

Abstract
By using techniques borrowed from statistical physics and neural networks, we determine the parameters, associated with a scoring function, that are chosen optimally to ensure complete success in threading tests in a training set of proteins. These parameters provide a quantitative measure of the propensities of amino acids to be buried or exposed and to be in a given secondary structure and are a good starting point for solving both the threading and design problems.
The principal objective of this paper is a demonstration of the viability of a framework, based on ideas from statistical physics and neural networks, for attacking the protein threading problem. Our work points to the difficulty associated with a commonly used statistical procedure for determining such parameters. We present the results of threading and design tests and present a singular value decomposition (SVD) analysis of the parameters that elucidate the interplay between degree of burial and secondary structure propensities in the folding problem.
The challenge of the protein folding problem (1–5) is to deduce the native state structure and thence the functionality of a protein from the knowledge of the sequence of amino acids. The successful completion of the human genome project has heightened interest in this problem. The information readily available as input are the sequences and native structures of a few thousand proteins (6). Given an entirely new sequence, one needs to have a sound strategy for determining its native state structure. A simpler problem, threading (7), relies on the belief that the total number of distinct folds in nature is only a few thousand (8) and attempts to match the new sequence with the best among a selection of possible native state structures. (A difficulty associated with threading is that, because of steric constraints, one may not be able to mount a given sequence on a piece of a native structure of a different sequence. See, for example, ref. 9.) To assess the fit of a given sequence with a putative native state structure, one might use a coarse grained representation of the amino acids in a sequence and postulate a scoring function with a simple functional form. Perhaps the simplest such function is one that characterizes the propensities of the various types of amino acids to be in different environments:
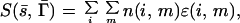 |
1 |
where S is the score function, which is a measure of the match of a sequence s̄ and target structure Γ̄, n(i,m) is the number of amino acids of type i in the environment m and ɛ(i,m) is the score associated with it (10). For a given amino acid i, each of the ɛ(i,m)s may be shifted by the same arbitrary constant so that, without loss of generality, one may set ∑mɛ(i,m) = 0. The advantages of such an environmental scoring function over pairwise interactions between amino acids are its simplicity and the far greater ease of incorporating gaps in both sequence and in structure.
Our focus is on determining the score quantifying the match of a sequence to a putative native state structure, for which the most common approach utilizes statistical considerations (10–13), based on counting the number of amino acids in a given environment in the native state. Pioneering work by Bowie et al. (10) has shown that a simple statistically based approach with an environmental score leads to excellent results for the inverse folding problem.
Our studies used a training set of 387 proteins (see Table 2, which is published as supporting information on the PNAS web site, www.pnas.org) from the PDBselect (6, 14) consisting of sequences varying in length from 44 to 1,017, with low sequence homology and covering many different three-dimensional-folds according to the Structural Classification of Proteins (SCOP) classification (15). Additional criteria used in selecting the proteins in the training set were as follows: (i) the protein structure was obtained through x-ray crystallography; (ii) the structures were monomeric; and (iii) the determined structures missed no more than two amino acids. The same criteria were used to obtain a test set of 213 distinct proteins (Table 3, which is published as supporting information on the PNAs web site), with lengths ranging between 54 and 869. For each structure, we used a simple environmental classification that consists of the local secondary structure (α-helix, β-strand, or other) and the exposed area evaluated as the ratio between the accessible area of each amino acid, X, of its native sequence (having this structure as its native state) and the corresponding area in a Gly-X-Gly extended structure. The values of the exposed area were divided into three categories of small, medium, and large exposures corresponding to <10%, 10−50%, and >50%, respectively. Thus, the scoring function consists of nine parameters for each amino acid corresponding to each of the nine environments that it might be found in.
