

Abstract
Purpose:
This study aimed to study whether cancer cells possess distinguishing metabolic features compared with surrounding normal cells, such as increased glutamine uptake. Given this, quantitative measures of glutamine uptake may reflect critical processes in oncology. Approximately, 10 % of patients with colorectal cancer (CRC) express BRAFV600E, which may be actionable with selective BRAF inhibitors or in combination with inhibitors of complementary signaling axes. Non-invasive and quantitative predictive measures of response to these targeted therapies remain poorly developed in this setting. The primary objective of this study was to explore 4-[18F]fluoroglutamine (4-[18F]F-GLN) positron emission tomography (PET) to predict response to BRAFV600E-targeted therapy in preclinical models of colon cancer.
Procedures:
Tumor microarrays from patients with primary human colon cancers (n = 115) and CRC liver metastases (n = 111) were used to evaluate the prevalence of ASCT2, the primary glutamine transporter in oncology, by immunohistochemistry. Subsequently, 4-[18F]F-GLN PET was evaluated in mouse models of human BRAFV600E-expressing and BRAF wild-type CRC.
Results:
Approximately 70 % of primary colon cancers and 53 % of metastases exhibited positive ASCT2 immunoreactivity, suggesting that [18F]4-F-GLN PET could be applicable to a majority of patients with colon cancer. ASCT2 expression was not associated selectively with the expression of mutant BRAF. Decreased 4-[18F]F-GLN predicted pharmacological response to single-agent BRAF and combination BRAF and PI3K/mTOR inhibition in BRAFV600E-mutant Colo-205 tumors. In contrast, a similar decrease was not observed in BRAF wild-type HCT-116 tumors, a setting where BRAFV600E-targeted therapies are ineffective.
Conclusions:
4-[18F]F-GLN PET selectively reflected pharmacodynamic response to BRAF inhibition when compared with 2-deoxy-2[18F]fluoro-D-glucose PET, which was decreased non-specifically for all treated cohorts, regardless of downstream pathway inhibition. These findings illustrate the utility of non-invasive PET imaging measures of glutamine uptake to selectively predict response to BRAF-targeted therapy in colon cancer and may suggest further opportunities to inform colon cancer clinical trials using targeted therapies against MAPK activation.
Keywords: Glutamine, Pet, Colon, Cancer, BRAF
Introduction
The metabolic profile of cancer cells can diverge significantly from that of normal cells. Energy production in cancer cells can be abnormally dependent on aerobic glycolysis [1, 2]. Consequently, positron emission tomography (PET) imaging using 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) is a mainstay of cancer diagnosis and staging. Despite the fact that glycolysis is enhanced in many tumors, it is not necessarily a tumor-specific molecular process [3, 4]. Like glucose, [18F]FDG accumulates in tissues that exhibit increased rates of glycolysis such as the brain; brown adipose tissue; rapidly-dividing tissues, such as cancers; and inflammation. Within this context, [18F]FDG PET is insufficient for the detection of tumors with lower metabolic rates, such as bladder, prostate, and renal cancer, in addition to small tumors and tumors of low cell density. Furthermore, [18F]FDG PET has been found to be indifferent to various therapeutics, such as predicting response to inhibitors of mutant BRAF in mutant BRAFV600E tumors [5, 6]. These limitations necessitate alternative targets for molecular imaging and corresponding PET imaging probes.
In addition to dependency on glycolysis, cancer cells can exhibit other atypical metabolic characteristics, such as increased dependency on the amino acid glutamine [7]. In cancer cells, both glucose and glutamine serve as key carbon sources for ATP production and biosynthesis. Glutamine also participates in protein synthesis and is a nitrogen source for the production of certain amino acids and nucleotides [8]. Emerging evidence suggests that glutamine uptake in cancer cells is controlled by oncogenic signaling pathways [9–13]. Using glioma cells, Wise and colleagues demonstrated that MYC regulates glutaminolysis and results in PI3K-independent glutamine addiction [14]. Other studies have revealed a role for mutant KRAS in the regulation of glutamine-derived cancer cell metabolism [11]. In pancreatic adenocarcinomas (PDACs), oncogenic KRAS has been shown to modulate the expression of key genes enabling the glutamine-utilizing transaminase pathway that is required for growth in PDAC, but not normal, cells [15]. Previously, alanine-serine-cysteine transporter 2 (ASCT2), a sodium-dependent neutral amino acid transporter, has been identified as the primary transporter of glutamine in many human cancers including breast, colon, lung, prostate, and pancreas [16–21]. In lung cancer, ASCT2 expression has been linked to poor survival. Preclinical studies in this setting have shown pharmacological inhibition of ASCT2 attenuates mTOR signaling and cell growth [18, 19]. Novel drugs targeting the glutamine metabolism pathway, such as glutaminase inhibitors, have shown promise in recent studies in triple-negative breast cancer [22]. Given this, quantitative measures of glutamine uptake may reflect critical processes in oncology that are difficult to measure using existing imaging metrics. Accordingly, PET imaging agents targeting glutamine uptake, such as [18F]-4-fluoroglutamine (4-[18F]F- GLN), have been reported and used in preclinical and clinical studies in oncology [23, 24]. In preclinical studies, 4-[18F]F-GLN was found to reflect ASCT2 expression in lung and CRC human xenograft tumors and has shown clinical potential in glioblastomas [23–26].
