

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a severe lung disease characterised by extensive pathological changes. The objective for this study was to identify the gene network and regulators underlying disease pathology in IPF and its association with lung function.
Methods
Lung Tissue Research Consortium dataset with 262 IPF and control subjects (GSE47460) was randomly divided into two non-overlapping groups for cross-validated differential gene expression analysis. Consensus weighted gene coexpression network analysis identified overlapping coexpressed gene modules between both IPF groups. Modules were correlated with lung function (diffusion capacity, DLCO; forced expiratory volume in 1 s, FEV1; forced vital capacity, FVC) and enrichment analyses used to identify biological function and transcription factors. Module correlation with miRNA data (GSE72967) identified associated regulators. Clinical relevance in IPF was assessed in a peripheral blood gene expression dataset (GSE93606) to identify modules related to survival.
Results
Correlation network analysis identified 16 modules in IPF. Upregulated modules were associated with cilia, DNA replication and repair, contractile fibres, B-cell and unfolded protein response, and extracellular matrix. Downregulated modules were associated with blood vessels, T-cell and interferon responses, leucocyte activation and degranulation, surfactant metabolism, and cellular metabolic and catabolic processes. Lung function correlated with nine modules (eight with DLCO, five with FVC). Intermodular network of transcription factors and miRNA showed clustering of fibrosis, immune response and contractile modules. The cilia-associated module was able to predict survival (p=0.0097) in an independent peripheral blood IPF cohort.
Conclusions
We identified a correlation gene expression network with associated regulators in IPF that provides novel insight into the pathological process of this disease.
Keywords: idiopathic pulmonary fibrosis
Key messages.
What is the key question?
While pathways relating to fibrosis have been associated and studied in-depth in idiopathic pulmonary fibrosis (IPF), other pathways, in particular involving the immune response or honeycombing have not been well identified or characterised in genomic studies.
What is the bottom line?
Pathways identified using gene coexpression network analysis were able to determine a regulatory framework for IPF and association with survival.
Why read on?
This study provides novel insight into the biological pathways in IPF and identified several pathways which warrant further research including dysfunctions in the immune response and blood vessel formation. Moreover, the epithelial signature could be detected in the blood and was related to survival.
Background
Idiopathic pulmonary fibrosis (IPF) is a severe and complex lung diseases characterised by fibrous destruction of the parenchyma and presence of usual interstitial pneumonia with honeycomb changes in the basal and peripheral regions of the lung. While fibrosis is the main feature of this disease and has been examined extensively in transcriptomic and histological studies, other pathways associated with IPF have not been well characterised.
Several studies on IPF have used transcriptome analysis that provided important insight into this disease.1–3 The methods used in these studies have generally been to test genes individually, while in vivo, genes function via networks of coexpressed genes with similar biological function. We hypothesised that identifying these coexpression patterns would provide additional insight into disease-associated biological pathways.
The present study aimed to apply weighted gene coexpression network analysis (WGCNA),4 a systems biology approach for identifying gene interactions in transcriptomic datasets, on differentially expressed genes in IPF previously generated by us (GSE47460)5–7 and stored on the Gene Expression Omnibus (GEO). These data were derived from lung tissue obtained through the National Institutes of Health - National Heart, Lung, and Blood Institute (NIH-NHLBI) funded Lung Tissue Research Consortium (LTRC) and consists of samples from IPF and control subjects who had undergone lung resection or transplantation. Lung function was correlated with gene modules and enrichment analyses used to determine biological function as has been reported in several studies on lung disease.8–10 Encode ChIP-seq enrichment was used to determine transcription factors associated with each module and miRNA expression (LTRC dataset GSE72967) was correlated with each module. Finally, the clinical relevance of these pathways was determined by an IPF peripheral blood RNA dataset (GSE93606) to identify modules related to survival.
Methods
Study population
All data were obtained from GEO datasets GSE47460, GSE72967 and GSE93606 (www.ncbi.nlm.nih.gov/geo). Lung tissue samples were composed of 268 patients who had undergone thoracic surgery. IPF samples (n=160) were from patients with interstitial lung disease (ILD) diagnosed with IPF by clinical history, CT scan and surgical pathology. Control samples (n=108) were from patients who had surgery for a suspected lung nodule but who otherwise had no diagnosis for chronic lung disease by CT or pathology. Blood samples comprised an independent cohort of 57 patients with IPF with follow-up until transplantation/death or forced vital capacity (FVC) decline >10% over a 6-month period. Available demographic data included age, sex, smoking status and lung function (diffusion capacity for carbon monoxide % predicted (DLCO), forced expiratory volume in 1 s % predicted (FEV1), and forced vital capacity % predicted (FVC).
The IPF and control lung tissue samples were randomly divided into two non-overlapping groups (IPF group 1, IPF group 2, control group 1 and control group 2) for cross-validated data analysis. The overall schematic of methods used in this study is shown in figure 1.
Figure 1.