Materials and Methods
We begin by applying the ideas of Bowie et al. (10) to the threading problem. The statistical score ɛs(i,m) associated with amino acid i in an environment m is readily deduced by using the expression
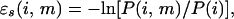 |
2 |
where P(i,m) is the probability of finding an amino acid of type i in the environment of type m and P(i) is the probability of finding an amino acid of type i in any environment. Both P(i,m) and P(i) are determined from a knowledge of the sequences and native state structures of the proteins in our training set. To assess the quality of the extracted scores, we carried out threading tests on all but the largest protein in the training set itself. Each protein sequence was mounted on its own native state structure and on every fragment (of the correct length chosen without insertions and deletions) of all of the larger proteins. The exposed area for the amino acid mounted on a fragment was assumed to be the same as that in the whole protein from which the fragment was extracted. As we shall see later, this may be a poor approximation when the size of the fragment is much smaller than the whole protein. In each case, Eq. 1 was used to determine the scoring function. Although the technique is simple, the results of gapless threading tests are only moderate; the native state structure is correctly recognized for 69% of the proteins. In a recent paper, Baud and Karlin (16) considered 418 proteins and determined the frequencies of occurrence of the twenty amino acids in nine environments that were defined in a way similar to ours. We have converted their frequencies into statistical scores (which turn out to be similar to the statistical scores derived from our training set) by using Eq. 2, and find 54 failures in our set of 213 proteins. This moderate performance may be due to the fact that the form of the scoring function is too simple. Support for this conclusion comes from earlier work that has pointed out the difficulty of determining the optimal interactions that stabilize the native state of even one protein (crambin) with a more complex scoring function involving 210 pairwise interactions (17). An alternative possibility, that the statistical approach is flawed (18), would be of more serious concern because such statistical schemes are commonly used in the protein folding problem.
We turn to a demonstration that an alternative strategy based on ideas originating in statistical physics and neural networks provides a powerful framework for tackling the threading problem. Following the pioneering work of Friedrichs and Wolynes (19) and especially Goldstein et al. (20), the basic idea is to postulate the form of a scoring function and to choose its parameters to ensure that the true native states of proteins with known structures (learning set) correspond to better (lower) scores than when the sequences are housed in competing decoy conformations (17–28). An important advantage of this procedure is that it can be used to verify whether the chosen form of the scoring function is equal to the task or not. Indeed, one may start with the simplest form of the scoring function and systematically expand the parameter space until the optimal interactions are learned. The statistical procedure considers proteins and their native state structures, whereas the learning procedure has information on competing structures as well. Our scheme is similar in spirit to that of previous work with the important differences that we consider an environmental scoring function instead of a pairwise contact potential and we optimize the energy gap without any normalization.
The total number of inequalities (one obtains the inequalities for each sequence in the training data set by considering as decoys all pieces of the native state structures of longer proteins in the training set) is over 13 million, making the problem technically difficult. For a given protein, each decoy leads to a linear inequality of the form
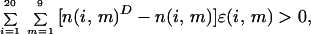 |
where n(i, m)D is the number of amino acids of type i found in the environment m in the given decoy. The perceptron procedure is a simple technique based on neural networks for simultaneously solving a set of such linear inequalities (29). We used this procedure to optimally choose the 180 parameters to ensure that the worst inequality (among the more than 13 millions) was satisfied as well as possible and that threading tests on the training set were 100% successful.
Results
We describe the results of several tests and a biological interpretation of these parameters.
Learning Procedure Versus Statistical Approach.
Fig. 1 shows a plot of the parameters determined by using the statistical approach vs. those deduced by the learning procedure. The poor correlation is consistent with the qualitatively different performance levels in threading. It underscores the fundamental difficulties of the statistical approach and points to the advantage of learning the optimized parameters in a systematically expanded parameter set.
Figure 1.

Plot of the optimal ɛ parameters vs. those determined by using a statistical scheme, ɛs, using a training set of 387 proteins.
Threading Tests.
