

Abstract
RNA interference (RNAi) is a convenient tool to identify and characterize biological functions in organisms. Recently, it has become an alternative to chemical insecticides as a biologically based control agent. This promising technology has the potential to avoid many problems associated with conventional chemical insecticides. In order for RNAi application to be practical for field use, a major hurdle is the development of a cost-effective system of double-stranded RNA (dsRNA) production for a large quantity of dsRNA. A handful of research reports has demonstrated microbial-based dsRNA production using L4440 vector and HT115 (DE3) Escherichia coli for application to vertebrate and invertebrate systems. However, the dsRNA yield, production efficiency, and biological purity from this in vitro system is still unclear. Thus, our study detailed biochemical and molecular tools for large-scale dsRNA production using the microbial system and investigated the production efficiency and yield of crude and purified dsRNAs. An unrelated insect gene, green fluorescent protein (GFP), and an insect neuropeptide gene, pyrokinin (PK) identified from Drosophila suzukii, were used to construct the recombinant L4440 to be expressed in the HT115 (DE3) cell. A considerable amount of dsRNA, 19.5 µg/mL of liquid culture, was isolated using ultrasonic disruption followed by phenol extraction. The sonication method was further evaluated to extract crude dsRNA without the additional phenol extraction and nuclease treatments and also to reduce potential bacterial viability. The results suggest that the ultrasonic method saved time and costs to isolate crude dsRNA directly from large volumes of cell culture without E coli contamination. We investigated whether the injection of PK dsRNA into flies resulted in increased adult mortality, but it was not statistically significant at 95% confidence level. In this study, the microbial-based dsRNA production has potential for applied RNAi technology to complement current insect pest management practices.
Keywords: RNAi, bacterial dsRNA production, pest control, spotted wing drosophila
Introduction
RNA interference (RNAi) is a post-transcriptional gene silencing mechanism that is initiated by the presence of double-stranded RNA (dsRNA). It was first reported in the nematode, Caenorhabditis elegans.1 During the past 2 decades, research by academic and commercial entities on the application of RNAi technology for insect pest management has led to great advancements. Two breakthrough reports demonstrated that insects feeding on transgenic plants engineered to produce specific dsRNA showed the suppression of the target gene expression, which led to increased mortality of the cotton bollworm, Helicoverpa armigera2 and the western corn rootworm, Diabrotica virgifera.3 RNAi technology provides a new avenue for insect pest management and has been applied to many insect groups including Diptera.4–6 Although RNAi technology is a new approach and promising tool for insect pest management, there are still technical challenges to successfully develop a next-generation pesticide. RNAi approach to pest management presents 3 major challenges: (1) identifying a suitable target gene and/or physiological system, (2) developing suitable RNAi delivery into the target pest, and (3) providing cost-effective dsRNA production.7–10
The identification of effective RNAi targets with a high level of gene silencing would result in insect developmental arrest and/or death when dsRNA of the target pest is delivered to the insect.11 Delivery of dsRNA to the target pest would be developed with plant-incorporated dsRNA expression, a transgenic method. An alternative delivery method, that is non-transgenic, would involve topical or bait-station application. This approach requires a large quantity of synthetic dsRNA to be effective. Initial evaluation of RNAi application can be achieved with a small-scale screening process using dsRNA that can be synthesized in vitro using commercially available kits. However, these kits are expensive and produce only a limited amount of dsRNA, thus the methods are not a practical means of dsRNA production for large-scale application needs. Therefore, a cost-effective means of dsRNA production is necessary for this technology to be implemented in the field.
Microbial-based dsRNA production by prokaryotic or eukaryotic cells is a sustainable strategy for providing large quantities of dsRNA. The bacteria- or yeast-expressed dsRNA has been developed to inhibit genes in worms,12,13 to control parasites of human14 or shrimp,15,16 and to control insect pests.17–21 In these studies, the dsRNA was orally delivered to the target pests, but the amount and quality of dsRNA that was actually transferred into the target organisms is unclear. The measurement of the quantity and quality of purified or crude extracted dsRNA would be helpful to evaluate the cost-effectiveness of biosynthesized dsRNA for field use.
