

Abstract
Protein thermal dynamics was evaluated by neutron scattering for halophilic malate dehydrogenase from Haloarcula marismortui (HmMalDH) and BSA under different solvent conditions. As a measure of thermal stability in each case, loss of secondary structure temperatures were determined by CD. HmMalDH requires molar salt and has different stability behavior in H2O, D2O, and in NaCl and KCl solvents. BSA remains soluble in molar NaCl. The neutron experiments provided values of mean-squared atomic fluctuations at the 0.1 ns time scale. Effective force constants, characterizing the mean resilience of the protein structure, were calculated from the variation of the mean-squared fluctuation with temperature. For HmMalDH, resilience increased progressively with increasing stability, from molar NaCl in H2O, via molar KCl in D2O, to molar NaCl in D2O. Surprisingly, however, the opposite was observed for BSA; its resilience is higher in H2O where it is less stable than in D2O. These results confirmed the complexity of dynamics–stability relationships in different proteins. Softer dynamics for BSA in D2O showed that the higher thermostability is associated with entropic fluctuations. In the halophilic protein, higher stability is associated with increased resilience showing the dominance of enthalpic terms arising from bonded interactions. From previous data, it is suggested that these are associated with hydrated ion binding stabilizing the protein in the high-salt solvent.
Solvent interactions provide a complex contribution to protein structure stabilization through hydration, van der Waals interactions, hydrogen bonds, ion binding, and the hydrophobic effect. Because the same forces control thermal fluctuations, a relation among solvent interactions, protein stabilization, and dynamics is expected intuitively, in which a softer, more flexible protein structure would be less stable. Stability, however, need not necessarily be associated with lower flexibility. Neutron-scattering experiments on α-amylase at room temperature have indicated larger amplitudes of motion for atoms in the thermophilic protein compared with the mesophilic homologue, suggesting that thermostability in this case is associated with entropic effects (1). Unfolding experiments on α-lytic protease have shown the existence of a partially unfolded state, I, which is favored entropically and has a lower free energy than the native state, N; under physiological conditions, N is not converted to I because of a very high activation-energy barrier (2). Where entropic terms are dominant, therefore, a more flexible protein could be more stable. Furthermore, measurements of flexibility and rigidity depend strongly on the experimental method used. They could relate to: thermal motions on very fast, ps to 100-ps time scales, measured by neutron scattering (1, 3–9); motions integrated up to the nanosecond or longer times, measured by NMR using isotope labeling (10); or slower conformational changes taking place in milliseconds, measured by hydrogen-exchange experiments (11, 12). The commonly accepted hypothesis in the case of thermophilic proteins, for example, is that their enhanced thermal stability results from enhanced conformational rigidity in the folded native state at room temperature (13). Some studies supported this hypothesis (11, 13), whereas others did not (1, 12). Thus, in this example, there is not a unique mechanism that is responsible for thermostability in the folded state.
Because the solvent environment plays a determinant role in protein stability at any given temperature, our interest in the present study was to evaluate solvent effects on stability, on the one hand, and protein dynamics, on the other hand, and to explore the relationships between them. In more specific terms, our aim was to assess whether there exists a correlation between protein rigidity (or resilience) calculated from dynamics data and secondary structure stability. The neutron-scattering experiments characterized how the mean-square atomic fluctuations in the protein native state varied with temperature in given solvent conditions; the kinetic stability measured by CD provided an indication of the activation free-energy barrier for unfolding (Fig. 1). In terms of Fig. 1, the question can be put in a different way: is the shape of the well in the native state (resilience) related to the height of the barrier (secondary structure stability)?
Figure 1.

Schematic diagram of free energy (ΔG) vs. folding showing atomic fluctuations u in the native state (N) and activation free energy (E∗) for secondary structure loss to give the unfolded state (U).
Heavy water (D2O) is often used as a solvent for
proteins in NMR, neutron-scattering, and spectroscopic studies. Also,
it is known to affect protein stabilization and has been used as probe
in work on protein folding. H2O and
D2O are molecules of almost identical dipole
moment, shape, size, and bond lengths. However, the different masses (D
has twice the mass of H), reduced masses, and moments of inertia make
their vibrational and librational frequencies substantially different
(14, 15). The origin of the different properties of
H2O and D2O with respect to
ionic solvation also lies in how the presence of ions affects the
frequencies of these modes. Zero-point frequencies of the modes in the
bulk solvents and at ions differ by an isotope factor of about
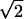 (the exact value depends on the mode) with corresponding
differences in zero-point energy. A smaller isotope effect is observed
in the intermolecular vibration along the hydrogen bond, where the
frequency is reduced by a factor of the square root of the molecular
mass ratio [(16 + 2)/(16 + 4)]1/2 = 0.948.