Flow chart of methodologies used in this study. GSE47460 was divided into two non-overlapping datasets for cross-validated differential gene expression analysis. This was followed by applying a consensus WGCNA on the two IPF groups using the differentially expressed genes as input. Identified modules were then analysed by association with lung function and enrichment analyses for biological function and transcription factor regulators. Samples in GSE72967 were matched to the IPF groups and correlated with the module eigengene to determine association with that module. GO, gene ontology; IPF, idiopathic pulmonary fibrosis; WGCNA, weighted gene coexpression network analysis.
Differential gene expression analysis
Microarray data were normalised using cyclic loess approach as previously described11 with the probe with the highest average signal selected per gene. A total of 15 180 genes per sample were available for analysis in the online matrix file. Hierarchical clustering of samples was used to identify outliers in each group (online supplementary figure S1). Differentially expressed genes were determined by comparing each IPF group to both control groups using a multivariate linear model controlling for age, sex and smoking status. Data were corrected for multiple comparisons by false discovery rate (FDR) adjustment and genes with FDR adjusted p values<0.05 in all four comparisons were considered differentially expressed. Principal component analysis of differentially expressed genes was performed to show clustering of disease and control samples.
thoraxjnl-2018-211929supp001.pdf (4.2MB, pdf)
Weighted gene coexpression network analysis
Consensus WGCNA was conducted using R/Bioconductor to determine coexpressed genes between the two IPF groups using only differentially expressed genes. Analysis setting included biweight midcorrelation (corType=‘bicor’) to account for outliers, sign of correlations between neighbours (TOMtype and networkType=‘signed’), and a more sensitive module detection parameter (deepSplit=3). Modules were identified by number according to module size. Module classification was applied to control data and showed good preservation of groupings based on module preservation statistics. Module eigengene (ME) was calculated as the first principal component of gene expression for the module and inter-relatedness of each module by eigengene network clustering (online supplementary figure S2). Up or downregulation of each module was determined by fold change of each gene, calculated as mean gene expression in IPF divided by mean gene expression in control.
MEs were compared with demographic data using Spearman’s correlation corrected for sex, age and smoking status and p values were adjusted for multiple comparisons by FDR. Consensus modules were defined as modules significantly correlated to lung function in both IPF groups and in the same direction (positive or negative correlation). Control MEs were correlated to lung function data as per the other datasets. Module membership, a measure of the association of a gene to its module, was determined by Pearson correlation of gene expression to ME and used to rank module connectivity.
Enrichment analysis for biological function and transcription factors
Module biological function was determined using gProfiler12 to determine enrichment for Biological Process gene ontology (GO) and Reactome pathways followed by enrichment mapping in Cytoscape (V.3.5.1) to define each functional cluster. Enrichment of cell types was determined by Enrichr using the Human Gene Atlas library.13
Transcription factors for each module were identified by iRegulon V.1.3 plugin in Cytoscape with a minimum Normalised Enrichment Score (NES) of 4.014 using data from 1120 ChIP-seq tracks in the Encode database. For each transcription factor, we show the number of genes regulated by that transcription factor identified within each module in the online supplementary table S1. The regulatory network of modules and transcription factors was plotted in Cytoscape to identify common regulators shared between modules.
miRNA correlation analysis
Subjects in both GSE47460 and GSE72967 were used to determine microRNA (miRNA) association. A total of 111 subjects were matched, 57 in IPF group 1 and 54 subjects in IPF group 2. MEs were correlated to the 338 miRNAs in the dataset using Pearson’s correlation with adjustment for multiple comparisons by FDR. Adjusted p values<0.005 in both groups were considered significant. Target genes associated with miRNAs for each of these modules were identified using the validated target dataset (miRTarBase) in the multi-miR database (http://multimir.ucdenver.edu/) and listed in the online supplementary table S2.15
Validation of datasets
Validation of differentially expressed genes in IPF was performed on two independent lung tissue datasets (GSE53845, GSE110147). The differently expressed genes were also used to show separation of IPF samples from control samples and other lung diseases, including chronic obstructive pulmonary disease (COPD), hypersensitivity pneumonitis, non-specific interstitial pneumonia and respiratory bronchiolitis ILD, that comprised the complete LRTC dataset.
Survival analysis
Clinical relevance of each module was determined using peripheral blood samples from patients with IPF at time of diagnosis with available RNA gene expression and survival data (GSE93606). Genes matched to each module were used to cluster patients into two groups using reversed graph embedding (DDRTree), a graph structure learning data reduction algorithm suited for ordering transcriptomic data by progressive changes16 and k-means clustering. A univariate Cox’s proportional-hazards model with Bonferroni correction for multiple comparisons was used to determine modules related with survival. Multivariate Cox’s proportional-hazards model, including FVC, age and sex, was then applied to the significant modules to determine the effects of confounding variables on module survival prediction.