In this study, we used green florescent protein (GFP), as a control, and pyrokinin (PK), which is a neuropeptide gene identified from the spotted wing drosophila (SWD), Drosophila suzukii,22 because neuropeptides have high potential for biological targets for insect pests.23–25 We constructed 2 recombinant expression vectors, one with GFP and one with PK, and then transformed them into an RNaseIII-deficient Escherichia coli strain. This bacterial-based system produced a large quantity of dsRNA. DsRNA yields obtained from crude and purified stages were determined and evaluated with different isolation methods used to extract dsRNA in cell culture. These methods of expression and isolation have potential toward the development of biologically based pest management.
Materials and Methods
Target genes
The pyrokinin (PK) gene of D. suzukii has been identified in a previous study.22 A partial sequence (305 nucleotides) of PK gene (GenBank Accession No: KX768139) was appended with a SmaI endonuclease sequence (underlined), and three adenines (AAA) at the 5′-ends that provide enough binding space for the restriction enzymes. The PK sequence was amplified with a primer set: PK-forward (5′-AAACCATGGATGTGTGGTCCCAGTTATTGC-3′) and PK-reverse (5′-AAACTGCAGTCCTGGCCCTGCGGCGGCACA-3′) using Phusion DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) and a Veriti Thermal Cycler (Thermo Fisher Scientific). The polymerase chain reaction (PCR) amplification was performed under the following condition: 98°C for 30 seconds, 35 cycles at 98°C for 10 seconds, 60°C for 20 seconds, and 72°C for 30 seconds, and then 72°C for 10 minutes. The PCR product was electrophoresed through a 1.2% agarose gel, and purified quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). As a control, a 448-bp fragment of green fluorescent protein (GFP) gene was amplified with a primer set appended with 2 different endonuclease sites (underlined) and 3 adenines (AAA): GFP-forward (5′-AAACCATGGGTAAACGGCCACAAGTTCAGC-3′) with NcoI and GFP-reverse (5′-AAACTGCAGGGTTGTGGCGGATCTTGAAG-3′) with PstI.
Vector construction and expression
The plasmid vector, L4440 (plasmid Addgene 1654) provided by Andrew Fire (Carnegie Institution for Science, Washington, DC, USA), contains two T7 promoters in inverted orientation flanking the multi-cloning sites indicated in Figure 1. Three DNA restriction enzymes, SmaI, NcoI, and PstI (New England BioLabs, Ipswich, MA, USA), were used to cut the PCR products and the L4440 vector prior to ligation. The SmaI enzyme was applied to cut the PK and the vector. The other two enzymes, NcoI and PstI, were applied to cut the GFP and the vector because the SmaI sequence is present in the middle of the GFP DNA sequence. The pre-digested target DNA and vector were ligated together using T4 DNA ligase (400 000 U/mL) (New England BioLabs) at 4°C for overnight (ca. 12 hours) per manufacturer’s instructions and then used directly for transformation into the host cell.
Figure 1.

Schematic diagram of the bacterial-expressed double-stranded RNA (dsRNA) production system. (A) The target gene fragment is inserted in the multiple-cloning site between two T7 promoter regions in inverted orientation in the expression vector (L4440), which is then transformed into the RNase III-deficient E coli strain HT115 (DE3). IPTG induces RNA transcription mediated by T7 promoter. The dsRNA produced can be purified either by phenol/chloroform/isoamyl alcohol extraction or by spin-column filtration. (B) The target dsRNAs isolated from bacterial culture were analyzed by separation through a 1.2% agarose gel by electrophoresis. White arrows indicate the target dsRNAs. AmpR indicates ampicillin resistance gene; GFP, green fluorescence gene dsRNA; IPTG, isopropyl β-d-1-thiogalactopyranoside; M, GeneRuler 1 kb DNA Ladder (Thermo Scientific); PK, pyrokinin gene dsRNA.
The host bacterium, E. coli HT115 (DE3) strain, was provided by CGC (Caenorhabditis Genetics Center, Minneapolis, MN, USA). The E. coli strain does not produce RNase III, a dsRNA degradation enzyme, and T7 RNA polymerase-mediated transcription is induced with isopropyl β-d-1-thiogalactopyranoside (IPTG).26,27 The recombinant L4440 vector was transformed into HT115 (DE3) by a standard transformation procedure. The cell mixture was then spread on LB agar plates containing both ampicillin (100 μg/mL) and tetracycline (12.5 μg/mL) and incubated overnight at 37°C. Using a colony PCR method, positive colonies were screened using primers pBluescriptKS and pBluescriptSK. The plasmid sequence isolated from selected colonies was confirmed by DNA sequencing.