The fundamental OH · · · O and OD · · · O hydrogen
bond stretch modes, however, show large frequency differences with
correspondingly large differences in zero-point energy of about 1.3
kcal/mol, which are the principal factors determining the different
properties of H2O and D2O.
D2O has a greater degree of structure than
H2O at a given temperature and displays a higher
temperature of maximum density, greater viscosity, and larger heat of
vaporization and sublimation when compared with
H2O. For proteins, this difference in properties
between H2O and D2O leads to stronger
hydration-bond interactions in D2O, as well as to
the solubility of apolar groups being lower in
D2O than in H2O, which
favors the hydrophobic interaction (14).
(the exact value depends on the mode) with corresponding
differences in zero-point energy. A smaller isotope effect is observed
in the intermolecular vibration along the hydrogen bond, where the
frequency is reduced by a factor of the square root of the molecular
mass ratio [(16 + 2)/(16 + 4)]1/2 = 0.948.
The fundamental OH · · · O and OD · · · O hydrogen
bond stretch modes, however, show large frequency differences with
correspondingly large differences in zero-point energy of about 1.3
kcal/mol, which are the principal factors determining the different
properties of H2O and D2O.
D2O has a greater degree of structure than
H2O at a given temperature and displays a higher
temperature of maximum density, greater viscosity, and larger heat of
vaporization and sublimation when compared with
H2O. For proteins, this difference in properties
between H2O and D2O leads to stronger
hydration-bond interactions in D2O, as well as to
the solubility of apolar groups being lower in
D2O than in H2O, which
favors the hydrophobic interaction (14).
Proteins from extreme halophilic Archaea are particularly well suited for the study of solvent–protein interactions because they are active and soluble in a wide range of solvent salt conditions with varying stability; their unfolding, for example, can be observed under normal temperatures in the absence of usual denaturing agents (16). Malate dehydrogenase from Haloarcula marismortui (HmMalDH) is the most extensively studied halophilic protein (17, 18). The enzyme is a tetramer of the lactate dehydrogenase (LDH)-like MalDH family (16, 19). It requires molar solvent salt concentrations for stability and solubility. In NaCl or KCl solutions, it binds exceptional amounts of hydrated salt ions (17). The crystal structure of HmMalDH shows intersubunit salt-bridge clusters, similar to hyperthermophilic proteins (20); the clusters in the halophilic enzyme seem to be stabilized by chloride ion binding (21), which may well be a feature of halophilic adaptation, because ion binding would be favored in the high salt conditions that might be expected to destabilize salt-bridge clusters. The kinetic stability of HmMalDH has been studied as a function of salt type and concentration in H2O and D2O solutions (16). Solvent-induced inactivation of the protein is due to concomitant dissociation of the tetramer and unfolding of monomers. It occurs as a first order reaction. In molar NaCl or KCl in H2O, enthalpic terms dominate. The protein is more stable in NaCl than in KCl, which was interpreted as caused by the higher hydration and binding energies of Na+ compared with K+. The protein is also more stable in D2O than in H2O (16). It has been shown that, under low-salt conditions, the stabilization of HmMalDH by D2O in the solvent arises from a larger entropic contribution to the activation-free energy of unfolding, which indicates a stronger hydrophobic effect than in H2O, rather than stronger hydration interactions (16). Because the stabilization of HmMalDH in various solvents has been studied extensively (16, 22, 23), the protein is well suited for the study of protein dynamics in corresponding conditions to explore a correlation between dynamics and stability. Also, KCl is selected universally as the dominant cytoplasmic salt, and considerable energy is consumed pumping Na+ ions out of cells; in addition, it was of particular interest to compare protein dynamics in NaCl and KCl solutions.
As a comparison with HmMalDH, we studied a nonhalophilic protein model. BSA was chosen because it is soluble and stable in NaCl concentrations up to the molar range, and its thermal unfolding has been characterized (24, 25). The structure of BSA is not known, but its sequence displays 80% homology with that of human serum albumin (26), whose crystal structure has been solved (27). BSA contains about 55% α-helix structure stabilized by 17 disulfide bonds (26). Raman optical activity measurements have indicated many helix-loop-helix motifs (28). A recent compressibility study confirmed the behavior of BSA native structure to be in the range of other native globular compact proteins (29).
We used neutron scattering to measure the dynamics of the halophilic and nonhalophilic protein as a function of temperature in various concentrations of NaCl or KCl in H2O or D2O solutions. The thermal secondary structure unfolding was measured by CD on the same solutions.