Results
Demographic data
IPF and control tissue samples were divided into two groups for cross-validated analysis. Following removal of outliers, 51 and 53 samples remained in each control group and 79 samples remained in both IPF groups. Age and sex were matched in all groups, lung function was also matched in both IPF groups. Demographic data on these subjects are presented in table 1.
Table 1.
Demographic data for all subjects used in this study
| Control (tissue) | IPF (tissue) | IPF (blood) | |||
| Group 1 | Group 2 | Group 1 | Group 2 | – | |
| N | 51 | 53 | 79 | 79 | 57 |
| Age (years) | 64.5±10.5 | 64.0±11.4 | 65.1±8.0 | 63.4±8.6 | 67.4±8.0 |
| Sex (%) | |||||
| Female | 29 (57) | 26 (49) | 23 (29) | 26 (33) | 19 (33) |
| Male | 22 (43) | 27 (51) | 56 (71) | 53 (67) | 38 (67) |
| Smoking status (%) | |||||
| Never | 12 (24) | 17 (32) | 26 (33) | 32 (41) | – |
| Former | 32 (63) | 31 (58) | 49 (62) | 45 (57) | – |
| Current | 2 (4) | 0 (0) | 0 (0) | 2 (3) | – |
| DLCO (% predicted) | 80.8±14.4 | 87.4±18.6 | 48.5±16.6 | 46.9±19.0 | 39.2±14.1 |
| FEV1 (% predicted) | 92.6±10.6 | 97.7±14.1 | 70.8±17.9 | 71.2±18.4 | – |
| FVC (% predicted) | 92.6±11.4 | 96.2±14.9 | 63.5±16.5 | 64.8±17.1 | 72.2±20.3 |
Data are presented as mean±SD.
FEV1, forced expiratory volume in one second; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis.
Differential gene expression analysis
Cross-validated comparison of IPF and control groups identified 6425 differentially expressed genes in IPF (figure 2A). Principal component analysis of these genes showed overlap of samples within the IPF or control groups and good separation between IPF and control (figure 2B). Differentially expressed genes were also used to plot the separation between control and IPF samples in two independent datasets as well as the complete LTRC dataset to show separation of IPF from other disease phenotypes such as COPD (online supplementary figure S3).
Figure 2.

(A) Euler diagram of the number of differentially expressed genes identified as significant (FDR adjusted p<0.05) for the cross-validated comparisons. Of 15 180 genes, 10 237 were identified as significant in at least one comparison and 6425 genes were significant in all four comparisons. (B) Principal component plot of the 6425 genes showing overlap between samples in each of the IPF or control groups and separation between disease and control. (C) Plot of fold change of genes in each module to determine overall upregulation or downregulation of the module. Labelled genes are those that have been shown to have a known association with IPF or with the biological function of that module. IPF, idiopathic pulmonary fibrosis.
WGCNA module identification
Consensus WGCNA identified 16 modules in the IPF cohort, 6 modules were upregulated in IPF and 10 were downregulated (figure 2C). The five most connected genes for each module are presented in table 2.
Table 2.
List of top five genes by connectivity within each module
| Module | Top five connected genes | ||||
| ME1 | DNAJA4 | SPA17 | SPATA18 | RBKS | C11orf70 |
| ME2 | NDRG4 | KIAA1462 | EPAS1 | CCDC85A | MYZAP |
| ME3 | IGF1 | COL14A1 | STEAP1 | COL15A1 | CTHRC1 |
| ME4 | CDCA5 | MELK | CCNB2 | TPX2 | CEP55 |
| ME5 | FKBP11 | SSR4 | STARD5 | PIM2 | SPAG4 |
| ME6 | COL1A2 | LOXL2 | COL3A1 | HEPH | GLI2 |
| ME7 | TRPV2 | DOK2 | CSF2RA | FLVCR2 | FCGR3A |
| ME8 | ARAF | TYK2 | FXR2 | EDC4 | PQLC1 |
| ME9 | GADD45B | IL6 | MAFF | CSRNP1 | FOSL2 |
| ME10 | CACNA2D2 | PLA2G4F | LGI3 | ABCA3 | HPN |
| ME11 | HLA-E | BIN2 | PSMB9 | IL12RB1 | PRF1 |
| ME12 | PKN1 | DBNL | CNPPD1 | KLC1 | AP1M1 |
| ME13 | KANK2 | AOC3 | RECK | PGR | FZD1 |
| ME14 | ACTA2 | CNN1 | ACTG2 | TPM2 | HSPB7 |
| ME15 | DLGAP4 | FMR1 | C6orf106 | PTPN14 | EXOC3 |
| ME16 | PRKCZ | NEDD4L | VSIG2 | RASSF7 | PPP1R9A |
Lung function correlated with nine modules in both IPF groups. DLCO was negatively correlated with ME3, ME4, ME5 and ME14 and positively correlated with ME2, ME10, ME13 and ME16. FVC was negatively correlated with ME4, ME5 and ME9 and positively correlated with ME10 and ME16. No significant correlations were present in control samples. Heatmap of correlated modules is shown in figure 3 (complete module–trait comparisons are in online supplementary figures S4–S7).