Expression of dsRNA
The E. coli HT115 containing the recombinant L4440 with GFP or PK was cultured in 4 mL LB medium amended with ampicillin (100 μg/mL) and tetracycline (12.5 μg/mL) at 37°C with shaking at 190 r/min overnight. Then, an aliquot (500 µL) from the overnight culture was added to 25 mL 2× YT medium (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl at pH 7.0) amended with the same antibiotics as above in a 125-mL Erlenmeyer flask. This culture was incubated at 37°C and 190 r/min for 4 hours until the mid-exponential phase (Log OD600 nm = ca. 0.4). To activate the T7 promoter for RNA transcription, IPTG was added at 1 mM concentration and then incubated for an additional 5 hours at the same conditions. Three flasks per target DNA were cultured separately.
Isolation of dsRNA using conventional method
The bacterial cultures (25 mL) were each transferred into a 50-mL Falcon tube and centrifuged at 5000g for 10 minutes at 4°C. After discarding the supernatant, the bacterial pellet was stored at −20°C until use, or resuspended in 2 mL NH4OAc (1 M) with 10 mM EDTA by pipetting rather than vortexing to avoid foaming. One milliliter of cell suspension was mixed with 700 µL of phenol/chloroform/isoamyl alcohol (25:24:1, v/v; Sigma-Aldrich), vortexed vigorously, incubated at 65°C for 30 minutes with occasional shaking, and then centrifuged at 12,000 g for 15 minutes at room temperature. The upper phase containing nucleic acids was carefully transferred to a new tube, mixed with 700 µL isopropanol (molecular biology grade, Sigma-Aldrich), stored at −20°C overnight, and then precipitated by centrifugation at 12,000 g for 30 minutes at 4°C. The nucleic acid pellet was carefully washed twice by adding 1 mL of ethanol (70%) and centrifugation at 7,500 g for 5 minutes at 4°C. After removing residual ethanol, the pellet was air-dried in a sterile hood for 5 to 10 minutes (nucleic acid pellet should turn clear when dried). The pellet was resuspended in 180 µL of pre-warmed nuclease-free water plus 20 µL 10× DNase buffer, and incubated at room temperature for 30 minutes. Then, after adding 2 µL Turbo DNase and 2 µL RNase (Thermo Scientific) to the tube, the mixture was incubated at 37°C for 1 hour to destroy remaining DNA and single-stranded RNA and then stored at −20°C until use. DsRNA was isolated using 200 µL of phenol/chloroform/isoamyl alcohol by vigorously vortexing, followed by centrifugation at 12,000 g for 15 minutes at room temperature. The upper phase containing dsRNA was transferred to a new tube and mixed with 100 µL of NH4OAc (7.5 M) and 100 µL of isopropanol by vortexing. After centrifugation at 12,000 g for 30 minutes at 4°C, the supernatant was discarded and the dsRNA pellet was carefully washed twice using 250 µL of ethanol (70%). DsRNA was air-dried in a sterile hood for 5 to 10 minutes and resuspended in 100 µL of pre-warmed nuclease-free water. The purified dsRNA was quantified using NanoDrop 2000 spectrophotometer and the integrity of the dsRNA was evaluated by electrophoresis through a 1.2% agarose gel and visualization under UV light.
Confirmation of dsRNA
The isolated dsRNA was confirmed using various nuclease enzymes. The GFP dsRNA (200 ng) per reaction was mixed with 1 U of RNase III, DNase I (New England BioLabs), and or RNase A (Thermo Scientific) in corresponding buffer. The mixture was incubated at 37°C for 1 hour and visualized after separation on a 1.2% agarose gel by electrophoresis.
Isolation of dsRNA using sonication or heat treatment
Sonication or heat (95°C) treatments were applied before isolating dsRNA from bacterial cells by conventional dsRNA purification process. One milliliter of the cell suspension (= 12.5 mL initial cell culture equivalent) was sonicated on ice using an ultrasonic cell disruptor (Microson Ultrasonic Cell Disrupter XL, Misonix, Wallkil, NY, USA) equipped with a microprobe. The sonication was performed at the strength of 5 to 6 W (outcome) on ice for 30 seconds 5 times with 30-second interval between sonications. The other 1 mL of cell suspension was heated at 95°C for 10 minutes with shaking at 350 r/min in ThermoMixer C (Eppendorf, Hauppauge, NY, USA). After sonication or heat treatment, dsRNA was extracted using the conventional method started with the 65°C incubation in the phenol/chloroform/isoamyl alcohol and quantified by NanoDrop 2000 spectrophotometer. The quality of the dsRNA was verified after separation by electrophoresis through a 1.2% agarose gel. To extract crude dsRNA, the cells lysed by sonication were centrifuged at 12,000 g for 30 minutes at 4°C. The supernatant was evaluated for dsRNA by gel electrophoresis and bacteria viability by spreading onto LB agar plates as described above.