The couplings ɛ387(i,m) obtained based on learning the native states of the 387 proteins in the training set were subjected to threading on the test set containing 213 distinct proteins (Table 3) and the decoys obtained from their native state structures. In contrast to the performance of the statistical parameters, for which one is unable to correctly recognize the native states of 76 of the 213 proteins, the number of failures when one uses the learned parameters is 23. The failed proteins are modest in size and have sequence lengths ranging between 54 and 131. The ranks of the native states, defined as the number of better performing decoys, of the failed proteins are plotted in Fig. 2 as a function of the sequence length for both sets of parameters (note the dramatically different scales of the y axis). For the poorest performer, by using the perceptron-based method, there are 102 decoys (of 37,617) that perform better than the native state (protein 1abq of length 56) whereas the corresponding number for the statistically derived parameters is 29,424 (of 31,436 decoys for protein 1vqb of length 86). We have checked that around half of the failures are spurious for the case of the learned parameters and arise because the exposed areas for the winning decoy, which is a piece of the native state structure of a longer protein, is quite different from that determined for the whole protein. This effect of an inaccurate assignment of the exposed area is strong only for small sized proteins. The remaining failures (a total of 5%) are likely due to the identification of genuine competitors to the native state or because the winning decoy is not a viable structure for the sequence under consideration (9).
Figure 2.

Results of the threading tests for 213 proteins arranged according to their length, N. Only the failed cases are shown. (Upper) A plot of the number of decoys that performed better than the native state structure vs. N; (Lower) A similar plot for the couplings that were determined statistically. Note the disparity in the scales of the y axes.
We also tested the ɛ387 parameters on all 600 proteins (Tables 2 and 3) and the decoys obtained by using all 600 native state structures. There were 57 failures whereas the statistically derived parameters resulted in 209 failures. We used the perceptron procedure (29) to learn the scoring parameters to ensure that the native state of all of the proteins in the training and test set were recognized with 100% success and the energies of all decoys were pushed up as much as possible compared with the native state energies. In the rest of the paper, we will use this refined set of optimal parameters ɛ(i,m) (Table 1) to carry out our further studies. Note that the sum of the first nine entries of each row in Table 1 is equal to zero and the sum of the squares of all such 180 entries has been chosen to be 180.
Table 1.
Environmental Scores
| Amino acid | α
|
β
|
Other
|
Si | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Small | Med. | Expo. | Small | Med. | Expo. | Small | Med. | Expo. | ||
| Cys (C) | −1.29 | 0.07 | 1.81 | −1.78 | −0.83 | 3.63 | −1.24 | −0.85 | 0.49 | −1.06 |
| Phe (F) | −0.90 | −0.35 | 2.33 | −1.77 | −1.02 | 1.51 | −0.26 | −0.28 | 0.74 | −0.73 |
| Trp (W) | 0.41 | 0.32 | 1.64 | −1.18 | −1.00 | −1.02 | 0.57 | 0.50 | 0.91 | −0.07 |
| Ile (I) | −0.50 | −0.27 | 0.38 | −0.25 | −0.39 | 0.61 | −1.05 | 0.56 | 0.92 | −0.29 |
| Val (V) | 0.42 | 0.06 | −0.12 | −1.48 | −0.64 | 0.89 | −0.28 | 0.57 | 0.58 | −0.31 |