The primary objective of this study was to explore 4- [18F]F-GLN) PET to predict response to BRAFV600E-targeted therapy in preclinical models of colon cancer. To this end, we evaluated the prevalence of ASCT2 expression in primary human colon cancer and CRC liver metastases. Subsequently, 4-[18F]F-GLN PET was evaluated as a measure of targeted therapeutic response in preclinical models of BRAFV600E-expressing and wild-type BRAF. Our results illustrate the utility of non-invasive PET imaging measures of glutamine uptake to selectively predict response to BRAF-targeted therapy in colon cancer and may further suggest an opportunity to inform the efficacy of colon cancer clinical trials targeting other portions of the canonical epidermal growth factor signaling cascade.
Materials and Methods
Human Colon Cancer Microarray
Samples from patients treated at Vanderbilt University Medical Center for primary (n = 115) and metastatic (n = 111) colon cancer along with adjacent and matched normal tissues were assembled into tumor microarrays by a GI pathologist (M. Kay Washington, M.D.). Immunohistochemistry for ASCT2 was carried out on 5-μm sections of formalin-fixed, paraffin-embedded tissue using the avidin-biotin complex method using the Vectastain Elite ABC kit (Vector Laboratories). Briefly, slides were placed on a Leica Bond Max IHC stainer and heat-induced antigen retrieval was performed using an Epitope Retrieval 2 solution for 20 min. Subsequently, the slides were placed in a Protein Block (×0909, DAKO) for 10 min and then incubated with anti-SLC1A5 (ASCT2; HPA035240, Sigma) for 1 h at a 1:5000 dilution. A Bond Polymer Refine detection system was used for visualization. Of the samples obtained from 115 patients with primary colon cancers and matching normal tissues and 111 metastatic colon cancers and matched normals, a somewhat smaller number contained sufficient quantities of evaluable tumor or matched normal tissue (n = 97 primary tumors plus normal, n = 84 metastatic tumors plus normal). ASCT2 levels were objectively quantitated in a blinded manner as the mean score of three independent reviewers on a scale from 0 (lowest immunoreactivity) to 3 (highest immunoreactivity).
Chemicals
Unless otherwise indicated, all other chemicals, reagents, and solvents were purchased from Sigma-Aldrich and used as received. For chemotherapeutic studies, BEZ-235 was obtained commercially (Selleckchem). PLX-4720 was synthesized analogously to previously reported methods [27].
Human Cell Line-Bearing Mouse Models
All studies involving small animals were conducted in accordance with both federal and institutional guidelines. C0L0–205 and HCT-116 cell line xenografts were generated by subcutaneously injecting 1 × 107 cells onto the right flank of 5- to 6-week-old female, athymic nude mice (Harlan Sprague-Dawley). Palpable tumors were observed within 1 to 2 weeks and 2 to 3 weeks, respectively, following inoculation. All therapeutic agents were administered via oral gavage for four continuous days as follows: BEZ-235 (35 mg/kg) once daily in 0.1 % Tween 80 and 0.5 % methyl cellulose; PLX-4720 (60 mg/kg) once daily in DMSO. For all combination treatments, agents were administered as separate doses. BEZ-235/PLX-4720 cohorts were treated initially with BEZ-235 followed by administration of PLX-4720 7 to 8 h later.
Tumor volumes were monitored every third day of treatment using high-resolution ultrasound imaging [28]. Dose preparation and administration, treatment duration, and imaging time points were determined based on our previous experience in these models [5, 6, 29].