Evaluation of cell viability after sonication
One milliliter of the cell pellet (= 12.5 mL cell culture equivalent) was sonicated as above. After sonication, the cell viability was measured by spreading an aliquot of serial dilutions onto LB agar plates containing ampicillin (100 μg/mL) and tetracycline (12.5 μg/mL). One milliliter of non-sonicated cell culture served as a control.
PK dsRNA injection into D. suzukii adults
PK dsRNA was purified and injected into the hemocoel of D. suzukii adults using a Nanoliter 2010 system using pulled borosilicate needles (World Precision Instruments, Sarasota, FL, USA) as demonstrated previously.24 PK dsRNA (1 µg/50 nL/fly) or GFP dsRNA (1 µg/50 nL/fly) was each injected into 5-day-old flies. Flies were observed for mortality for 3 days post-injection. Each treatment was composed of 20 flies and replicated 5 or more times.
Results and Discussion
Oral delivery of dsRNA through ingestion is advantageous for the ability to save both time and labor. Ingestion would be a convenient way to transfer more dsRNA to a target organism, but has the disadvantage of requiring a large quantity of dsRNA to be effective.4,28 Therefore, a low-cost means of producing dsRNA is an essential first step for successful application of non-transgenic RNAi technology for pest control. Biologically based systems should provide a sustainable method for large-scale production of dsRNA. In this study, we demonstrate the technical methodology for the biosynthesis of dsRNA in bacterial cells, including cell culture conditions, and the isolation and purification of dsRNA using conventional and simplified methods. Purified and crude extracted dsRNAs were evaluated for yields, and the potential use of crude dsRNA for a practical RNAi application was discussed.
Outline of bacterial-based dsRNA production system
The system of dsRNA production using the L4440 vector expressed in the E. coli HT115, outlined in Figure 1A, can be used with any RNAi target genes of interest. The two vectors, L4440-GFP and L4440-PK, were successfully constructed. The PK gene was cut with two restriction enzymes, NcoI and PstI, between 2 T7 promoters that resulted in an additional 82 and 62 nucleotides of the vector appended to the ends of the PK dsRNA. The TA-cloning method is also an option to construct a recombinant vector containing an RNAi target gene. However, this method would add even more extra nucleotides in the target dsRNA because the total length of multi-cloning sites of L4440 vector is 185 nucleotides between two inverted T7 promoters. The GFP gene was cut using a single enzyme SmaI, which resulted in an additional 114 and 74 nucleotides of the vector appended to both ends of the GFP dsRNA. To avoid or minimize non-specific sequences, the cloning method and sites should be carefully considered when designing the vector construction.
The E. coli HT115 were transformed with vectors of L4440-GFP and L4440-PK, grown to log phase and induced with IPTG to produce the target dsRNA (Figure 1B). The result indicated that the T7 promoter-mediated RNA transcription was activated by IPTG to transcribe two complementary single-stranded RNAs that formed the dsRNA in the cell. The first report of the recombinant vector L4440 was to express the GFP dsRNA ingested by C. elegans to suppress GFP messenger RNA (mRNA).26 The L4440-HT115 E. coli system has also been used to produce dsRNA for down-regulation of a gene in the protozoan, Entamoeba histolytica.14 The utility of the system for an insect pest was first demonstrated for a lepidopteran, Spodoptera exigua, by ingestion of dsRNA of the chitin synthase gene.20
IPTG induction and optimum concentration
The growth of HT115-L4440-GFP showed a standard mode of bacterial growth during the initial lag phase (0-2 hours), an exponential phase (2-7 hours), and a stationary phase (after 7 hours) (Figure 2A). Once RNA transcription was induced by IPTG at the mid-exponential phase (4 hours, Log OD600nm = ca. 0.4), the growth was reduced slightly compared with no IPTG induction, but it was not different among the various IPTG concentrations (Figure 2A).
Figure 2.

