
Figure 6. A model comparing early atherogenesis and foam cell formation in humans and ApoE−/− mice.
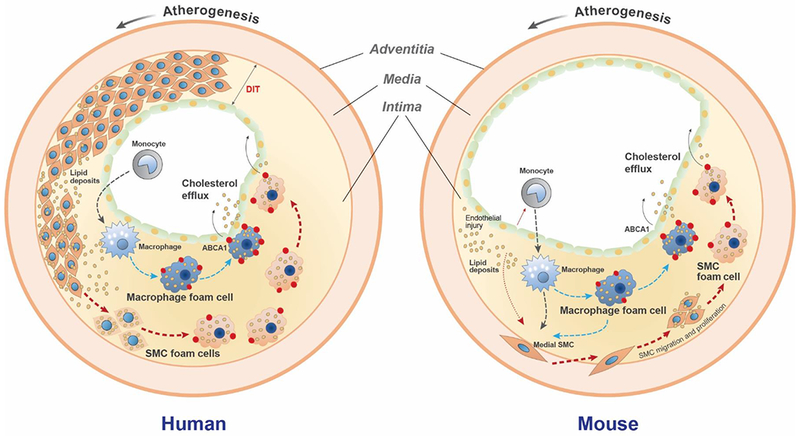
In humans, a thick diffuse intimal thickening (DIT) layer of SMCs is present in atherosclerosis-prone arteries beginning from birth8. At the onset of atherosclerosis lipoproteins that diffuse into the artery wall are trapped through a charge-charge interaction with SMC-secreted proteoglycans, primarily in the deep intima7. These trapped lipoproteins are modified and aggregated, becoming a substrate for uptake by the surrounding SMCs. At this stage monocyte/macrophages are located primarily in the subendothelial region away from the deposited lipids9. Over time both SMCs and macrophages take up modified lipoproteins to become foam cells independently and/or interactively. ABCA1 (red dots), the rate-limiting cholesterol efflux promoter, is robustly upregulated in macrophage foam cells, but less so in SMC foam cells. Increased ABCA1 expression facilitates cholesterol removal out of macrophage-derived foam cells, while impaired ABCA1 expression reduces cholesterol efflux from SMC foam cells, resulting in SMCs being the predominant lineage among total foam cells. In ApoE−/− mice, no DIT SMC layer is present. Atherogenic lipoproteins induce endothelial injury and monocyte binding and infiltration into the intima, where they mature into macrophages that take up the modified intimal lipids. Cytokines released by injured endothelial cells and monocytes/macrophages, including PDGF, TNF-α, and IL-1β induce SMC migration from the media into the intima. SMCs proliferate in the intima and take up modified lipoproteins to become foam cells. Similar to human lesions, the SMC foam cells exhibit reduced ABCA1 expression compared to macrophages. Over time the total contribution to foam cells shifts from macrophages to SMCs, due at least in part to impaired ABCA1 expression by the SMC foam cells.