There is an overlap between the time scales of neutron-scattering experiments and those of NMR. Neutron-scattering experiments, however, can be performed on proteins of any size to provide time-dependent atomic fluctuation amplitudes in absolute units. Neutron-scattering experiments were performed on a spectrometer that allows one to examine atomic motions in the space and time window of ≈1 Å in 0.1 ns. All motions that are outside the window, such as the diffusion of bulk water (≈10 Å in 0.1 ns), for example, did not contribute to the scattering signal. In a set of so-called “elastic incoherent” neutron-scattering experiments, the global mean-square fluctuation 〈u2〉 is measured as a function of temperature. Two parameters providing information on dynamics can be analyzed from such a scan: the accurate value of the global mean-square fluctuation 〈u2〉, in Å2 and the mean resilience, or rigidity of the structure calculated as an effective mean force constant (in Newtons per meter) from the slope of 〈u2〉 vs. T (3).
Various biophysical methods have been applied to probe the hierarchical events that occur during protein unfolding. We chose the CD method as a simple and rapid method to provide a qualitative measure of protein stability by monitoring secondary structure as a function of temperature in the various solutions. The CD method also is efficient in that small amounts of sample are required.
We present results for HmMalDH and BSA in H2O and D2O solutions containing different salts. The neutron results combined with CD data in corresponding conditions established complex correlations between dynamics and stability, which are different for the halophilic and nonhalophilic proteins. We discuss these observations in terms of entropy or enthalpy dominated mechanisms for stabilization of a protein in a given solvent environment.
Materials and Methods
Sample Preparation.
Halophilic MalDH was expressed in E. coli and purified according to the protocol of Cendrin et al. (30). It was stored in 4 M NaCl and 50 mM Tris⋅HCl (pH 8) at 4°C. Before the neutron-scattering experiments, the protein was concentrated by ultrafiltration over Centricon 30 membranes (Amicon) to ≈220 mg/ml. It was dialyzed at 4°C for 2 days with four changes of the solvent in 2 M NaCl in D2O (2 M NaCl⋅D2O), 2 M KCl in D2O (2 M KCl⋅D2O) and 2 M NaCl in H2O (2 M NaCl⋅H2O), respectively. The final concentration of 200 mg/ml for the neutron-scattering experiments was obtained by dilution with the corresponding solvent. HmMalDH concentration was determined by spectrophotometry by using an extinction coefficient E280 of 0.85 ml/mg/cm (17).
BSA was obtained from Bayer (BSA crystallized). Solutions of 200 mg/ml were dialyzed at 4°C for 2 days with four changes of the solvent in 200 mM KCl in D2O, 200 mM KCl in H2O, and 2 M NaCl in D2O, respectively. Approximate BSA concentration was determined by weight.
Before the CD experiments, BSA and HmMalDH solutions were diluted with the corresponding solvent, to obtain a final concentration of 0.5 mg/ml.
BSA and HmMalDH samples for neutron-scattering and CD experiments were buffered with 10 mM Tris⋅HCl at pH 8.
Neutron-Scattering Theory.
The technique of neutron scattering is uniquely suited to measure protein dynamics because neutron wavelengths and energies, respectively, match the amplitudes and energies of macromolecular thermal fluctuations (3, 4). Because of their large incoherent scattering cross section, the motions of H nuclei dominate the observations (5); the experiments, nevertheless, provide information on protein dynamics because, in the time scale examined (up to ≈0.1 ns), H atoms reflect the motions of the side chains and backbone atoms to which they are bound (6). There are multiple contributions to these motions in the space-time window examined, such as local side chain motions (but not the rotation of buried aromatic groups, which is very slow) and collective motions of ∝-helix and β-sheet secondary structure elements; cooperative “breathing” domain motions may be too slow to contribute, depending on the size of the domain (6). The incoherent scattering of deuterium, D(2H), also is weak with respect to that of 1H; so far, protein dynamics have been studied predominantly in D2O to reduce the scattering contribution of the solvent (6). Neutron spectrometers are characterized by their energy resolution, Δω, scattering vector, Q, and range, corresponding to time and space windows related to 1/Δω and 1/Q, respectively. The energy resolution on back-scattering spectrometers is better than 10 μeV, corresponding to a time window of about 0.1 ns. The Q values are of the order 1 Å−1, corresponding to a window for 〈u2〉 of about 1 Å2. The motion can be considered as localized when the atomic fluctuations are inside the time and space window. For Q2 〈u2〉 ≈ 2, the incoherent elastic scattered intensity can be analyzed according to a Gaussian approximation (6):
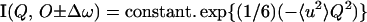 |
1 |
Q is given by 4πsinθ/λ, where 2θ is the scattering angle, and λ is the incident neutron wavelength; 〈u2〉 values include all contributions to motions in the accessible space and time windows. In this approximation, 〈u2〉 includes all displacements in the time scale, vibrational fluctuations (usually expressed as a Debye-Waller factor), as well as diffusional motions. An effective mean force constant 〈k′〉 can be calculated from the derivative of 〈u2〉 plotted vs. temperature, T (3, 7):
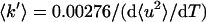 |
2 |
The numerical constants allow one to express 〈k′〉 in newtons/meter (N/m) when 〈u2〉 is in Å2 and T is Kelvin.