| Met (M) | −0.26 | −0.36 | 0.65 | −0.52 | 0.71 | 1.26 | −0.24 | −0.77 | −0.47 | −0.30 |
| Leu (L) | −0.33 | −0.16 | 0.09 | −0.32 | 0.83 | −0.76 | −0.54 | 0.77 | 0.41 | −0.10 |
| Gly (G) | 0.36 | 1.16 | 0.73 | −0.05 | 0.16 | 0.14 | −0.49 | −0.95 | −1.06 | −0.48 |
| Tyr (Y) | 0.13 | 0.83 | −0.06 | −0.42 | −1.18 | −0.23 | 0.23 | 0.08 | 0.63 | 0.00 |
| Ala (A) | −0.40 | −0.05 | −0.13 | 0.27 | 0.50 | −0.15 | −0.23 | 0.35 | −0.25 | −0.06 |
| His (H) | 1.05 | −0.60 | −0.82 | 0.62 | 0.56 | 0.14 | −0.29 | −0.08 | −0.57 | −0.09 |
| Asp (D) | −0.29 | −0.79 | −0.90 | 1.31 | 0.93 | 1.32 | 0.59 | −0.99 | −1.17 | −0.60 |
| Ser (S) | −0.31 | −0.01 | −0.98 | 0.48 | 0.78 | −0.75 | 1.00 | −0.32 | −0.10 | 0.03 |
| Thr (T) | 0.80 | 0.49 | −0.46 | 0.55 | −0.50 | −0.80 | 0.74 | −0.34 | −0.48 | 0.01 |
| Asn (N) | 0.67 | −0.66 | −0.66 | 1.34 | 0.60 | −0.06 | 0.55 | −0.48 | −1.30 | −0.39 |
| Pro (P) | 2.35 | −0.28 | −0.88 | 1.32 | 1.03 | −0.30 | −1.02 | −0.62 | −1.61 | −0.65 |
| Gln (Q) | 1.74 | −0.84 | −1.24 | 0.94 | −0.87 | −1.07 | 1.32 | 0.01 | 0.01 | −0.26 |
| Glu (E) | 0.83 | −0.81 | −1.28 | 1.67 | −0.21 | −0.67 | 1.60 | 0.04 | −1.16 | −0.53 |
| Arg (R) | 2.29 | −0.80 | −1.37 | 1.37 | −1.16 | −1.35 | 1.82 | 0.13 | −0.94 | −0.38 |
| Lys (K) | 1.20 | −1.13 | −1.77 | 4.32 | −1.43 | −1.91 | 2.38 | −0.32 | −1.35 | −1.11 |
Table of ɛ, the nine environmental scores for each amino acid. Large negative values indicate a strong preference for the particular environment whereas large positive values indicate an aversion. The last column shows Si, which is a measure of the average contribution of each amino acid to the native state score and provides an estimate of the expectation of the contribution of a given amino acid to the native state score.
The native state of crambin (1crn), which was not part of the training set, is recognized in threading. This result is encouraging because of earlier difficulties in learning pairwise parameters for this protein (19). It should be noted, however, that a single amino acid mutation of 1crn, the protein 1cbn, was present in the basic learning set of 387 proteins.
As a further test, we selected 16 globin proteins from the PDB (6) (http://www.rcsb.org/pdb/), which were in the DEOXY form, which were not mutated, and whose structures are resolved well. Strikingly, 13 of the 16 proteins correctly picked their own native state from among the millions of decoy conformations obtained from the fragments of the 600 proteins in the training and test sets described previously. For the 3 other cases, fragments from the globin family were picked to be the best structure. Indeed, the scores of the globin proteins on fragments of other globin structures were generally lower than on fragments of structures of unrelated proteins, underscoring the quality of our scoring function.
Biological Interpretation of Learned Parameters.
Let us begin with a geometrical picture of ɛ(i,m), considered to be 20 vectors of nine components each. For a given amino acid i, the components of the nine-dimensional vector, labeled by the index m, capture the propensities of that amino acid to be in each of nine environments. Each environment may be thought of as representing an axis in an orthogonal 9-dimensional space. SVD (28, 30) affords a simple prescription for dimensional reduction by the optimal choice of a new set of orthogonal axes. In this new reference frame, the original vectors span a lower-dimensional space, and the axes may be conveniently rank-ordered in importance.