PET Imaging and Analysis
Animal handling methods in preparation for and during all PET imaging studies, including [18F]FDG and 4-[18F]F-Gln, were derived from protocol standards established for [18F]FDG [30–32]. Prior to imaging, animals were fasted between 6 to 8 h and allowed to acclimate to facility environment for at least 1 h in a warmed chamber at 31.5 °C. Animals were administered 10.4–11.8 MBq of PET imaging agent via intravenous injection and imaged using a dedicated Concorde Microsystems Focus 220 microPET scanner (Siemens Preclinical Solutions). Animals were maintained under 2 % isoflurane anesthesia in 100 % oxygen at 2 l/ min and kept warm for the duration of the PET scan.
4-[18F]F-Gln PET images in xenograft-bearing mice were acquired as 20-min static data sets following a 40-min uptake period and anesthetization. Similarly, [18F]FDG PET images were collected as 10-min static data sets following a 50-min uptake period and anesthetization. Animals were conscious, allowed free access to water, and kept in the 31.5 °C warmed chamber for the duration of the uptake period. Imaging in models of therapeutic response was performed on the fourth day of treatment within 3 to 4 h following single-agent administration. For combination cohorts, imaging was performed approximately 4 to 5 h and 2 to 3 h following the first and second therapeutic treatments, respectively.
PET data were reconstructed using a three-dimensional (3D) ordered subset expectation maximization/maximum a posteriori (OSEM3D/MAP) algorithm. The resulting three-dimensional reconstructions had an x-y voxel size of 0.474 mm and inter-slice distance of 0.796 mm. ASIPro software (Siemens Preclinical Solutions) was used to manually draw 3D regions of interest (ROIs) surrounding the entire tumor volume. PET agent uptake was quantified as the percentage of the injected dose per gram of tissue (%ID/g).
Statistical Methods
Experimental replicates of %ID/g and percent change in volume are reported as the arithmetic mean ± standard deviation. The Wilcoxon rank sum (Mann-Whitney U) test was used to test between-group statistical significance for both in vitro and in vivo data sets. Biomarker expression is reported in frequency tables (Tables 1 and 2). Differences in the frequency of ASCT2 positivity between human tissue types from tumor microarrays were tested using the Pearson chi-square test. Analyses were conducted using the GraphPad Prism 6.01 and R 3.1.1 software packages. Comparisons are considered statistically significant if p < 0.05.
Table 1.
Frequency of ASCT2 expression in primary human colon cancers
| Number | (+) N =40 |
(−) N =57 |
Test statistic | |
|---|---|---|---|---|
| Tissue type | 115 | |||
| P < 0.001 | ||||
| Normal | 8 % (3) | 72 % (41) | ||
| Primary | 92 % (37) | 28 % (16) | ||
Normal vs primary samples. ASCT2 immunoreactivity was evaluated in tissues samples derived from 115 patients with primary colon cancer. Among tissues from patients with primary colon cancer, a total of 53 distinct colon cancers and 44 adjacent normal colon samples were evaluated. Across all normal colon tissue samples (n = 88), greater than 90 % of the samples were negative for ASCT2 immunoreactivity. In contrast, we found that approximately 70 % of primary colon cancers exhibited positive ASCT2 immunoreactivity
Table 2.
Frequency of ASCT2 expression in human colon cancers liver metastases
| N | (+) N =24 |
(−) N =60 |
Test Statistic | |
|---|---|---|---|---|
| Tissue Type | 111 | |||
| P < 0.001 | ||||
| Normal | 12 % (3) | 72 % (41) | ||
| Metastatic | 88 % (21) | 28 % (19) | ||
Normal vs metastatic samples. ASCT2 immunoreactivity was evaluated in tissues samples derived from 111 patients with metastatic colon cancer. Among tissues from patients with metastatic colon cancer, a total of 40 metastases and 44 adjacent normal colon samples were evaluated Across all normal colon tissue samples (n = 88), greater than 90 % of the samples were negative for ASCT2 immunoreactivity. In contrast, we found that approximately 53 % of colon cancer metastases exhibited positive ASCT2 immunoreactivity
Results
Evaluation of ASCT2 Expression in Human Colon Cancers
ASCT2 immunoreactivity was evaluated in tissue samples derived from 115 patients with primary colon cancer and 111 patients with metastatic colon cancer (Fig. 1). Among tissues from patients with primary colon cancer, a total of 53 distinct colon cancers and 44 adjacent normal colon samples were evaluated. Among tissues from patients with metastatic colon cancer, a total of 40 metastases and 44 adjacent normal colon samples were evaluated. Across all normal colon tissue samples (n = 88), greater than 90 % of the samples were negative for ASCT2 immunoreactivity. In contrast, we found that approximately 70 % of primary colon cancers and 53 % of metastases exhibited positive ASCT2 immunoreactivity. Given that 4-[18F]F-GLN PET has been shown to reflect ASCT2 levels [31], these data suggest that 4-[18F]F-GLN PET could be applicable in a large proportion of patients with colon cancer. To determine if ASCT2 expression was associated with expression of mutant BRAF in CRC, we queried the cancer genome atlas (TCGA). We found no statistically significant association between expression of mutant BRAF and SLC1A5 in CRC, while a similar association was also not found for mutant BRAF in melanoma, we did observe a statistically significant association between SLC1A5 expression and BRAFV600E in thyroid cancer (see supporting information) [33].
