
Figure 1.
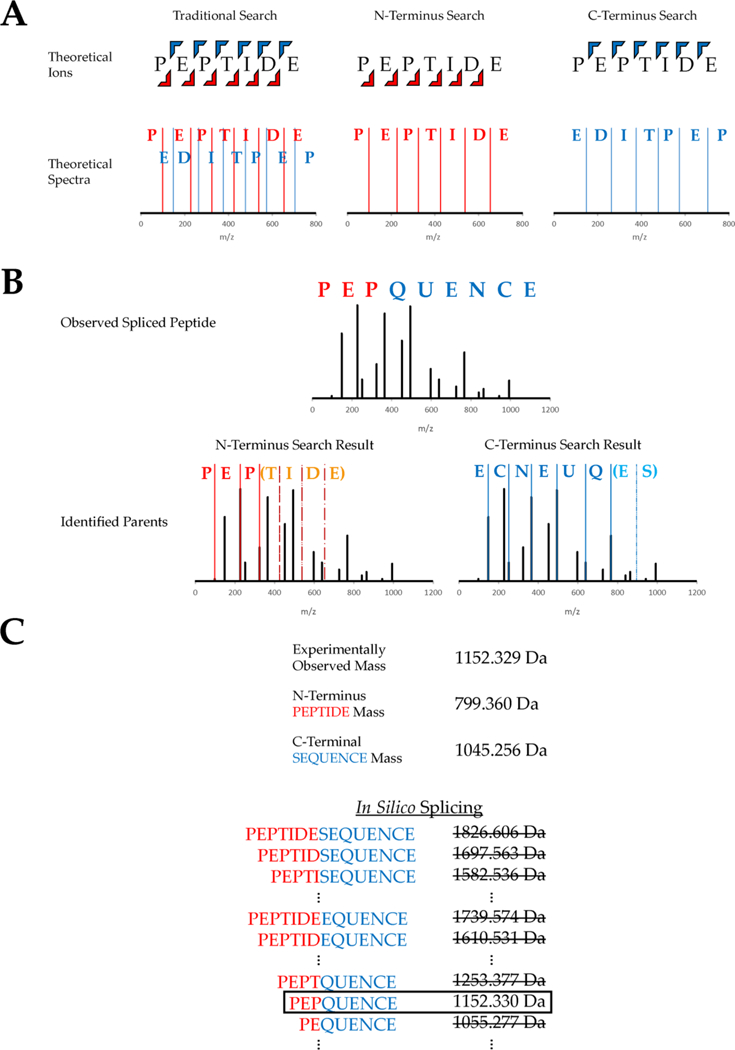
Workflow of the database search algorithm used to identify spliced peptides. (A) Two separate databases are generated containing only N-terminal ions or C-terminal ions. (B) Experimental spectra are compared against both databases in an open mass tolerance search, and the highest scoring match from each search is recorded. This example shows an experimental spectrum for the spliced peptide “PEP-QUENCE, derived from parent peptides “PEPTIDE” and “SEQUENCE”. Both parents are identified from the separate ion database searches. (C) The two parent peptide assignments have different precursor masses than the experimental spectrum, and in silico splicing is used to identify the full spliced sequence. This is accomplished through a narrow mass tolerance comparison between the observed precursor mass and all theoretical spliced precursor masses that can be obtained from the two parents.