, Vermeer PD, Wieking BG, Vermeer DW, Lee JH. CD137 enhancement of HPV positive head and neck squamous cell carcinoma tumor clearance. Vaccines (Basel) 2014;2:841–53. doi: 10.3390/vaccines2040841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19:1035–43. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sathawane D, Kharat RS, Halder S, Roy S, Swami R, Patel R, et al. Monocyte CD40 expression in head and neck squamous cell carcinoma (HNSCC) Hum Immunol. 2013;74:1–5. doi: 10.1016/j.humimm.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Cao W, Cavacini LA, Tillman KC, Posner MR. CD40 function in squamous cell cancer of the head and neck. Oral Oncol. 2005;41:462–9. doi: 10.1016/j.oraloncology.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Posner MR, Cavacini LA, Upton MP, Tillman KC, Gornstein ER, Norris CM., Jr Surface membrane-expressed CD40 is present on tumor cells from squamous cell cancer of the head and neck in vitro and in vivo and regulates cell growth in tumor cell lines. Clin Cancer Res. 1999;5:2261–70. [PubMed] [Google Scholar]
- 26.Bergstrom RT, Silverman DA, Chambers K, Kim JA. CD40 monoclonal antibody activation of antigen-presenting cells improves therapeutic efficacy of tumor-specific T cells. Otolaryngol Head Neck Surg. 2004;130:94–103. doi: 10.1016/j.otohns.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann TK, Meidenbauer N, Muller-Berghaus J, Storkus WJ, Whiteside TL. Proinflammatory cytokines and CD40 ligand enhance cross-presentation and cross-priming capability of human dendritic cells internalizing apoptotic cancer cells. J Immunother. 2001;24:162–71. doi: 10.1097/00002371-200103000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med. 1999;5:774–9. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- 29.Vonderheide RH, Dutcher JP, Anderson JE, Eckhardt SG, Stephans KF, Razvillas B, et al. Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol. 2001;19:3280–7. doi: 10.1200/JCO.2001.19.13.3280. [DOI] [PubMed] [Google Scholar]
- 30.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–95. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and biological effects of an agonist anti-CD40 antibody a cancer research UK phase I study. Clin Cancer Res. 2015;21:1321–8. doi: 10.1158/1078-0432.CCR-14-2355. [DOI] [PubMed] [Google Scholar]
- 32.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033. doi: 10.4161/onci.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vetto JT, Lum S, Morris A, Sicotte M, Davis J, Lemon M, et al. Presence of the T-cell activation marker OX-40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am J Surg. 1997;174:258–65. doi: 10.1016/S0002-9610(97)00139-6. [DOI] [PubMed] [Google Scholar]
- 34.Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–9. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 35.Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med. 2009;206:1103–16. doi: 10.1084/jem.20082205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33:798–809. doi: 10.1097/CJI.0b013e3181ee7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–86. doi: 10.1038/nrclinonc.2016.58. [DOI] [PubMed] [Google Scholar]
- 39.Ferris R. PD-1 targeting in cancer immunotherapy. Cancer. 2013;119:E1–3. doi: 10.1002/cncr.27832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–5. [PubMed] [Google Scholar]
- 41.Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73:1733–41. doi: 10.1158/0008-5472.CAN-12-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73:128–38. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- 43.Schoppy DW, Sunwoo JB. Immunotherapy for Head and Neck Squamous Cell Carcinoma. Hematol Oncol Clin North Am. 2015;29:1033–43. doi: 10.1016/j.hoc.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 44.Mehra R, Seiwert TY, Mahipal A, Weiss J, Berger R, Eder JP, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC): Pooled analyses after long-term follow-up in KEYNOTE-012. J Clin Oncol. 34;2016 (suppl; abstr 6012)
- 45.Gillison ML, Blumenschein GJ, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Proceedings of the Annual Meeting of the American Association for Cancer Research. New Orleans: AACR; 2016 Abstract nr CT099; 2016. Nivolumab (nivo) vs investigator’s choice (IC) for recurrent or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): CheckMate-141 [abstract] [Google Scholar]
- 46.Ferris RL, Blumenschein GR, Fayette J, Guigay J, Colevas AD, Licitra LF, et al. Further evaluations of nivolumab (nivo) versus investigator’s choice (IC) chemotherapy for recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CheckMate 141. J Clin Oncol. 34, 2016 (suppl; abstr 6009).