Fig. 1.
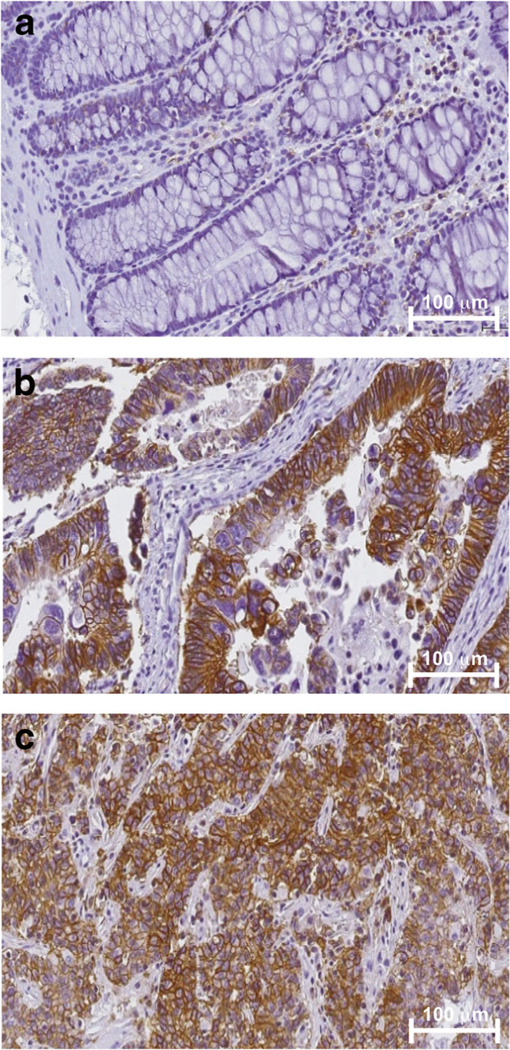
The glutamine transporter ASCT2 is highly expressed in primary colon tumors and colon cancer liver metastases. Representative images of IHC staining for ASCT2 protein in a tissue microarray (TMA) constructed from archival colon tissue sections collected from 115 colon cancer patients representing primary and 111 metastatic disease sections in the liver. a Representative micrograph of sections of normal colon tissue from a patient with colon cancer which stained negative for ASCT2. Representative micrographs are presented of b primary colon cancer sections and c matched liver metastases from a patient with colon cancer.
PET Imaging of Therapeutic Response in BRAFV600E Colon Cancer
In vivo 4-[18F]F-Gln PET was explored as a means to reflect response to targeted therapies in BRAFV600E C0L0–205 xenograft-bearing mice (Fig. 2a). 4-[18F]F-Gln tumor accumulation in vehicle-treated mice averaged 6.34 ± 0.93%ID/ g. When treated with a dual inhibitor of PI3K and mTOR activity (BEZ-235), PET tracer accumulation in tumor tissue remained unchanged (6.07 ± 0.72%ID/g, p = 0.5161). A significant reduction in 4-[18F]F-Gln tumor uptake (5.30 ± 0.91%ID/g) was observed in tumors treated with an inhibitor of mutant BRAF (PLX-4720, p = 0.0185). 4- [18F]F-Gln accumulation was also found to be significantly reduced (4.76 ± 1.07%ID/g, p = 0.0025) when C0L0–205 xenograft tumors were treated with a combination of BEZ- 235 and PLX-4720; however, the addition of the PI3K/mT0R inhibitor did not appear to have an additive effect. In contrast, [18F]FDG PET was non-selectively decreased in all the treatment cohorts (Fig. 2b). Tumor tissues harvested immediately following PET imaging on treatment day 4 were evaluated by western blot to demonstrate the anticipated therapy-induced effects on signaling pathway activity with drug exposure (Fig. 2c). As expected, PLX-4720 exposure led to decreased pERK levels in Colo-205 xenografts, while BEZ-235 exposure led to decreased pAKT. Similarly, combination treatment led to decreases in both pERK and pAKT levels. Importantly, decreased 4- [18F]F-Gln PET in PLX4720-treated animals collected at treatment day 4 selectively predicted the tumor volumes measured at treatment day 10, where significantly decreased tumor volumes were observed in single-agent PLX-4720-treated animals or in animals treated with combination therapy that included PLX-4720 (Fig. 2d). In contrast, similarly collected day 4 [18F]FDG PET did not predict day 10 tumor volumes.