Bacterial growth and induction of dsRNA production by IPTG. (A) Various concentrations of IPTG were treated at the mid-exponential phase (arrow). (B) GFP-dsRNA extracted from different IPTG treatments were run by 1.2% agarose gel electrophoresis. M indicates TrackIt 1Kb Plus DNA Ladder (Invitrogen). (C) Digestion of dsRNA by various nucleases. The purified bacterial GFP-dsRNA (Lane 1) was degraded by RNase III (Lane 2), but not by DNase I (Lane 3) or RNase A (Lane 4). dsRNA indicates double-stranded RNA; GFP, green fluorescent protein; IPTG, isopropyl β-d-1-thiogalactopyranoside; M, GeneRuler 1 kb DNA Ladder (Thermo Scientific).
The cells were harvested 5 hours after IPTG induction, 9-hour total incubation, for all concentrations of IPTG tested. It was found that the quantities of dsRNA produced in different IPTG treatments (0.1-3.0 mM) were not significantly different and thus seems independent of IPTG concentration. But, dsRNA was not found in the control without IPTG (Figure 2B), indicating that IPTG is essential to induce dsRNA production. Various IPTG concentrations (0.4-2.0 mM) have been used in previous studies to produce dsRNAs in bacterial systems.13,14,17,19,20 However, the IPTG amounts or concentrations used for dsRNA production have not been explained or justified. There is a report that the dsRNA production of a plant virus showed no difference with 0.4 mM versus 1 mM IPTG.29 Results from the previous and current studies, therefore, suggest IPTG concentration is not correlated to dsRNA production in the range of 0.1 to 3.0 mM. However, isopropyl β-d-1-thiogalactopyranoside is needed at some concentration to induce RNA transcription, initiated by T7 promoter, but more tests at concentrations below 0.1 mM are required to identify a critical threshold. In this study, we decided 1 mM IPTG as a reasonable concentration for dsRNA production unless stated.
Confirmation of dsRNA
The expected GFP-dsRNA, isolated from bacterial culture, was treated with the following nuclease enzymes: RNase III, DNase I, and RNase A. The dsRNA was shown to be degraded by RNase III only (Figure 2C), which is a double-stranded RNA-specific endonuclease cleaving it into small interfering RNA (siRNA),30 but not by the other two enzymes. The result indicates the purified product was double-stranded RNAs, not DNA or single-stranded RNA. Although the process of enzymatic degradation might not be time and cost effective, it would be required to obtain a high-quality dsRNA without other nucleic acid contamination.
Yield of dsRNA from different isolation methods
The “conventional or (=standard)” isolation procedure for cell lysis is to incubate with phenol/chloroform/isoamyl alcohol at 65°C for 10 minutes. In this study, two pre-treatments, sonication and heating, were applied prior to proceeding with the conventional isolation procedure, and the dsRNA yields were compared, using the conventional method without pre-treatment as a control.
After sonication or heat treatment of the cells, the dsRNA yields were increased by 2.5- to 5-fold compared with the control method without the pre-treatments (Figure 3). The estimated total dsRNA isolated by sonication was 488 μg per 25 mL culture (19.5 μg/mL), whereas heating and conventional control were 239 μg per 25 mL culture (9.6 μg/mL) and 97 μg per 25 mL culture (3.9 μg/mL), respectively (Figure 3). The dsRNA amount obtained from the sonication method was 5 or 2 times more than those amounts from the heating or the conventional (= control) methods, respectively. The dsRNAs isolated by sonication were relatively smeared with smaller bands on the gel electrophoresis, indicating that the sonication treatment resulted in some shearing of the dsRNA to smaller fragments. Further work is required to optimize the sonication intensity and duration to increase intact dsRNA without small fragments and maximize RNAi effect. Sonication has been shown to have the greatest RNAi effect on S. exigua.17 The previous and current results indicate that the sonication treatment is the most effective means to extract dsRNA from bacterial cells. Therefore, the sonication treatment was adapted in the isolation process for dsRNA produced in the bacteria.
Figure 3.