Figure 3.
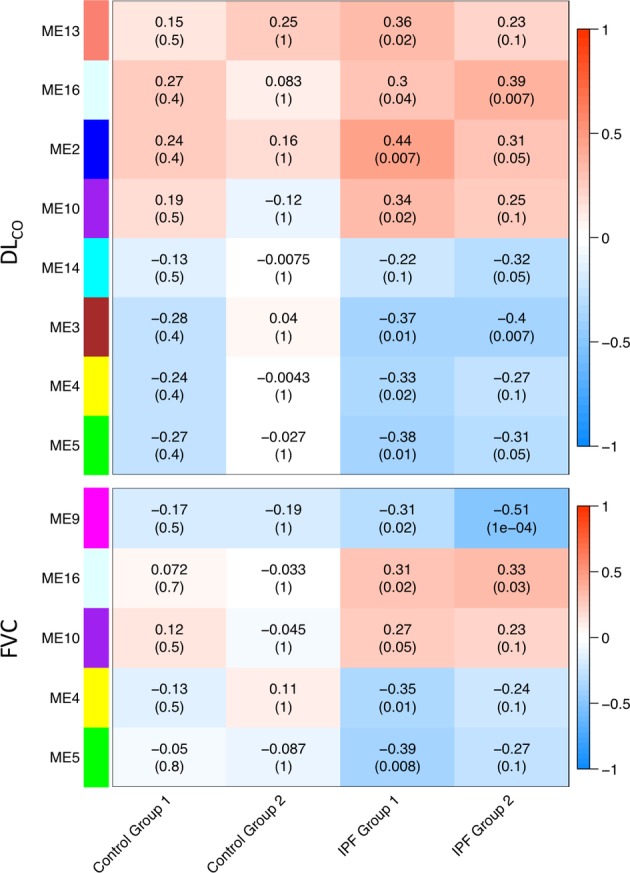
Consensus modules for both IPF groups that were significantly correlated with DLCO (top) or FVC (bottom). No significant correlations were found in the control group. Number is the R-value for each correlation. Number in parenthesis is the FDR corrected p value. DLCO, diffusion capacity; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis; ME, module eigengene.
Module biological function
Modules were grouped by pathway enrichment into several categories: immune response (ME5, ME7, ME9, ME11, ME12); extracellular matrix or contractile fibres (ME3, ME6, ME13, ME14); developmental pathways of specific lung structures (ME1, ME2); cell division, DNA replication and DNA repair (ME4); cellular metabolic and catabolic processes (ME8, ME15, ME16) and surfactant metabolism (ME10). Biological function enrichment for these categories is presented in table 3 and key modules are summarised below. We confirmed the validity of GO pathway identification of modules by localising the most highly connected genes of each module to specific cluster of cells that match its respective GO pathway using a single cell lung tissue dataset we had previously generated17 (online supplementary figure S8).
Table 3.
Representative biological functions for each module based on enrichment map clustering of gene ontology: biological process and Reactome terms
| Module | Expression in disease | No genes in module | Biological function |
| ME1 | UP | 1579 | Cilia organisation |
| ME2 | DOWN | 1404 | Blood vessel development |
| ME3 | UP | 396 | Extracellular Matrix organisation |
| ME4 | UP | 380 | Cell division/DNA replication/DNA repair |
| ME5 | UP | 371 | B-cell activation/unfolded protein response |
| ME6 | UP | 207 | Extracellular Matrix organisation |
| ME7 | DOWN | 204 | Leucocyte activation/degranulation |
| ME8 | DOWN | 188 | Cellular metabolic process |
| ME9 | DOWN | 163 | Response to bacteria/apoptosis/RNA transcription |
| ME10 | DOWN | 158 | Surfactant metabolism |
| ME11 | DOWN | 116 | T-cell activation/Major histocompatability complex (MHC) class I activity/interferon response |
| ME12 | DOWN | 107 | Leucocyte activation/degranulation |
| ME13 | DOWN | 80 | Muscle contraction/organ development |
| ME14 | UP | 75 | Contractile fibre/cell-extracellular matrix interactions |
| ME15 | DOWN | 58 | Cellular protein catabolic mechanism |
| ME16 | DOWN | 39 | Cell-cell junction organisation |
Immune response
Modules related to the immune response displayed specific inflammatory pathways and gene atlas cell types. Downregulated modules include ME7, ME12 and ME11. ME7 was enriched for leucocyte activation (GO:0045321, p=2.55×10−10), degranulation (GO:0043299, pP=4.75×10−8) and CD14 +monocytes (p=7.96×10−11) and CD33 +myeloid cells (p=6.08×10−7). ME12 was enriched for myeloid leucocyte activation (GO:0002274, p=8.31×10−4) and leucocyte degranulation (GO:0043299, p=0.00171) but was not associated with a specific cell type by gene atlas. ME11 was related to T-cell activation (GO:0042110, p=2.53×10−8), interferon signalling (REAC:913531, p=1.38×10−15), response to virus (GO:0009615, p=9.0×10−9) and Class I MHC mediated antigen processing and presentation (REAC: 983169, p=2.34×10−5), suggestive of a T-cell-mediated antiviral phenotype. Human gene atlas showed enrichment for CD56 +Natural Killer cells (p=7.32×10−18), CD8 +T cells (p=2.5×10−8) and CD4 +T cells (p=4.32×10−6).