It was shown by comparing experiments on proteins in solution and in hydrated powders that, at the protein concentration used here, solvent viscosity could shift 〈u2〉 values but has no effect on the slope of 〈u2〉 vs. T (8).
Incoherent Neutron-Scattering Experiments.
Experiments were performed on the backscattering spectrometer IN13 at the Institute Laue Langevin Grenoble, France (information on the Institute and the instrument is available on the web at: www.ill.fr). The instrument is managed by a French–Italian Collaborating Research Group. The energy resolution was 8 μeV with a neutron wavelength λ = 2.23 Å. Samples were contained in Teflon-coated aluminum sample holders. The elastic incoherent neutron scattering within the energy resolution was analyzed. The scattering of the dialysis buffer alone was much lower than the protein solution scattering, barely above the scattering of the aluminum container. It (the scattering of the dialysis buffer) was subtracted from the data with no correction for protein-excluded volume. The data were divided by the scattering of a vanadium sample to correct for detector response. The transmissions of all samples were about 0.9 and corrections for self-absorption using Paalman-Pings coefficients were carried out with standard programs. Resulting intensities were analyzed according to Eq. 1. The 〈u2〉 values were calculated from linear fits of ln[Iinc(Q, 0 ± Δω, T)] versus Q2 using the Marquart-Levenberg algorithm (gnufit under gnuplot software on UNIX), in the Q-range 1.2–2.2 Å−1, where the Gaussian approximation is valid. The often-used correction for coherent scattering (dividing the data by that at very low temperature) ≅ 20 K (9) was not applied, because 〈u2〉 values at 280 K were relatively large, and the coherent scattering in the range examined was negligible. To avoid damage from freezing and denaturation and to maintain conditions in which the samples retain biological activity, experiments were performed in the restricted temperature range 280–320K (7°C–47°C).
CD Experiments.
A Jobin Yvon CD6 spectropolarimeter with a thermostated sample holder (Haake thermostat) was used. Aliquots of protein solutions were incubated during 30 min at the required temperature. Incubation times, at each temperature, were kept sufficiently long to reach the final state of denaturation. Data were recorded at 25°C, in the wavelength range of 180–260 nm with an interval of 1 nm and an integration time of 2 s using 0.1-cm quartz cuvettes. BSA and HmMalDH concentrations were ≈0.5 mg/ml. The mean residue ellipticity, θ (deg·cm2·dmol−1), was calculated according to:
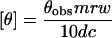 |
3 |
where θobs is the observed dichroic absorption, mrw is the mean residue weight (110), d is the optical path length in centimeters, and c is the enzyme concentration in mg/ml.
The thermal denaturation curves were measured by monitoring the change in the CD values at 222 nm, and the normalized ellipticity was determined according to:
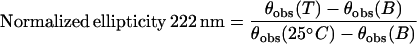 |
4 |
where θobs(T), θobs(25°C), and θobs(B) are the observed dichroic absorptions of the protein solutions incubated at the temperature T, at 25C and of the buffer respectively.
In the present study, we defined “Stability” by secondary structure resistance to thermal denaturation.
Results and Discussion
CD from D2O and H2O Solutions.
Typical CD scans from D2O and H2O protein solutions are in Fig. 2. To assess thermal loss of secondary structure, the value at 222 nm was plotted as a function of temperature (in Fig. 3, and see Fig. 5 below).
Figure 2.

Far-UV CD spectra of BSA recorded at 25°C in 200 mM KCl H2O (A) and 200 mM KCl D2O (B) at pH 8. The protein concentration was 0.5 mg/ml. Spectra obtained after different temperature incubations [25°C (⋄), 55°C (○), 65°C (▿), 70°C (×), 80°C (+), 85°C (▵), and buffer at 25°C (●)] are presented. Experimental details are in Materials and Methods.
Figure 3.

Normalized ellipticities at 222 nm plotted as a function of accurate temperature. HmMalDH curves in 2 M NaCl D2O (■), 2 M KCl D2O (□), and 2 M NaCl H2O (▿) at pH 8. HmMalDH samples in 2 M NaCl D2O and in 2 M KCl D2O were aggregated at temperature incubation greater than 70°C. Protein concentrations were 0.5 mg/ml. Experimental details are in Materials and Methods.
Figure 5.

Normalized ellipticities at 222 nm are plotted as a function of accurate temperature. Experimental details are given in Materials and Methods. BSA curves in 200 mM KCl H2O (⋄) and 200 mM KCl D2O (♦) at pH 8. Protein concentrations were 0.5 mg/ml.
Incoherent Neutron Scattering from D2O and H2O Solutions.