The SVD theorem (30) states that the 20 × 9 (non-square) matrix ɛ can be written as
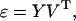 |
3 |
where Y is a 20 × 9 dimensional matrix and V is a 9 × 9 dimensional matrix. The superscript T denotes the transpose matrix. The matrix Y is given by Y = UΣ, where Σ is a 20 × 9 dimensional matrix whose elements are all zero except for the diagonal terms, Σn,n, n = 1, … , 9. These diagonal terms are equal to the square roots, σn, of the common eigenvalues of ɛɛT and ɛTɛ. The σn's are called singular values and are assumed to be rank ordered so that σ1 is the largest. Here, they are as follows: 10.59, 5.02, 3.98, 3.42, 2.57, 2.09, 1.77, 0.95, and 0.0. The columns of V, denoted by Vk, are the eigenvectors corresponding to the rank ordered eigenvalues of the matrix ɛTɛ, and the columns of the 20 × 20 matrix U, denoted as Uk, k = 1, … , 20, are determined by the formula
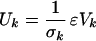 |
(when the singular values are non-zero; the other cases are
irrelevant for the reconstruction of the ɛ parameters). The result of
the SVD transformation is that ɛ(i,m) may now
be represented as a sum of contributions that diminish in an overall
sense as one considers smaller singular values. The nth
contribution is given by
y(n)(i)
v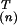 (m),
where the first factor depends on the amino acid and the second on the
environmental index. Thus
v
(m),
where the first factor depends on the amino acid and the second on the
environmental index. Thus
v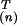 are the new
orthogonal and normalized directions—or modes—in the space of
environments.
are the new
orthogonal and normalized directions—or modes—in the space of
environments.
Fig. 3 shows the three most dominant contributions, corresponding to the top three singular eigenvalues. Each contribution is displayed in two panels. The Upper panels show the mode as a function of the nine environmental parameters. The Lower panels show the corresponding amplitudes y(n) plotted so that y(n) increases monotonically.
Figure 3.

The top three contributions to ɛ(i,m)
as emerging from the SVD analysis. The numbers in the ovals indicate
the mode number. The letters at the top left of each segment of two
panels indicate amino acids (in the single letter code) for which this
particular mode is dominant. The Upper panels in each
segment show the modes—the values of
v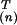 for the nine
values of the environmental variable m. For each kind of
secondary structure, the environments are listed in order from the
small to large exposure. The Lower panels show the amino
acid-dependent weights y(n) with which the
displayed mode contributes to the score in a given environment.
for the nine
values of the environmental variable m. For each kind of
secondary structure, the environments are listed in order from the
small to large exposure. The Lower panels show the amino
acid-dependent weights y(n) with which the
displayed mode contributes to the score in a given environment.
The first mode is dominant for 13 aa: C, F, I, V, H, S, T, N, P, Q, E, R, and K. The second mode is the leader for W, M, Y, and D, and the third for L and A. The remaining amino acid, G, is dominated by the fifth mode. The first mode provides the overall dominant behavior and strongly distinguishes between the buried and exposed environments in a monotonic way regardless of the secondary structure; one may arrange the amino acids into buried and exposed groups depending on whether y(1) is large and positive or large and negative. One may further subdivide the two basic groups of buried (B) and exposed (E) amino acid into subgroups: B1, B2, B3, E1, and E2. The division is illustrated in Fig. 3 and corresponds to occurrences of more rapid variations in y(1) as one moves from one amino acid to the next. The key point is that the amino acids in B1 have a strong tendency to be buried and the charged amino acid K in E2 has a strong tendency to be exposed, and most of the amino acids are more sensitive to the degree of burial than to other considerations. This tendency for burial is usually associated with hydrophobicity in the protein folding problem (31–33). The hydrophobic amino acids F, I, V, L, and A do belong to group B, but this group also contains polar amino acids. Cysteine, C, shows the strongest propensity to be buried. It should be noted that a pair of C's may form a strong contact by establishing a disulfide bridge. Of the 896 C–C contacts generated in our study of 600 proteins, 402 had both C's buried whereas only in four cases were both of the C's exposed (independent of the secondary structure). This tendency alone yields a high statistical score for C being buried. (Note that 37% of the structural sites of the 600 proteins are classified as buried, 40% as medium, and 33% as exposed.) The learned score is even further accentuated because most of the decoys correspond to C being not buried and stability of the native state with respect to decoys is enhanced by such an adjustment.
The remaining modes break the symmetry between the secondary structures. The second mode is neutral to α and favors (disfavors) β (loop) when the coefficient y(2) is negative. It shows a strong preference for amino acids, such as W, with a large negative y(2) to be in a β-strand with a large exposed area and for amino acids, such as D, with a large positive y(2) to be in loops with a large exposed area. The third mode introduces a preference for C, F, K, etc. to be in β-strands with medium exposure and for L, P, and A, etc. to stay either in exposed β-strands or in buried loops and avoid exposed helices.