- 47.Honeychurch J, Cheadle EJ, Dovedi SJ, Illidge TM. Immuno-regulatory antibodies for the treatment of cancer. Expert Opin Biol Ther. 2015;15:787–801. doi: 10.1517/14712598.2015.1036737. [DOI] [PubMed] [Google Scholar]
- 48.Strauss L, Bergmann C, Gooding W, Johnson J. T., Whiteside, T. L. The frequency and suppressor function of CD4 + CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–6311. doi: 10.1158/1078-0432.CCR-07-1403. [DOI] [PubMed] [Google Scholar]
- 49.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science (80-) 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 50.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80-) 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 51.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 53.Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, Ferris RL. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109:2629–2635. doi: 10.1038/bjc.2013.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sega EI, Leveson-Gower DB, Florek M, Schneidawind D, Luong RH, Negrin RS. Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PLoS One. 2014;9:e86551. doi: 10.1371/journal.pone.0086551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, Anders R, Netto G, Getnet D, Bruno TC, Goldberg MV, Pardoll DM, Drake CG. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117:3383–92. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DAA. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–13. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 57.Huang R-Y, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359–77. doi: 10.18632/oncotarget.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7:e30676. doi: 10.1371/journal.pone.0030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193:1525–30. doi: 10.4049/jimmunol.1400557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 63.Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269–74. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 64.Suh W-K, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. 2003;4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- 65.Prasad DVR, Nguyen T, Li Z, Yang Y, Duong J, Wang Y, et al. Murine B7-H3 is a negative regulator of T cells. J Immunol. 2004;173:2500–6. doi: 10.4049/jimmunol.173.4.2500. [DOI] [PubMed] [Google Scholar]
- 66.Katayama A, Takahara M, Kishibe K, Nagato T, Kunibe I, Katada A, et al. Expression of B7-H3 in hypopharyngeal squamous cell carcinoma as a predictive indicator for tumor metastasis and prognosis. Int J Oncol. 2011;38:1219–26. doi: 10.3892/ijo.2011.949. [DOI] [PubMed] [Google Scholar]
- 67.Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. 2012;18:3834–45. doi: 10.1158/1078-0432.CCR-12-0715. [DOI] [PubMed] [Google Scholar]
- 68.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–72. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li J, Srivastava RM, Ettyreddy A, Ferris RL. Cetuximab ameliorates suppressive phenotypes of myeloid antigen presenting cells in head and neck cancer patients. J Immunother Cancer. 2015;3:54. doi: 10.1186/s40425-015-0097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, Mueller A, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124:2668–82. doi: 10.1172/JCI73014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Houot R, Kohrt H. CD137 stimulation enhances the vaccinal effect of anti-tumor antibodies. Oncoimmunology. 2014;3:e941740. doi: 10.4161/21624011.2014.941740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015;75:2200–10. doi: 10.1158/0008-5472.CAN-14-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27:190–9. doi: 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- 75.Lan T, Kandimalla ER, Yu D, Bhagat L, Li Y, Wang D, et al. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc Natl Acad Sci U S A. 2007;104:13750–5. doi: 10.1073/pnas.0706059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–4. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 77.Lu H, Dietsch GN, Matthews MA, Yang Y, Ghanekar S, Inokuma M, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- 78.Stephenson RM, Lim CM, Matthews M, Dietsch G, Hershberg R, Ferris RL. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother. 2013;62:1347–57. doi: 10.1007/s00262-013-1437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Northfelt DW, Ramanathan RK, Cohen PA, Von Hoff DD, Weiss GJ, Dietsch GN, et al. A phase I dose-finding study of the novel Toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin Cancer Res. 2014;20:3683–91. doi: 10.1158/1078-0432.CCR-14-0392. [DOI] [PubMed] [Google Scholar]
- 80.Seiwert TY, Haddad RI, Gupta S, Mehra R, Tahara M, Berger R, et al. Antitumor activity and safety of pembrolizumab in patients (pts) with advanced squamous cell carcinoma of the head and neck (SCCHN): Preliminary results from KEYNOTE-012 expansion cohort. J Clin Oncol. 33, 2015 (suppl; abstr LBA6008)
- 81.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci. 2010;107:4275–80. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rizvi N, Chaft J, Balmanoukian A, Goldberg SB, Sanborn RE, Steele KE, et al. Tumor response from durvalumab (MEDI4736) + tremelimumab treatment in patients with advanced non-small cell lung cancer (NSCLC) is observed regardless of PD-L1 status. J Immunother Cancer. 2015;3:193. doi: 10.1186/2051-1426-3-S2-P193. [DOI] [Google Scholar]
- 85.Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014;2:142–53. doi: 10.1158/2326-6066.CIR-13-0031-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai M, Wei H, Yip YY, Feng Q, He K, Popov V, et al. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J Immunother. 2013;36:248–57. doi: 10.1097/CJI.0b013e3182943549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20:5384–91. doi: 10.1158/1078-0432.CCR-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hato SV, Khong A, de Vries IJM, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res. 2014;20:2831–7. doi: 10.1158/1078-0432.CCR-13-3141. [DOI] [PubMed] [Google Scholar]
- 89.Dovedi SJ, Illidge TM. The antitumor immune response generated by fractionated radiation therapy may be limited by tumor cell adaptive resistance and can be circumvented by PD-L1 blockade. Oncoimmunology. 2015;4:e1016709. doi: 10.1080/2162402X.2015.1016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmitt NC, Cash H, Ferris RL, Van Waes C, Allen CT. Cisplatin Increases Expression of Antigen Presentation Machinery and Programmed Death Ligand 1 in Head and Neck Squamous Cell Carcinoma Cells. Abstract for 9th International Conference on Head and Neck Cancer, American Head and Neck Society, 2016.
- 91.Sridharan V, Margalit DN, Curreri SA, Severgnini M, Hodi FS, Haddad RI, et al. Systemic Immunologic Effects of Definitive Radiation in Head and Neck Cancer. Int J Radiat Oncol. 2016;94:864. doi: 10.1016/j.ijrobp.2015.12.023. [DOI] [Google Scholar]
- 92.Parikh F, Duluc D, Imai N, Clark A, Misiukiewicz K, Bonomi M, et al. Chemoradiotherapy-induced upregulation of PD-1 antagonizes immunity to HPV-related oropharyngeal cancer. Cancer Res. 2014;74:7205–16. doi: 10.1158/0008-5472.CAN-14-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.