
Figure 4. Haplotype variation in a diploid human genome.
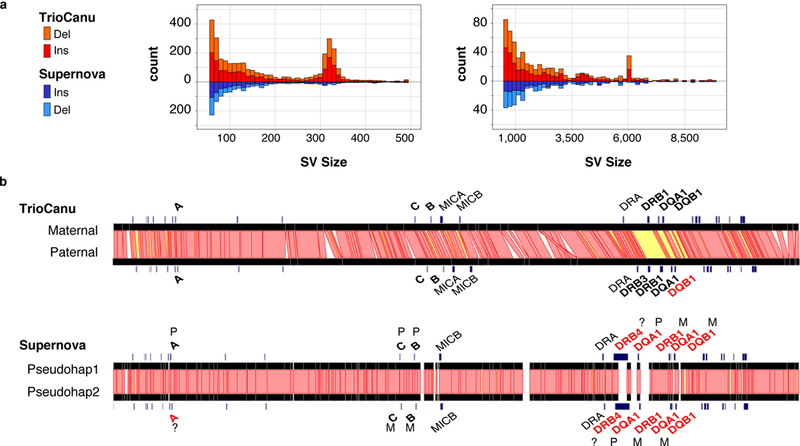
a) Counts of structural variants between NA12878 haplotypes across the entire genome as reported by Assemblytics 34. Canu haplotypes (top, red) showed a balance of insertions and deletions, with peaks at ~300 bp and ~6 kbp corresponding to human Alu and LINE elements, respectively. In comparison, the Supernova pseudo-haplotypes (bottom, blue) were missing these larger structural variants. b) Ribbon visualization 56 of MHC haplotypes for human reference sample NA12878 as assembled by TrioCanu from PacBio data (top) and Supernova from 10X Genomics data (bottom). Red bands indicate >95% identity between haplotypes; yellow bands <95% identity; and unaligned in white (gaps and indels). Genes are annotated in black if matching the known truth without error. TrioCanu captured more haplotype variation than Supernova, especially in the highly variable MHC class II region, which contains a long stretch of high sequence divergence (yellow). In addition to phasing the entire region, TrioCanu perfectly reconstructed all typed MHC genes on both haplotypes, with the exception of the paternal DQB1, which contained a single base indel (Supplementary Table 4). Supernova produced an overly homozygous reconstruction that incorrectly assembled a majority of genes and introduced false gene duplications (Supplementary Table 5). FALCON-Unzip correctly reconstructed the MHC genes but with a higher edit distance than TrioCanu (Supplementary Table 6). Canu (without binning) correctly reconstructed the more heterozygous class II genes but collapsed the class I genes (Supplementary Table 7).