We recall that previous neutron experiments on protein dynamics have been performed predominantly in D2O, to reduce the scattering contribution of the solvent. However, it is well known that D2O has important effects on protein stability and solubility (14, 16), so that observations made in D2O solutions cannot be assumed to reflect behavior in H2O. We have succeeded in measuring protein dynamics by neutron scattering in H2O solutions without being bothered by the scattering contribution of solvent H nuclei, because bulk solvent and protein motions are in different space-time windows. In the case of the backscattering spectrometer IN13, the space-time window corresponds to ≈1 Å in 0.1 ns. The scattering of atoms that diffuse out of this window will be limited essentially to Q = 0 and will not contribute to the observed data. This is the case for bulk water, which has a diffusion coefficient of 2·10−5 cm2/s at 2°C, corresponding to a 〈u2〉 value of 100 Å2 in 0.1 ns, well outside the IN13 window.
Ionic and D2O Effects on the Native-Unfolded Transition of HmMalDH.
HmMalDH was studied in 2 M NaCl⋅H2O, 2 M NaCl⋅D2O, and 2 M KCl⋅D2O solutions, where it is stable. HmMalDH denatures with faster first-order kinetics in KCl than in NaCl solvents (23) and with faster first-order kinetics in H2O than in D2O solution (16). Denaturation of HmMalDH in “low” salt has been shown to be caused by dissociation of the tetramer and monomer unfolding, with concomitant loss of secondary and tertiary structure (18). In this case, therefore, the CD measurement characterizes a unique unfolding temperature.
A measure of the “stability” of HmMalDH in each solvent condition studied by neutron scattering was obtained by monitoring the thermal loss of secondary structure by CD (Fig. 3). We note, however, that these experiments (as well as the ones on BSA discussed below) were intended to provide only a qualitative indication of the height of the denaturation barrier in Fig. 1, because of the aggregation observed in some of the conditions. Also, protein concentration itself is known to have a stabilizing effect on the native fold, and for the CD experiments, protein concentration was more than two orders of magnitude lower than for the neutron experiments.
Ionic Effects.
The normalized ellipticity in 2 M KCl⋅D2O decreases in the explored temperature range, whereas that in 2 M NaCl⋅D2O remains more or less constant, showing the protein to be more thermostable in the latter condition (Fig. 3). This difference in stability between HmMalDH in 2 M KCl⋅D2O and in 2 M NaCl⋅D2O is small but significant. The higher stability in NaCl as compared with KCl has been interpreted in terms of a hydration shell model in which the protein binds hydrated solvent ions (16). The increased stability of the protein in NaCl would result because the hydration shell energy of Na+ is larger than that of K+ (31).
D2O Effects.
In NaCl solvents, denaturation of HmMalDH is significantly faster in H2O than in D2O (16). The CD in 2 M NaCl⋅D2O and 2 M NaCl⋅H2O results follow this trend (Fig. 3). A denaturation curve showing a melting temperature of about 48°C was observed in 2 M NaCl⋅H2O, whereas in D2O full denaturation does not occur up to aggregation at about 7°C. The higher stability of the protein in D2O compared with H2O could be interpreted in terms of the hydrophobic effect as well as the stronger ion–water–protein D-bonds in the hydration shell (14, 16).
Ionic and D2O Effects on the Dynamics of Native HmMalDH.
The mean-square fluctuations, 〈u2〉, of HmMalDH in the various solvents are plotted as a function of temperature in Fig. 4. The 〈u2〉 values were fitted by straight lines between 280 K and 320 K. The CD data showed the protein in 2 M NaCl⋅H2O to be denatured at 320 K (47°C) (Fig. 3). The mean amplitudes of fast protein motions have been shown by neutron scattering to increase significantly when the protein unfolds (32). By scaling the 〈u2〉 values according to reference (32), we expected a value between 3 and 4 Å2 at 320 K. The measured value for 2 M NaCl⋅H2O at 320 K, however, falls on the straight line, suggesting that the protein is still in its native, folded state at this temperature. This stabilization could well be the result of the high protein concentration in the neutron experiments (about 200 g/liter, compared with 0.5 g/liter for the CD experiments). The inverse of the slope of the 〈u2〉 vs. temperature corresponds to the mean resilience of the protein structure, characterized by an effective mean force constant, 〈k′〉 (Eq. 2). A smaller slope would indicate a more resilient (rigid) protein structure and vice-versa. Error bars are somewhat larger for the 2 M NaCl⋅H2O condition, because of higher solvent background scattering. Significant differences in mean resilience are observed for the protein in the different solvent conditions. For comparison, we recall that the resilience of myoglobin measured in D2O solution or as a D2O-hydrated powder was 0.3 N/m (8). The mean resilience is larger in 2 M NaCl⋅D2O (0.505 ± 0.049 N/m) than in 2 M KCl⋅D2O (0.205 ± 0.04 N/m), indicating a more rigid protein structure. We recall that HmMalDH is more stable in NaCl than in KCl. In this case, therefore, thermal stability is correlated with higher resilience.