ME9 was downregulated in IPF but was the only module that showed a reverse trend in relation to lung function with increased ME9 associated with a decline in FVC. It was enriched for response to bacterium (GO:0009617, p=5.52×10−7), apoptotic process (GO:0006915, p=7.2×10−6), regulation of gene expression (GO:0010468, p=8.75×10−6) and the CD33 +myeloid cell type (p=2.46×10−9).
The only immune response module that was increased in disease and negatively correlated with DLCO and FVC lung function measurements was ME5, which was associated with B-cell activation (GO:0042113, p=3.95×10−5) and the unfolded protein response pathway (REAC:381119, p=2.64×10−6). Gene atlas identified this module as enriched for CD19 +B cells (p=5.95×10−5) and included genes associated with development of B-cells into germinal centres and plasma cells such as POU2AF1 and MZB1.
Fibrotic response
Extracellular matrix organisation pathway was enriched in ME3 (REAC:1474244, p=5.11×10−11) and ME6 (REAC:1474244, p=3.34×10−7). ME6 was composed of the collagen markers COL1A2 and COL3A1 while ME3 included COL14A1, COL15A1 and TGFB3. While both modules were upregulated in IPF, only ME3 was negatively correlated with decline in DLCO. Closely associated to the ECM modules by eigengene clustering was ME14 which was associated with muscle contraction (REAC:397014, p=4.56×10−8) and alpha-smooth muscle actin. The combination of contractile fibres and extracellular matrix suggest these modules are related to a myofibroblast signature.
Developmental response
ME2 associated with vasculature development (GO:0001944, p=5.39×10−15) and cholesterol biosynthesis pathways (REAC:191273, p=0.00701). This module was downregulated in IPF and positively correlated with DLCO. A large number of genes was identified for this module including DISP1, required for effective hedgehog signalling which is an important pathway in angiogenesis,18 and CAV1, which regulates VEGF stimulated angiogenesis.19 This module also included the WNT genes WNT3A and WNT7A.
ME1 was upregulated in IPF and was strongly associated with cilium organisation (GO:0044782, p=9.88×10−43). Cilia in the lungs is mainly present on ciliated bronchial epithelial cells suggesting this module may be related to airway pathology including the development of the bronchiolar structures in honeycomb cysts.20 Interestingly, this module was also enriched for genes related to viral gene expression (GO:0019080, p=6.24×10−6).
Module regulators
The 16 modules were enriched for 25 transcription factors and 21 miRNAs (figure 4). The strongest enrichment scores were for transcription factors associated with the downregulated viral immune response module ME11, STAT2 (NES=9.068) and IRF1 (NES=9.789), the upregulated cilia-related module ME1, ZBTB7B (NES=9.935) and the DNA replication module ME4, E2F4 (NES=10.072). Several transcription factors were also included in their associated module suggesting a positive feedback loop; these regulators include FOXM1 (ME4), MYBL2 (ME4), IRF1 (ME11), STAT1 (ME11) and TCF12 (ME14). Overall, these transcription factors are able to directly regulate 44% of the genes identified in these modules (online supplementary table S1).
Figure 4.

Network of enriched transcription factors (white) and correlated miRNA (yellow) with each module. Modules upregulated in disease are shown as rectangles and modules downregulated in disease are shown as hexagons. Increased normalised enrichment scores are denoted by increased edge thickness. Biological function was categorised for immune response (blue) and fibrosis (green), with remaining modules labelled purple. The transcription factor EP300 (red lines) and TCF12 (orange lines) were highlighted as forming a network linking the fibrosis and immune response associated modules.
Module ME1 had the greatest number and strongest correlations of miRNAs of all modules. These correlated miRNAs were mainly from two families, miR-34/449 (miR-34b: p=1.91×10−8, r=0.80, miR-34c-3p, miR-34c-5p, miR-449a, miR-449b) and miR-200/429 (miR-200a: p=6.00×10−9, r=0.80; miR-200b: p=5.55×10−13, r=0.86; miR-429: p=7.49×10−8, r=0.73). The miRNAs miR-205 (p=8.86×10−10, r=0.84) and miR-31 (p=3.09×10−9, r=0.82) were also strongly correlated and linked this module with ME2 (miR-205: 3.83×10–3, r=−0.68; miR-31: p=6.76×10−4, r=−0.66). The miR-30s correlated with ME2 (miR-30a: p=1.87×10−6, r=0.68; miR-30b: p=1.10×10−4, r=0.72), ME3 (miR-30a: p=3.19×10−4, r=−0.62; miR-30b: p=3.19×10−4, r=−0.66) and ME5 (miR-30a: p=2.47×10−4, r=−0.59; miR-30b: p=2.47×10−4, r=−0.70), further supporting a link between fibrosis and B-cells. The contractile fibre module ME14 was also correlated with two miRNAs (miR-133b: p=1.08×10−4, r=0.77 and miR-143: p=2.65×10−3, r=0.64).