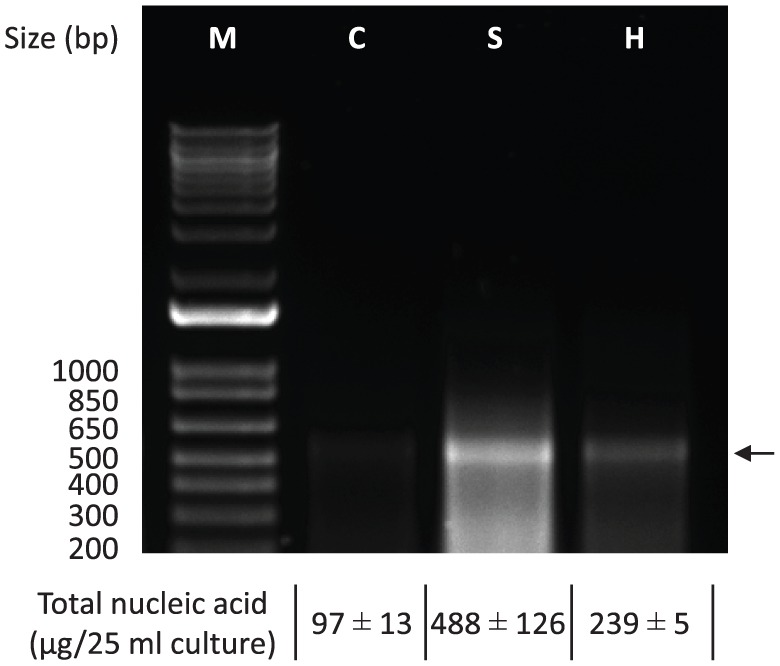
Effect of pre-treatments on cell lysis and dsRNA isolation by different lysis methods. Sonication or heating was performed before the conventional dsRNA isolation using phenol/chloroform/isoamyl alcohol. The dsRNA crude extracts were diluted 100 times and 10 μL aliquots were run in 1.2% agarose gel electrophoresis. Arrow indicates the target dsRNA (GFP-dsRNA). dsRNA indicates double-stranded RNA; GFP, green fluorescent protein; M, TrackIt 1Kb Plus DNA Ladder (Invitrogen); C, control; S, sonication; H, heating.
Evaluation of cell viability after sonication
DsRNA production and isolation needs to be cost effective in order for RNAi technology to advance but it also needs to consider the potential of bacterial contamination for it to be a feasible practice for field use. The experiment tested whether sonication is an efficient means to extract dsRNA directly without further isolation procedures, and whether it was an effective treatment to lyse the cells thus making them nonviable. With just sonication applied to the cell suspension, the lysis efficiencies were measured by turbidity (OD 600 nm) and by colony-forming unit (CFU), which were 89.3% and 98.4%, respectively. Bacterial colonies were not detected in the supernatant of the cell suspension when centrifugation followed sonication (Table 1 and Figure 4D). However, few bacterial colonies were detected in the cell suspension with sonication (Figure 4B) and the centrifuged supernatant of the cell suspension without sonication (Figure 4C). The results indicate the centrifugation or sonication only may not be enough, and both procedures are needed to avoid bacterial contamination for the file use.
Table 1.
Cell lysis of Escherichia coli HT115-GFP by sonication.
| Sonication | Lysis efficiency (%) | ||
|---|---|---|---|
| – | + | ||
| Cell suspension | |||
| Turbidity (OD600 nm) | 40.3 ± 1.5 | 4.3 ± 1.0 | 89.3 |
| CFU/mL | 49 × 105 | 0.8 × 105 | 98.4 |
| Supernatant | |||
| CFU/mL | 250 ± 9.7 | 0 ± 0.0 | 100.0 |
| Nucleic acid (ng/µL)a | 431 ± 20 | 2787 ± 64 | – |
Abbreviation: CFU, colony-forming unit.
Total nucleic acid concentration was measured by NanoDrop 2000 using the final dsRNA extract (100 μL) from 12.5 mL bacterial culture.
Figure 4.
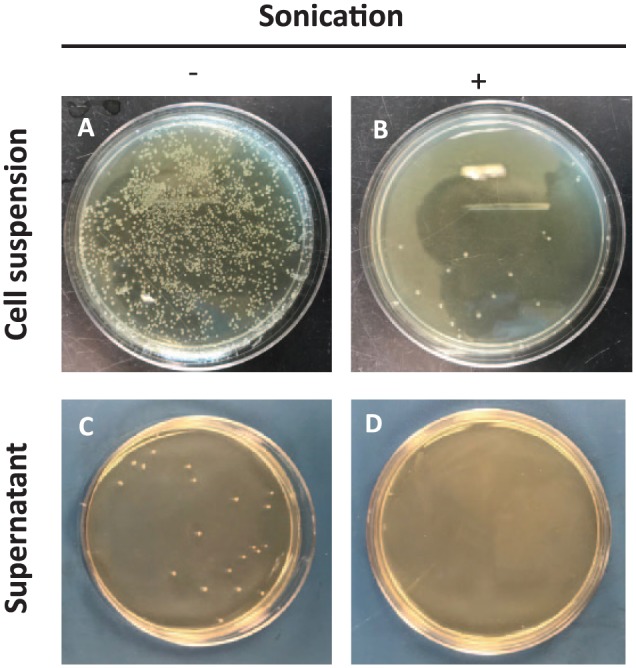
Bacterial cell lysis before and after sonication. An aliquot (100 μL) of 1/100 dilution of cell suspension or supernatant was spread on LB agar plate supplemented with ampicillin and tetracycline. (A) Cell suspension without sonication, (B) cell suspension with sonication, (C) supernatant of the cell suspension without sonication, and (D) supernatant of the cell suspension with sonication.