Figure 4.

Global mean-square fluctuations 〈u2〉 in HmMalDH were calculated from the scattered elastic incoherent intensity and plotted as a function of temperature; mean resilience, which is characterized by an effective force constants, 〈k′〉, was calculated from the slopes of the straight-line fits to the data. The error bars correspond to statistical errors. Experimental details are in Materials and Methods. In 2 M NaCl D2O (○), 2 M KCl D2O (▿), and 2 M NaCl H2O (⋄) at pH 8. Protein concentration was 200 mg/ml. Calculated k′ values were 0.113 ± 0.007 N/m in 2 M NaCl⋅H2O, 0.205 ± 0.04 N/m in 2 M KCl⋅D2O, and 0.505 ± 0.049 N/m in 2 M NaCl⋅D2O.
Parameters describing the dynamics of HmMalDH in 2 M NaCl⋅D2O and in 2 M NaCl⋅H2O are significantly different. The mean resilience in 2 M NaCl⋅D2O (0.505 ± 0.049 N/m), where HmMalDH is significantly more stable, is larger than in 2 M NaCl⋅H2O (0.113 ± 0.007 N/m). Here, also, higher stability is correlated with higher resilience, suggesting that in the 2 M NaCl conditions, the stronger D-bonds in the hydration shell dominate, rather than a stronger hydrophobic effect. We recall that in low-salt D2O solvents, the entropic term has been found to be dominant in the activation-free energy of denaturation, suggesting stabilization by the hydrophobic effect in these conditions (16).
In conclusion, the resilience of HmMalDH increases progressively with increasing secondary structure thermostability with 〈k′〉 values varying from 0.113 ± 0.007 N/m in 2 M NaCl⋅H2O through 0.205 ± 0.04 N/m in 2 M KCl⋅D2O to 0.505 ± 0.049 N/m in 2 M NaCl⋅D2O. The correlation with the strength of hydration ion-binding in the molar-salt solutions studied (16) suggests the enthalpic contribution of the hydration shell dominates the dynamics behavior of the halophilic protein.
Ionic and D2O Effects on the Native-Unfolded Transition of BSA.
BSA was studied in low-salt H2O and D2O solvents and in 2 M NaCl⋅D2O.
Ionic Effects.
In comparative experiments to measure salt and water binding to proteins in molar salt solvents, it has been shown that, unlike HmMalDH, BSA does not bind salt but displays usual solvation by about 0.2 g water per g of protein (22). The mild stabilizing effect of molar NaCl solvent on BSA then would be caused by “salting out” and would be related to a strengthening of the hydrophobic effect (33).
D2O Effects.
The normalized ellipticity of BSA in H2O starts to decrease significantly from about 55°C, whereas that of BSA in D2O is maintained to about 65°C (Fig. 5). Thus, as observed with other proteins (16), D2O increases the stability of BSA. BSA is known to form dimers under certain conditions. We addressed the question whether D2O favored dimer formation in the solvent conditions of the experiments by analytical ultracentrifugation (data not shown). Protein concentration was in the order of 1 g/liter; sedimentation profiles were measured for the protein in three solvents: 200 mM NaCl in H2O, 200 mM NaCl in D2O, and 2 M NaCl in D2O. Profiles were fitted well by a single species in all cases, with a normalized sedimentation coefficient, S20,W of 4.6 s, corresponding to the BSA monomer.
Ionic and D2O Effects on the Dynamics of Native BSA.
The global mean-square fluctuations, 〈u2〉, of BSA in the various solvents are shown as a function of temperature in Fig. 6. The error bars are larger for the BSA in D2O sample, because the experimental measuring time was half as long as for the other samples. The resilience values for BSA in D2O and in 2 M NaCl⋅D2O are similar (0.085 ± 0.012 and 0.125 ± 0.008 N/m, respectively). The 〈u2〉 values for BSA in 2 M NaCl⋅D2O, however, are lower by about 0.7 Å2 with respect to BSA in D2O, probably due to the higher viscosity of the 2 M NaCl solvent. A concentration of 2 M NaCl in D2O solvent, therefore, does not significantly modify the mean resilience of the BSA structure when compared with the low-salt condition.
Figure 6.

Global mean-square fluctuations 〈u2〉 in BSA were calculated from the scattered elastic incoherent intensity and plotted as a function of accurate temperature; mean resilience, which is characterized by an effective force constants, 〈k′〉, were calculated from the slopes of the straight-line fits to the data. The error bars correspond to statistical errors. Experimental details are in Materials and Methods. In 200 mM KCl H2O (⋄), 200 mM KCl D2O (♦), and 2 M NaCl D2O (●) at pH 8. Protein concentration was 200 mg/ml. Calculated k′ values were 0.085 ± 0.012 N/m in 200 mM KCl D2O, 0.125 ± 0.008 N/m in 2 M NaCl D2O, and 0.55 ± 0.246 N/m in 200 mM KCl H2O.