Protein Design.
We turn now to an extension of our studies to protein design or the inverse folding problem. In analogy with equilibrium statistical mechanics, the probability that a sequence s̄ is housed in a structure Γ̄ is given by (34–37)
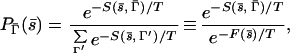 |
4 |
where T, here, is a fictitious temperature, the score S has been assumed to play a role analogous to the energy, and F, the free score, plays the role of the free energy. The key point is that, in the limit of T→0 and when Γ̄ is the native state structure of s̄, P→1. In this limit, therefore, the “free score,” which is a function of the sequence alone, approaches the score of the sequence in its native state. The last column of Table 1 shows the average contribution to the native state scores, Si, from each type of amino acid in the various environments. It is defined by
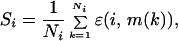 |
where the sum is over the Ni occurrences of amino acid i in the native state of all 600 proteins in the training and test sets. The zero “temperature” free score of a sequence may then be readily deduced without any knowledge of the structure, by adding up these contributions for the amino acids in the sequence. Fig. 4 shows a plot of the native state score vs. the sequence free score for all 600 proteins. The latter, which has no structure dependent information, provides a reasonable approximation to the actual native state score. We have verified that both are linearly proportional to the protein length and that, for the longer proteins, the native state score is somewhat higher than the free score because of the increasing tendency toward frustration as the sequence length increases. For design purposes, the free score provides a measure of the score one is entitled to expect in a typical native state structure, and the lower the score in the target native state structure with reference to the free score, the better is the design.
Figure 4.

Plot of the zero “temperature” free score and the native state score of each of the proteins in the training and test sets.
Stability of Cold Shock Proteins.
We used the learned parameters to provide a molecular interpretation of the different thermal stabilities of a pair of cold shock proteins (38), one of which is mesophilic Bacillus subtilis (Bs-CspB: 1csp) and the other thermophilic Bacillus caldolyticus (Bc-Csp: 1c9o). The former has a score of −34.80 in the native state, whereas the latter is more stable, with a score of −41.64. More strikingly, the free scores are −28.64 and −27.97, respectively, underscoring the much better design of the thermophilic protein. We also used the conformation space of all decoys to estimate the “heat capacity” of the two proteins as a function of temperature. The heat capacity, which is a measure of the fluctuations in the score (viewed as an energy), shows a peak as a function of the temperature in both cases. The peak temperature, which is a measure of the folding transition temperature of a protein for 1c9o is higher than that of 1csp, and reflects the better thermal stability of 1c9o in accord with the experimentally observed behavior (38).
Conclusion
In summary, we have shown that a straightforward learning scheme leads to the determination of excellent environmental parameters that can be used in simple threading tests. Our results point to the danger of using statistical procedures for estimating these values. The learned parameters capture information on the environments in the competing structures in addition to that in the native state structures and allows for a stabilization of the native state with respect to decoy structures. Our procedure validates the notion that, in the simplest cases we have studied here, a simple environmental scoring function is sufficient for capturing the essential features of protein threading. Our method has the distinct advantage of ease of expanding the parameter space and opens up the possibility of using the scoring parameters determined here as a starting point for learning the penalty parameters characterizing insertion and deletion.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Aeronautics and Space Administration, Istituto Nazionale per la Fisica della Materia (INFM), and Ministero dell'Universitá e della Ricerca Scientifica e Tecnologica (MURST; Italy), the Donors of the Petroleum Research Fund administered by the American Chemical Society, the Pusan National University (PNU) research fund, and Komitet Badaǹ Naukowych (KBN; Grant 2P03B-146-18).