We found many of these regulators have been shown in previous studies to have a significant role with their associated modules. With regard to module ME1, these models have confirmed an important role for the miR-34/449 family in ciliogenesis and the miR-200/429 family as being upregulated under hypoxic conditions.21 22 Module ME14 was enriched for transcription factors SRF and TCF12 that have both been found to be required for myofibroblast differentiation and contractile activity in fibroblasts.23 24 Of particular note is the transcription ZBTB7B which was strongly enriched as a regulator for module ME1. This gene is normally associated with lineage commitment of T-cells to the CD4 phenotype25 but not has not been previously shown to regulate the epithelium. Examining the human protein atlas, we found ZBTB7B to be highly expressed in all epithelial cells types (skin and digestive tract) and the protein highly expressed in bronchial epithelial cells. Furthermore, examination using the lung single cell dataset found ZBTB7B to be highly expressed in epithelial cells further supporting its role in epithelial cell development (online supplementary figure S9).
Survival analysis
Clinical relevance of these modules was evaluated using an independent cohort of 57 patient with IPF peripheral blood RNA expression profiles with multivariate Cox’s proportional-hazard modelling used to evaluate the influence of each module in predicting survival. Median gene expression for genes comprising each module was assessed to determine if gene signature was detectable in the blood samples (online supplementary figure S10). Of the 16 modules we identified, four modules were found to be significantly associated with survival after Bonferroni adjustment for multiple comparisons (ME1 p=0.038, ME8 p=0.008, ME9 p=0.042, ME12 p=0.041). Multivariate Cox’s proportional-hazards model to adjust for FVC, age and sex was applied to these four modules and showed ME1 had the greatest association with survival with an overall concordance of 0.777, an adjusted log-rank test p value of 0.001, and a ME1 HR of 2.73 (95% CI 1.28 to 5.87; p=0.0097) (figure 5).
Figure 5.

(A) Discriminative dimension reduction (DDR) graph of GSE93606 IPF peripheral blood RNA data using module ME1 (cilia) gene list. K-means clustering was used to separate samples into two groups (yellow and purple). (B) Heatmap of gene expression sorted by DDR score. (C) Kaplan-Meier curves for groups defined by DDR k-means clustering of ME1 genes in peripheral blood dataset. Multivariate Cox’s proportional hazard modelling of module groups, FVC, sex and age showed ME1 had significant association with survival (p=0.0097). IPF, idiopathic pulmonary fibrosis; ME, module eigengene.
Discussion
Consensus network analysis was used to identify differentially expressed modules with associated biological function and transcriptional regulators expressed in IPF with relation to decline in lung function. Similar to previous studies that have used high-throughput datasets to examine IPF, these modules were enriched for pathways related to the immune response, fibrosis and development. Regulators cross-linking these modules include the transcriptional coactivator p300 and TCF12, as well as the miRNAs miR-205 and miR-30s.
Several observations can be derived from these data. For the first, IPF is shown to have a dysfunctional immune response highlighted by the decrease in interferon, MHC class I presentation, and T-cell activation and decrease in pathways related to activation and degranulation of leucocytes. Interestingly, while the module related to detection of bacteria was downregulated in IPF, it was negatively correlated with lung function decline. It has been shown that patients with a more rapid decline in lung function have an increased lung bacterial burden26 which suggests that despite a downregulated antibacterial immune response, the lung is responding to the presence of these micro-organisms. Viral infections have also thought to be involved in IPF but evidence are lacking.27 28 Overall, the dysfunctional immune responses may be related to the poor clearance of micro-organisms resulting in their increased presence in IPF. These downregulated immune responses would also be exacerbated by treatment with anti-inflammatory drugs and may explain the increased mortality in patients with IPF when treated with corticosteroids.