Fig. 2.
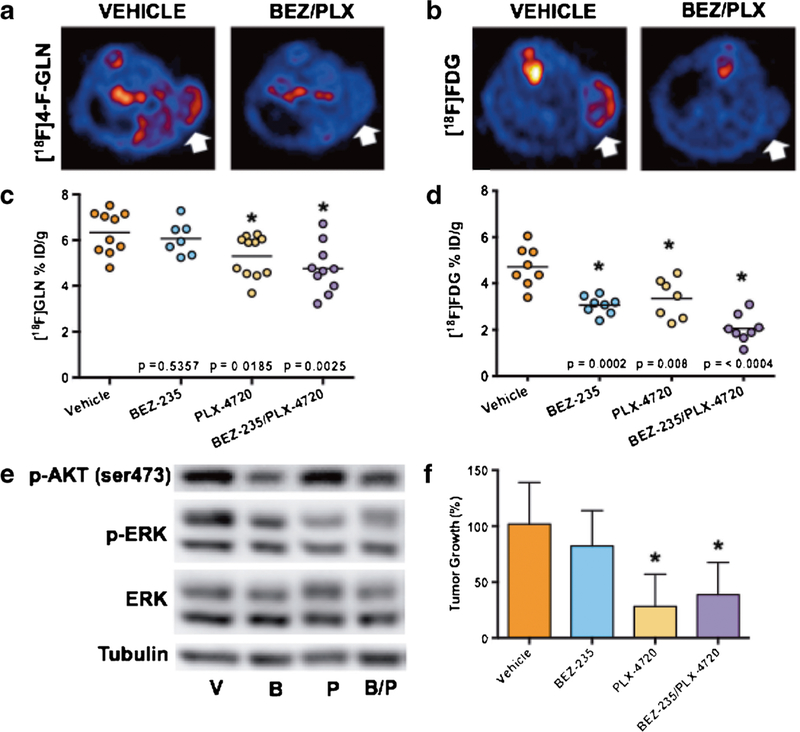
4-[18F]F-Gln uptake reflects molecular response to mutant BRAF therapy in vivo. Representative transverse and coronal a 4-[18F]F-Gln, and b [18F]FDG PET images of COLO-205 xenograft tumor-bearing vehicle or BEZ-235/PLX-4720-treated mice; tumors are denoted by white arrows. PET quantification of tissue %ID/g revealed a significant difference between vehicle and PLX-4720-single-agent and BEZ-235/PLX-4720 combination-treated xenografts only for c 4-[18F]F-Gln while d [18F]FDG PET was significantly reduced non-specifically in all the treated cohorts. PLX-4720 exposure led to decreased pERK levels in Colo- 205 xenografts while BEZ-235 exposure led to decreased pAKT. e Similarly, combination treatment led to decreases in both pERK and pAKT levels. d Changes in C0L0–205 tumor volume by the tenth day of treatment (N ≥ 8 for all cohorts), shown as percent change from day one baseline, revealed a significant reduction in size from vehicle-treated mice for PLX-470 and BEZ- 235/PLX-4720-treated tumors (p = 0.0001 and p = 0.0005, respectively).
PET Imaging of Therapeutic Response in WT BRAF Colon Cancer
As a control, parallel 4-[18F]F-Gln PET imaging studies included a model of WT BRAF colon cancer (HCT-116; Fig. 3). 4-[18F]F-Gln tumor accumulation in vehicle-treated mice was found to average 3.66 ± 0.71%ID/g. In this setting, 4-[18F]F-Gln uptake in all drug-treated cohorts was comparable to that observed in the vehicle-treated cohort: 4.26 ± 0.78%ID/g, p = 0.1783 (BEZ-235); 4.48 ± 1.56%ID/g, p = 0.3983 (PLX-4720); 4.15 ± 0.71%ID/g, p = 0.3337 (BEZ-235/PLX-4720) (Fig. 3a), which supported a lack of pERK inhibition previously observed in mutant BRAF, Colo-205 xenografts. 4-[18F]F-Gln PET in this setting also agreed with a lack of treatment effect observed by day 10 tumor volumes (Fig. 3d). Analogous the Colo-205 model, 4-[18F]F-Gln PET was not diminished with exposure to BEZ-235, despite inhibition of pAKT (Fig. 3c), suggesting potential sensitivity of 4-[18F]F-Gln PET to selectively reflect MAPK-pathway inhibition, but not PI3K/mTOR pathway inhibition. Similar to the Colo-205 model, treatment day 4 [18F]FDG PET was decreased in all HCT-116 treatment cohorts and did not predict day 10 tumor volumes. To understand the relationship between 4-[18F]F-Gln PET uptake and ASCT2 expression in tissues, we evaluated ASCT2 levels in treated tumor tissue compared to vehicle-treated cohorts using immunohistochemistry. We found modestly decreased levels of ASCT2 in BRAFV600E tumors treated with PLX-4720 and BEZ-235/PLX-4720 combination therapies (see supporting information).