After sonication, the total amount of nucleic acids extracted from the bacterial cells was 6.5-fold greater than without sonication (control) (Table 1). It is possible that the results of RNAi may be affected by the quality of dsRNA used, whether it is in crude or pure form. Previous work on S. exigua larvae demonstrated that crude dsRNA, extracted by sonication, significantly enhanced larval mortality.17 Even though crude dsRNA is mixed with other nucleic acids, cell lysis by sonication followed by centrifugation led to a greater quantity of dsRNA being extracted from the cell suspension. This low-cost simple procedure could be adapted to dsRNA production, making RNAi application a feasible option for field use.
Phenotypic impacts on fly adults
Injection of flies with PK dsRNA resulted in increased almost double adult mortality when compared with the control (PK 19.0% vs GFP 9.6%), but it was not statistically significant at 95% confidence level (PK vs GFP: Mann Whitney test, 2-tailed, n ⩾ 100, P = .0543) (Figure S1). The RNAi using pheromone biosynthesis activating neuropeptide (PBAN)/PK family genes was first applied to the corn earworm, Helicoverpa zea, and the fire ant, Solenopsis invicta, showing various negative phenotypic impacts including mortality in two insect species.24 Recently, the RNAi treatments on immature moths that had ingested the specific dsRNA starting at the first instar larva through pupation resulted in delay of larval growth, interference of pupal development, and mortality in the two moth species.25 The previous and current RNAi studies indicate that phenotypic impacts using even the same RNAi targets could vary because they might have different physiological functions from different developmental stages and/or Orders. Identifying biological functions of target genes in insects would be a better approach to apply RNAi.
Supplemental Material
Supplemental material, Figure_S1_R2_xyz1607784970975_(4) for Microbial-Based Double-Stranded RNA Production to Develop Cost-Effective RNA Interference Application for Insect Pest Management by Seung-Joon Ahn, Kelly Donahue, Youngho Koh, Robert R. Martin and Man-Yeon Choi in International Journal of Insect Science
Acknowledgments
We thank Addgene for L4440 plasmid, Caenorhabditis Genetics Center (CGC) for HT115 (DE3) cell, Ann You for technical support, and anonymous reviewers for valuable comments on the manuscript.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by research grants from Northwest Center for Small Fruits Research and base funding from USDA-ARS CRIS 2072-22000-040-00D, the United States of America.
Declaration of conflicting interests:The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S-JA, KD, YK, RRM, and M-YC have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Author Contributions: S-JA, RM, and M-YC conceived the research and analyzed the data. S-JA, KD, and M-YC conducted the experiments. S-JA, KD, YK, and RRM wrote the manuscript. All authors read and approved the manuscript.
Supplemental Material: Supplemental material for this article is available online.