Despite a general downregulation in the immune response, the humoral immune response was upregulated, specifically related to activation of B-cells. This is supported by the previously reported increased presence and number of B-cells and tertiary lymphoid follicles in IPF29 as well as a transcriptomic study where increased B-cell but not T-cell activation in IPF was observed.30 While the role of B-cells in IPF remains unknown, the plasma cell marker, MZB1, has been associated with numerous fibrotic diseases.31 Further supporting a link between B cells and fibrosis are the miR-30s which were correlated with the B cell module and ME3, one of the extracellular matrix associated modules. These miRNAs have been shown to be involved in both fibrosis and epithelial–mesenchymal transition32 and repressing B-cell activating factor (BAFF) expression in B-cells.33
Second, our data show that two parallel fibrotic processes are active. The most highly connected genes in module ME6 are collagen 1 and 3 (COL1A2 and COL3A1), which form the primary structures in the ECM. ME3 was highly connected with collagen 14 (COL14A1) which has a role in cross-linking collagen 1 and the development of more advanced fibrotic structures. While both modules were upregulated in IPF, only ME3 was correlated (negatively) with lung function decline. It remains unknown which of these pathways are affected by antifibrotics, warranting further research in this area.
Finally, the largest module was associated with cilia (ME1). Previous studies have identified a cilia signature in IPF and found it to be related with microscopic honeycombing.1 3 In comparing our modules with an independent dataset, we showed ME1 as significantly improving survival models in patients with IPF. Serum protein epithelial biomarkers have previously been reported to predict survival in patients with IPF.34–36 However, this is the first report that we are aware of that used an RNA epithelial signature in the blood as a biomarker in IPF. While the detection of an epithelial signature in the blood may seem counter-intuitive, there is precedence in cancer studies where circulating epithelial cancer cells are present in blood samples.37 Circulating epithelial cells have also been detected in chronic inflammatory bowel diseases suggesting a similar process may be present in IPF.38
One limitation of the study was that while we performed a cross-validated analysis between two non-overlapping groups of samples, it was not truly independent. Our dataset consisted of 160 IPF lung tissue samples and there does not exist a dataset of equal size to use for a proper replication cohort with most datasets composed of less than 15 samples. (See online supplementary table S3 for complete listing of IPF-related datasets.) Rather, we believe that our cross-validated and consensus-based approach provides sufficient validation to be considered robust. Also, while our study was able to identify a number of novel pathways in IPF, it was limited by the available clinical data and biases inherent in enrichment analyses. Lung function data were available for most samples, but smoking history was limited to smoking status (never, current or ever-smoker) without regard to pack-years. Enrichment analysis (GO or Human Cell Atlas) was also limited in identifying pathways or cell-types as it is based on validated gene lists that result in over-representation of well-characterised pathways. This may be reflected in the number of immune response modules we have identified warranting further studies to validate the role of these pathways in IPF and to determine its functional significance in disease progression.
In conclusion, these data demonstrate several pathways in IPF consistent with current knowledge of the pathology of this disease. We believe that this hypothesis generating study provides novel insight into the biological pathways in IPF and identifies several candidate regulators as targets for intervention.
Footnotes
Contributors: JEM conceived and performed the study. BT contributed the single cell dataset. NK, JCH, BMV and WW were involved in writing and editing the manuscript.
Funding: Funded by grants from KULeuven (C24/15/30) and NIH (R01HL127349). JEM was funded by a fellowship from the ERS (RESPIRE2-2015-9192).
Competing interests: NK is an inventor on a pending patent on use of thyroid hormone as an antifibrotic agent (licensed), as well as a patent on novel biomarkers in IPF (not licensed). NK consulted Biogen Idec, Boehringer Ingelheim, Numedii, MMI, Pliant, Third Rock, Indaloo and Samumed. NK has an ongoing collaboration with MiRagen but no fund exchange.
Patient consent: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Yang IV, Coldren CD, Leach SM, et al. . Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 2013;68:1114–21. 10.1136/thoraxjnl-2012-202943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DePianto DJ, Chandriani S, Abbas AR, et al. . Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015;70:48–56. 10.1136/thoraxjnl-2013-204596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Yella J, Chen J, et al. . Unsupervised gene expression analyses identify IPF-severity correlated signatures, associated genes and biomarkers. BMC Pulm Med 2017;17:133 10.1186/s12890-017-0472-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bauer Y, Tedrow J, de Bernard S, et al. . A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015;52:217–31. 10.1165/rcmb.2013-0310OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S, Herazo-Maya JD, Kang DD, et al. . Integrative phenotyping framework (iPF): integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genomics 2015;16:924 10.1186/s12864-015-2170-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kusko RL, Brothers JF, Tedrow J, et al. . Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;194:948–60. 10.1164/rccm.201510-2026OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Modena BD, Bleecker ER, Busse WW, et al. . Gene expression correlated with severe asthma characteristics reveals heterogeneous mechanisms of severe disease. Am J Respir Crit Care Med 2017;195:1449–63. 