Fig. 3.
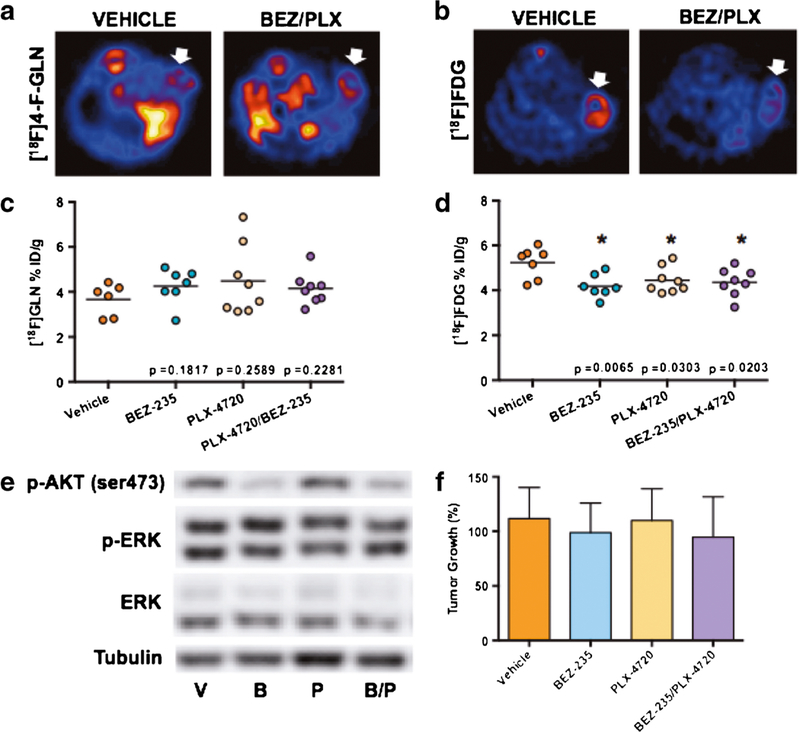
4-[18F]F-Gln uptake is not affected by BRAF-targeted therapy in wild-type BRAF, HCT-116 colon cancer xenografts. Representative transverse and coronal a 4-[18F]F-Gln and b [18F]FDG PET images of HCT-116 xenograft tumor-bearing vehicle or BEZ-235/PLX-4720-treated mice; tumors are denoted by white arrows. PET quantification of tissue %ID/g revealed no significant difference between vehicle and any of the treated cohorts for c 4-[18F]F-Gln while d [18F]FDG PET was significantly reduced non-selectively in all the treated cohorts. e As expected, PLX-4720 exposure did not result in any difference in pERK levels in HCT-116 xenografts while single-agent BEZ-235 and BEZ-235/PLX-4720 combination treatment led to decreased pAKT. f Changes in HCT-116 tumor volume by the tenth day of treatment (N ≥ 8 for all cohorts), shown as percent change from day 1 baseline, revealed no significant reduction in size from vehicle-treated mice for any of the treated tumors.
Discussion
Emerging evidence has implicated oncogenic signaling with metabolic reprogramming in cancer cells [34, 35]. For example, cellular uptake of glucose can be controlled by activation of oncogenic drivers [35], such as PI3K, RAS, BRAF, and MYC, while glutamine dependency has been linked to activations in MYC [36–39] and KRAS [11]. An observed consequence of oncogene-induced glutaminolysis has been the enhanced expression of a sodium-dependent transporter of glutamine, ASCT2 (gene symbol SLC1A5). ASCT2 has been suggested to play a role in metabolism, growth, and survival of lung cancer [18,19] with comparable observations being reported for clinical CRC tissue samples [1, 24]. Additionally, the activity of the mitochondrial enzyme glutaminase (GLS1), which is responsible for metabolism of glutamine to glutamate, has been shown to be induced by ligand-dependent activation of EGFR and subsequent signal transduction through the Raf/Mek/Erk pathway [40]. Therefore, linking glutamine PET with inhibition of downstream MAPK target could represent a clinically translatable modality.