Abbreviation
- SVD
singular value decomposition
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Anfinsen C. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Wolynes P G, Onuchic J N, Thirumalai D. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 3.Dill K A, Chan H S. Nat. Struct. Biol. 1997. 410–419. [DOI] [PubMed] [Google Scholar]
- 4.Fersht A P. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1998. [Google Scholar]
- 5.Baker D A. Nature (London) 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 6.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones D T, Taylor W R, Thornton J M. Nature (London) 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 8.Chothia C. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran G N, Sasisekharan V. Adv Protein Chem. 1968;28:283–437. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 10.Bowie J, Lüthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Scheraga H A. Macromolecules. 1976;9:945–950. doi: 10.1021/ma60054a013. [DOI] [PubMed] [Google Scholar]
- 12.Miyazawa S, Jernigan R L. Macromolecules. 1985;18:534–552. [Google Scholar]
- 13.Zhang C, Kim S. Proc Natl Acad Sci USA. 2000;97:2550–2555. doi: 10.1073/pnas.040573597. . (First Published March 7, 2000; 10.1073/pnas.040573597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hobohm U, Sander C. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murzin A G, Brenner S E, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 16.Baud F, Karlin S. Proc Natl Acad Sci USA. 1999;96:12494–12499. doi: 10.1073/pnas.96.22.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vendruscolo M, Najmanovich R, Domany E. Phys Rev Lett. 1999;82:656–659. [Google Scholar]
- 18.Dima R I, Banavar J R, Maritan A. Protein Sci. 2000;9:812–819. doi: 10.1110/ps.9.4.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedrichs M S, Wolynes P G. Science. 1989;246:371–373. doi: 10.1126/science.246.4928.371. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein R, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koretke K K, Luthey-Schulten Z A, Wolynes P G. Protein Sci. 1996;5:1043–1059. doi: 10.1002/pro.5560050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maiorov V N, Crippen G M. J Mol Biol. 1992;227:876–888. doi: 10.1016/0022-2836(92)90228-c. [DOI] [PubMed] [Google Scholar]
- 23.Mirny L A, Shakhnovich E I. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 24.Clementi C, Maritan A, Banavar J R. Phys Rev Lett. 1998;81:3287–3290. [Google Scholar]
- 25.Dima R I, Settanni G, Micheletti C, Banavar J R, Maritan A. J Chem Phys. 2000;112:9151–9166. [Google Scholar]
- 26.Vendruscolo M, Mirny L A, Shakhnovich E I, Domany E. Proteins Struct Funct Genet. 2000;41:192–201. [PubMed] [Google Scholar]
- 27.Tobi D, Elber R. Proteins Struct Funct Genet. 2000;41:40–46. [PubMed] [Google Scholar]
- 28.Tobi D, Shafran G, Linial N, Elber R. Proteins Struct Funct Genet. 2000;40:71–85. doi: 10.1002/(sici)1097-0134(20000701)40:1<71::aid-prot90>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Krauth W, Mezard M. J Phys A. 1987;20:L745–L752. [Google Scholar]
- 30.Watkins D S. Fundamentals of Matrix Computations. New York: Wiley; 1991. [Google Scholar]
- 31.Kauzmann W. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 32.Dill K A. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 33.Kamtekar S, Schiffer J M, Xiong H Y, Babik J M, Hecht M H. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 34.Deutsch J M, Kurosky T. Phys Rev Lett. 1996;76:323–326. doi: 10.1103/PhysRevLett.76.323. [DOI] [PubMed] [Google Scholar]
- 35.Seno F, Vendruscolo M, Maritan A, Banavar J R. Phys Rev Lett. 1996;77:1901–1904. doi: 10.1103/PhysRevLett.77.1901. [DOI] [PubMed] [Google Scholar]
- 36.Dima R I, Banavar J R, Cieplak M, Maritan A. Proc Natl Acad Sci USA. 1999;96:4904–4907. doi: 10.1073/pnas.96.9.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Micheletti C, Maritan A, Banavar J R. J Chem Phys. 1999;110:9730–9738. [Google Scholar]
- 38.Perl D, Mueller U, Heinemann U, Schmid F. Nat Struct Biol. 2000;7:380–383. doi: 10.1038/75151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.