10.1164/rccm.201607-1407OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Molyneaux PL, Willis-Owen SAG, Cox MJ, et al. . Host-microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2017;195:1640–50. 10.1164/rccm.201607-1408OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Obeidat M, Nie Y, Chen V, et al. . Network-based analysis reveals novel gene signatures in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Res 2017;18:72 10.1186/s12931-017-0558-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu W, Dave N, Tseng GC, et al. . Comparison of normalization methods for CodeLink Bioarray data. BMC Bioinformatics 2005;6:309 10.1186/1471-2105-6-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reimand J, Arak T, Adler P, et al. . g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 2016;44:W83–W89. 10.1093/nar/gkw199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuleshov MV, Jones MR, Rouillard AD, et al. . Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janky R, Verfaillie A, Imrichová H, et al. . iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 2014;10:e1003731 10.1371/journal.pcbi.1003731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ru Y, Kechris KJ, Tabakoff B, et al. . The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res 2014;42:e133 10.1093/nar/gku631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mao Q, Wang L, Goodison S, et al. . Dimensionality Reduction Via Graph Structure Learning Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York: NY, USA: ACM, 2015:765–74. [Google Scholar]
- 17. Lambrechts D, Wauters E, Boeckx B, et al. . Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277–89. 10.1038/s41591-018-0096-5 [DOI] [PubMed] [Google Scholar]
- 18. Renault MA, Robbesyn F, Chapouly C, et al. . Hedgehog-dependent regulation of angiogenesis and myogenesis is impaired in aged mice. Arterioscler Thromb Vasc Biol 2013;33:2858–66. 10.1161/ATVBAHA.113.302494 [DOI] [PubMed] [Google Scholar]
- 19. Tahir SA, Park S, Thompson TC. Caveolin-1 regulates VEGF-stimulated angiogenic activities in prostate cancer and endothelial cells. Cancer Biol Ther 2009;8:2284–94. 10.4161/cbt.8.23.10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seibold MA, Smith RW, Urbanek C, et al. . The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One 2013;8:e58658 10.1371/journal.pone.0058658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song R, Walentek P, Sponer N, et al. . miR-34/449 miRNAs are required for motile ciliogenesis by repressing cp110 . Nature 2014;510:115–20. 10.1038/nature13413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartoszewska S, Kamysz W, Jakiela B, et al. . miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol Biol Lett 2017;22:23 10.1186/s11658-017-0054-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chai J, Norng M, Tarnawski AS, et al. . A critical role of serum response factor in myofibroblast differentiation during experimental oesophageal ulcer healing in rats. Gut 2007;56:621–30. 10.1136/gut.2006.106674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang X, Hou Y, Yang G, et al. . Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ 2016;23:132–45. 10.1038/cdd.2015.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang L, Wildt KF, Castro E, et al. . The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity 2008;29:876–87. 10.1016/j.immuni.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Molyneaux PL, Cox MJ, Willis-Owen SA, et al. . The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014;190:906–13. 10.1164/rccm.201403-0541OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wootton SC, Kim DS, Kondoh Y, et al. . Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:1698–702. 10.1164/rccm.201010-1752OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore BB, Moore TA. Viruses in idiopathic pulmonary fibrosis. etiology and exacerbation. Ann Am Thorac Soc 2015;12 Suppl 2(Suppl 2):S186–92. 10.1513/AnnalsATS.201502-088AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, et al. . Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol 2006;176:5735–9. 10.4049/jimmunol.176.10.5735 [DOI] [PubMed] [Google Scholar]
- 30. Zuo F, Kaminski N, Eugui E, et al. . Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A 2002;99:6292–7. 10.1073/pnas.092134099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schiller HB, Mayr CH, Leuschner G, et al. . Deep proteome profiling reveals common prevalence of mzb1-positive plasma b cells in human lung and skin fibrosis. Am J Respir Crit Care Med 2017;196:1298–310. 10.1164/rccm.201611-2263OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duisters RF, Tijsen AJ, Schroen B, et al. . miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 2009;104:170–8. 10.1161/CIRCRESAHA.108.182535 [DOI] [PubMed] [Google Scholar]
- 33. Alsaleh G, François A, Philippe L, et al. . MiR-30a-3p negatively regulates BAFF synthesis in systemic sclerosis and rheumatoid arthritis fibroblasts. PLoS One 2014;9:e111266 10.1371/journal.pone.0111266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kinder BW, Brown KK, McCormack FX, et al. . Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 2009;135:1557–63. 10.1378/chest.08-2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barlo NP, van Moorsel CH, Ruven HJ, et al. . Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2009;26:155–61. [PubMed] [Google Scholar]
- 36. Maher TM, Oballa E, Simpson JK, et al. . An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med 2017;5:946–55. 10.1016/S2213-2600(17)30430-7 [DOI] [PubMed] [Google Scholar]
- 37. Tanaka F, Yoneda K, Kondo N, et al. . Circulating tumor cell as a diagnostic marker in primary lung cancer. Clin Cancer Res 2009;15:6980–6. 10.1158/1078-0432.CCR-09-1095 [DOI] [PubMed] [Google Scholar]
- 38. Pantel K, Denève E, Nocca D, et al. . Circulating epithelial cells in patients with benign colon diseases. Clin Chem 2012;58:936–40. 10.1373/clinchem.2011.175570 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2018-211929supp001.pdf (4.2MB, pdf)