As shown in this study, ASCT2 expression is significantly elevated in primary colon tumor tissue compared to normal colon tissue (p < 0.001) and metastatic tissue compared to normal tissue (p < 0.001). Given the cellular and molecular implications of cancer metabolic reprogramming beyond glycolysis and the Warburg effect, there is a pressing need to characterize and evaluate non-invasive imaging metrics that could serve as supplementary or even alternative tools to conventional [18F]FDG PET imaging. Toward this end, these studies sought to elucidate the feasibility of 4-[18F]F-Gln PET to report inhibition of mutant BRAF in CRC.
A multidrug therapeutic regimen that includes an inhibitor of mutant BRAF and a PBK/mTOR inhibitor has been shown to be a rational therapeutic approach for BRAFV600E melanoma [39, 40] and CRC [5, 6]. Activation of the PI3K/ mTOR signing pathway has been shown to be a potential escape mechanism used by mutant BRAF tumors and thus combined targeting of these two axes may result in improved tumor response [6]. Our previous work in this setting illustrated [18F]FDG PET to be insufficient for observing activation of prosurvival signaling pathways that were detectable by [18F]FLT PET [6] when using BRAF/PI3K targeted therapy in BRAFV600E mutant CRC preclinical models. Analogously, a novel imaging agent for the detection of caspase activity in vivo, [18F]FB-VAD-FMK, was capable of reflecting apoptotic cell death induced from combined inhibition of mutant BRAF and PI3K/mTOR whereas single-agent therapeutics were ineffective [24].
Toward this end, 4-[18F]F-Gln PET was used to evaluate response to single and multidrug therapy using PLX-4720, an inhibitor of mutant BRAF, and BEZ-235, a dual PI3K/ mTOR inhibitor. Both BRAFV600E-expressing COLO-205 xenograft-bearing mice and WT BRAF HCT-116 xenograft- bearing mice were evaluated. In vivo, decreased 4-[18F]F-Gln PET was observed in COLO-205 xenograft tumors treated with a BRAF inhibitor as a single agent or in combination with a PI3K/mTOR inhibitor. However, the same effect on 4-[18F]F-Gln PET was not observed for HCT-116 xenografts. Our data suggest that 4-[18F]F-Gln PET may serve as a sensitive measure of events downstream of BRAF inhibition, namely, p-ERK activity. Uniquely, 4- [18F]F-Gln PET was capable of reflecting pERK inhibition following PLX-4720 selectively, in contrast to PI3K/mTOR inhibition. Furthermore, the magnitude of 4-[18F]F-Gln accumulation collected shortly after the initiation of treatment predicted tumor volumes measured at treatment day 10. In agreement with 4-[18F]F-Gln PET data, we found that inhibition of BRAFV600E had modestly decreased ASCT2 levels in tumors. Notably, [18F]FDG PET did not selectively reflect inhibition of pERK nor was it capable of predicting day 10 tumor volumes in this studies.
Conclusions
These findings illustrate the utility of non-invasive PET imaging measures of glutamine uptake to selectively predict response to BRAF-targeted therapy in colon cancer and may suggest further opportunities to inform colon cancer clinical trials testing other targeted therapies that inhibit MAPK-pathway activity.
Supplementary Material
Acknowledgments.
These studies were supported by grants from the National Institutes of Health (R01-CA140628, P50-CA95103, 5P30-DK058404, P30-CA068485), the Kleberg Foundation, and the Vanderbilt Center for Molecular Probes. The authors wish to thank Ping Zhao for her assistance with the animal experiments, Md. Noor Tantaway for his assistance with the small animal PET acquisition, and Michael Nickels for his assistance with radio tracer production.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest.
Compliance with Ethical Standards
Electronic supplementary material The online version of this article (doi:10.1007/s11307–016-1008-z) contains supplementary material, which is available to authorized users.