The dynamics of the protein in D2O and H2O solvents were found to be significantly different (Fig. 6). The mean resilience of BSA in H2O (〈k′〉 = 0.55 ± 0.246 N/m) is a factor of six greater than that of BSA in D2O (〈k′〉 = 0.085 ± 0.012 N/m). The more stable BSA structure, therefore, seems to be the least resilient, suggesting that, in this case, the stabilizing effect of D2O is dominated by entropic terms, i.e., an increase in conformational freedom, not only in the solvent but also in the protein. Such an increase in protein-fluctuation amplitudes has been observed in other experiments that used H2O/D2O or D2O solvents, where the effects of hydrophobic ligand binding on protein dynamics were analyzed by NMR (34), and in a comparison of motion amplitudes in thermophilic and mesophilic α-amylases by quasi-elastic neutron scattering (1). Unfolding experiments on α-lytic protease have shown that an intermediate has a lower free energy than the native state because it is favored entropically (2).
In conclusion, in the case of BSA, stability is not correlated with higher resilience.
Conclusions
Neutron Scattering from H2O and D2O Solutions.
Most previous incoherent neutron-scattering experiments on proteins have been performed in D2O solvents to have a good signal-on-background ratio, the incoherent neutron cross section of D being much lower than that of H. Heavy water solvents, however, have significant effects on protein stability and dynamics. Such experiments should not be interpreted without taking these effects into account. We showed that by using the space-time resolution of a spectrometer to filter out bulk-water motions, it is possible to examine protein dynamics in H2O solution.
Comparing BSA and HmMalDH.
The 〈u2〉 found for BSA and HmMalDH in H2O solution at 300 K are similar at about 2 Å2; 〈u2〉 values at other temperatures and in D2O solution depend on the protein resilience in each condition. The mean resilience of BSA is higher in H2O solvents than in D2O. In this case, the less resilient protein structure is more thermostable. A higher activation energy barrier is associated with a wider well in Fig. 1. This fact shows a dominance of entropic effects in stabilization, the native protein sampling different conformational substates. On the contrary, enthalpic terms seem to dominate HmMalDH behavior. The most stabilizing solvent conditions (including H2O–D2O exchange) correspond to increasing resilience. In this case, a higher activation energy barrier is associated with a narrower well (Fig. 1). The order of rising mean resilience values for the HmMalDH in 2 M NaCl⋅H2O, 2 M KCl⋅D2O, and 2 M NaCl⋅D2O is correlated with that of a variety of thermodynamic and spectroscopic parameters characteristic of the effect of ions and of the isotope effect of D2O on H-bond structure, indicating a stronger bonding in the order H2O to D2O and K+ to Na+ (15). We could expect protein ion interactions in the hydration shell to follow the same ranking, suggesting that bonded interactions in the hydration shell of the halophilic protein dominate its stability and its dynamics.
The results in this paper confirmed that the relationship between dynamics and stability in proteins is more complex than expected intuitively. A more stable native protein structure is associated with lower resilience in the mesophilic globular protein, whereas in the halophilic protein, higher resilience that was observed to be associated with stability seemed to be dominated by bonded protein–solvent interactions.
Acknowledgments
We thank Dr. Christine Ebel, Dr. Anna Mitraki, Dr. Bruno Franzetti, Dr. Lawrence Cosenza, and Adriana Irimia for very fruitful discussions. M.T. is supported by a doctorate grant from the Région Rhône-Alpes, France.
Abbreviations
- HmMalDH
malate dehydrogenase from Haloarcula marismortui
- LDH
lactate dehydrogenase
- BSA
bovine serum albumin
References
- 1.Fitter J, Heberle J. Biophys J. 2000;79:1629–1636. doi: 10.1016/S0006-3495(00)76413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sohl J L, Jaswal S S, Agard D A. Nature (London) 1998;395:817–819. doi: 10.1038/27470. [DOI] [PubMed] [Google Scholar]
- 3.Zaccai G. Science. 2000;288:1604–1607. doi: 10.1126/science.288.5471.1604. [DOI] [PubMed] [Google Scholar]
- 4.Bu Z, Neumann D A, Lee S H, Brown C M, Engelman D M, Han C C. J Mol Biol. 2000;301:525–536. doi: 10.1006/jmbi.2000.3978. [DOI] [PubMed] [Google Scholar]
- 5.Bée M. Quasielastic Neutron Scattering: Principles and Applications in Solid State Chemistry, Biology and Materials Science. Philadelphia, PA: Adam Hilger; 1988. [Google Scholar]
- 6.Smith J C. Q Rev Biophys. 1991;24:227–291. doi: 10.1017/s0033583500003723. [DOI] [PubMed] [Google Scholar]
- 7.Bicout D J, Zaccai G. Biophys J. 2001;80:1115–1123. doi: 10.1016/S0006-3495(01)76089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaccai, G., Tehei, M., Scherbakova, I., Serdyuk, I., Gerez, C. & Pfister, C. (2000) J. Phys. IV [French]10, Pr7-283–287.