References
- 1.Zhao Y, Butler EB, Tan M (2013) Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 4:e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707 [DOI] [PubMed] [Google Scholar]
- 3.Ide M (2006) Cancer screening with FDG-PET. Q J Nucl Med Mol Imaging 50:23–27 [PubMed] [Google Scholar]
- 4.Tagliabue L, Del Sole A (2014) Appropriate use of positron emission tomography with [(18)F]fluorodeoxyglucose for staging of oncology patients. Eur J Intern Med 25:6–11 [DOI] [PubMed] [Google Scholar]
- 5.McKinley ET, Smith RA, Zhao P et al. (2013) 3’-Deoxy-3’−18F- fluorothymidine PET predicts response to (V600E)BRAF-targeted therapy in preclinical models of colorectal cancer. J Nucl Med 54:424–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKinley ET, Zhao P, Coffey RJ et al. (2014) 3’-Deoxy-3’-[18F]- fluorothymidine PET imaging reflects PI3K-mTOR-mediated prosurvival response to targeted therapy in colorectal cancer. PLoS One 9:e108193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wellen KE, Lu C, Mancuso A et al. (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24:2784–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123:3678–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu W, Le A, Hancock C et al. (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A 109:8983–8988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaglio D, Metallo CM, Gameiro PA et al. (2011) Oncogenic K-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wise DR, Ward PS, Shay JE et al. (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation ofalpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 108:19611–19616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JB, Erickson JW, Fuji R et al. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wise DR, DeBerardinis RJ, Mancuso A et al. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. ProcNatlAcad Sci U SA 105:18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Son J, Lyssiotis CA, Ying H et al. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Geldermaisen M, Wang Q, Nagarajah R et al. (2016) ASCT2/ SLC1A5 controls glutamine uptake and tumour growth in triplenegative basal-like breast cancer. Oncogene 35:3201–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witte D, Ali N, Carlson N et al. (2002) Overexpression of the neutral amino acid transporter ASCT2 in human colorectal adenocarcinoma. Anticancer Res 22:2555–2557 [PubMed] [Google Scholar]
- 18.Hassanein M, Hoeksema MD, Shiota M et al. (2013) SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res 19:560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassanein M, Quan J, Hoeksema MD et al. (2015) Targeting SLC1A5- mediated glutamine dependence in non-small cell lung cancer. Int J Cancer 137:1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Hardie RA, Hoy AJ et al. (2015) Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol 236:278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaira K, Sunrose Y, Arakawa K et al. (2015) Clinicopathological significance of ASC amino acid transporter-2 expression in pancreatic ductal carcinoma. Histopathology 66:234–243 [DOI] [PubMed] [Google Scholar]
- 22.Gross MI, Demo SD, Dennison JB et al. (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 13:890–901 [DOI] [PubMed] [Google Scholar]
- 23.Lieberman BP, Ploessl K, Wang L et al. (2011) PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4-fluoroglutamine. J Nucl Med 52:1947–1955 [DOI] [PubMed] [Google Scholar]
- 24.Qu W, Zha Z, Ploessl K et al. (2011) Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J Am Chem Soc 133:1122–1133 [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Zha Z, Li G et al. (2014) 18F](2S,4S)-4-(3-Fluoropropyl)glut-amine as a tumor imaging agent. Mol Pharm 11:3852–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassanein M, Hight MR, Buck JR et al. (2016) Preclinical evaluation of 4-[(18)F]fluoroglutamine PET to assess ASCT2 expression in lung cancer. Mol Imaging Biol 18:18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buck JR, Saleh S, Uddin MI et al. (2012) Rapid, microwave-assisted organic synthesis of selective (V600E)BRAF inhibitors for preclinical cancer research. Tetrahedron Lett 53:4161–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayers GD, McKinley ET, Zhao P et al. (2010) Volume of preclinical xenograft tumors is more accurately assessed by ultrasound imaging than manual caliper measurements. J Ultrasound Med 29:891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hight MR, Cheung YY, Nickels ML et al. (2014) A peptide-based positron emission tomography probe for in vivo detection of caspase activity in apoptotic cells. Clin Cancer Res 20:2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fueger BJ, Czernin J, Hildebrandt I et al. (2006) Impact of animal handling on the results of 18F-FDG PET studies in mice. J Nucl Med 47:999–1006 [PubMed] [Google Scholar]
- 31.Dandekar M, Tseng JR, Gambhir SS (2007) Reproducibility of 18F- FDG microPET studies in mouse tumor xenografts. J Nucl Med 48:602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinley ET, Bugaj JE, Zhao P et al. (2011) 18FDG-PET predicts pharmacodynamic response to OSI-906, a dual IGF-1R/IR inhibitor, in preclinical mouse models of lung cancer. Clin Cancer Res 17:3332–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine AJ, Puzio-Kuter AM (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330:1340–1344 [DOI] [PubMed] [Google Scholar]
- 35.Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao P, Tchernyshyov I, Chang TC et al. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang CV (2010) Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 70:859–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang CV, Le A, Gao P (2009) MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 15:6479–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordan JD, Thompson CB, Simon MC (2007) HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12:108–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thangavelu K, Pan CQ, Karlberg T et al. (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. PNAS 109:7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.