- 9.Reat V, Patzelt H, Ferrand M, Pfister C, Oesterhelt D, Zaccai G. Proc Natl Acad Sci USA. 1998;95:4970–4975. doi: 10.1073/pnas.95.9.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee A L, Wand A J. Nature (London) 2001;411:501–504. doi: 10.1038/35078119. [DOI] [PubMed] [Google Scholar]
- 11.Zavodszky P, Kardos J, Svingor, Petsko G A. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez G, Jenney F E, Jr, Adams M W, LeMaster D M. Proc Natl Acad Sci USA. 2000;97:3166–3170. doi: 10.1073/pnas.040569697. . (First Published March 14, 2000; 10.1073/pnas.040569697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaenicke R. Naturwissenschaften. 1996;83:544–554. doi: 10.1007/BF01141979. [DOI] [PubMed] [Google Scholar]
- 14.Némethy G, Scheraga H A. J Chem Phys. 1964;41:680–689. [Google Scholar]
- 15.Conway B E. Studies in Physical and Theoretical Chemistry. Vol. 12. Amsterdam, The Netherlands: Elsevier Science; 1981. [Google Scholar]
- 16.Bonnete F, Madern D, Zaccai G. J Mol Biol. 1994;244:436–447. doi: 10.1006/jmbi.1994.1741. [DOI] [PubMed] [Google Scholar]
- 17.Bonneté F, Ebel C, Zaccai G, Eisenberg H. J Chem Soc Faraday Trans. 1993;89:2659–2666. [Google Scholar]
- 18.Madern D, Ebel C, Zaccai G. Extremophiles. 2000;4:91–98. doi: 10.1007/s007920050142. [DOI] [PubMed] [Google Scholar]
- 19.Madern D, Ebel C, Mevarech M, Richard S B, Pfister C, Zaccai G. Biochemistry. 2000;39:1001–1010. doi: 10.1021/bi9910023. [DOI] [PubMed] [Google Scholar]
- 20.Dym O, Mevarech M, Sussman J L. Science. 1995;267:1344–1346. doi: 10.1126/science.267.5202.1344. [DOI] [PubMed] [Google Scholar]
- 21.Richard S B, Madern D, Garcin E, Zaccai G. Biochemistry. 2000;39:992–1000. doi: 10.1021/bi991001a. [DOI] [PubMed] [Google Scholar]
- 22.Pundak S, Eisenberg H. Eur J Biochem. 1981;118:463–470. doi: 10.1111/j.1432-1033.1981.tb05542.x. [DOI] [PubMed] [Google Scholar]
- 23.Pundak S, Aloni H, Eisenberg H. Eur J Biochem. 1981;118:471–477. doi: 10.1111/j.1432-1033.1981.tb05543.x. [DOI] [PubMed] [Google Scholar]
- 24.Privalov P L. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 25.Bulone D, Martorana V, San Biagio P L. Biophys Chem. 2001;91:61–69. doi: 10.1016/s0301-4622(01)00155-7. [DOI] [PubMed] [Google Scholar]
- 26.Peters T., Jr Adv Protein Chem. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- 27.He X M, Carter D C. Nature (London) 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 28.Wen Z Q, Hecht L, Barron L D. J Am Chem Soc. 1994;116:443–445. [Google Scholar]
- 29.Katzhendler I, Priev A, Friedman M. J Controlled Release. 2000;67:261–274. doi: 10.1016/s0168-3659(00)00211-x. [DOI] [PubMed] [Google Scholar]
- 30.Cendrin F, Jouve H M, Gaillard J, Thibault P, Zaccai G. Biochim Biophys Acta. 1994;1209:1–9. doi: 10.1016/0167-4838(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 31.Collins K D. Biophys J. 1997;72:65–76. doi: 10.1016/S0006-3495(97)78647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Receveur V, Calmettes P, Smith J C, Desmadril M, Coddens G, Durand D. Proteins. 1997;28:380–387. [PubMed] [Google Scholar]
- 33.Von Hippel P, Schleich T. In: Structure of Biological Macromolecules. Timasheff S N, Fasman G D, editors. Vol. 2. New York: Dekker; 1969. pp. 417–575. [Google Scholar]
- 34.Zidek L, Novotny M V, Stone M J. Nat Struct Biol. 1999;6:1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]