ORCID iDs: Seung-Joon Ahn  https://orcid.org/0000-0002-5980-106X
https://orcid.org/0000-0002-5980-106X
Man-Yeon Choi
https://orcid.org/0000-0003-0769-380X
References
- 1. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. [DOI] [PubMed] [Google Scholar]
- 2. Mao YB, Cai WJ, Wang JW, et al. Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol. Nat Biotechnol. 2007;25:1307–1313. [DOI] [PubMed] [Google Scholar]
- 3. Baum JA, Bogaert T, Clinton W, et al. Control of coleopteran insect pests through RNA interference. Nat Biotechnol. 2007;25:1322–1326. [DOI] [PubMed] [Google Scholar]
- 4. Zotti MJ, Smagghe G. RNAi technology for insect management and protection of beneficial insects from diseases: lessons, challenges and risk assessments. Neotrop Entomol. 2015;44:197–213. [DOI] [PubMed] [Google Scholar]
- 5. Gundersen-Rindal DE, Adrianos SL, Allen ML, et al. Arthropod genomics research in the United States Department of Agriculture Agricultural Research Service: applications of RNA interference and CRISPR gene editing technologies in pest control. Trends Entomol. 2017;13:109–137. [Google Scholar]
- 6. Pridgeon JW, Zhao L, Becnel JJ, Strickman DA, Clark GG, Linthicum KJ. Topically applied AaeIAP1 double-stranded RNA kills female adults of Aedes aegypti. J Med Entomol. 2008;45:414–420. [DOI] [PubMed] [Google Scholar]
- 7. Huvenne H, Smagghe G. Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: a review. J Insect Physiol. 2010;56:227–235. [DOI] [PubMed] [Google Scholar]
- 8. Asokan R. RNA interference (RNAi): a novel strategy in insect pest management. Curr Sci India. 2008;94:1119. [Google Scholar]
- 9. Andrade EC, Hunter WB. RNA interference. In: Abdurakhmonov IY, ed. Natural Gene-Based Technology for Highly Specific Pest Control (HiSPeC). London: InTech; 2016:391–409. [Google Scholar]
- 10. Ghosh SKB, Hunter WB, Park AL, Gundersen-Rindal DE. Double-stranded RNA oral delivery methods to induce RNA interference in phloem and plant-sap-feeding hemipteran insects. J Vis Exp. 2018;135:e57390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang H, Li HC, Miao XX. Feasibility, limitation and possible solutions of RNAi-based technology for insect pest control. Insect Sci. 2013;20:15–30. [DOI] [PubMed] [Google Scholar]
- 12. Newmark PA, Reddien PW, Cebria F, Sanchez Alvarado A. Ingestion of bacterially expressed double-stranded RNA inhibits gene expression in planarians. Proc Natl Acad Sci U S A. 2003;100:11861–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. [DOI] [PubMed] [Google Scholar]
- 14. Solis CF, Santi-Rocca J, Perdomo D, Weber C, Guillen N. Use of bacterially expressed dsRNA to downregulate Entamoeba histolytica gene expression. PLoS ONE. 2009;4:e8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ongvarrasopone C, Roshorm Y, Panyim S. A simple and cost effective method to generate dsRNA for RNAi studies in invertebrates. ScienceAsia. 2007;33:35–39. [Google Scholar]
- 16. Attasart P, Namramoon O, Kongphom U, Chimwai C, Panyim S. Ingestion of bacteria expressing dsRNA triggers specific RNA silencing in shrimp. Virus Res. 2013;171:252–256. [DOI] [PubMed] [Google Scholar]
- 17. Kim E, Park Y, Kim Y. A transformed bacterium expressing double-stranded RNA specific to integrin beta1 enhances Bt toxin efficacy against a polyphagous insect pest, Spodoptera exigua. PLoS ONE. 2015;10:e0132631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murphy KA, Tabuloc CA, Cervantes KR, Chiu JC. Ingestion of genetically modified yeast symbiont reduces fitness of an insect pest via RNA interference. Sci Rep. 2016;6:22587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ratzka C, Gross R, Feldhaar H. Systemic gene knockdown in Camponotus floridanus workers by feeding of dsRNA. Insect Soc. 2013;60:475–484. [Google Scholar]
- 20. Tian H, Peng H, Yao Q, et al. Developmental control of a lepidopteran pest Spodoptera exigua by ingestion of bacteria expressing dsRNA of a non-midgut gene. PLoS ONE. 2009;4:e6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu J, Dong YC, Li P, Niu CY. The effect of silencing 20E biosynthesis relative genes by feeding bacterially expressed dsRNA on the larval development of Chilo suppressalis. Sci Rep. 2016;6:28697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi M-Y, Ahn S-J, Kim AY, Koh Y. Identification and characterization of pyrokinin and CAPA peptides, and corresponding GPCRs from spotted wing drosophila, Drosophila suzukii. Gen Comp Endocrinol. 2017;246:354–362. [DOI] [PubMed] [Google Scholar]
- 23. Audsley N, Down RE, Isaac RE. Genomic and peptidomic analyses of the neuropeptides from the emerging pest, Drosophila suzukii. Peptides. 2015;68:33–42. [DOI] [PubMed] [Google Scholar]
- 24. Choi MY, Vander Meer RK, Coy M, Scharf ME. Phenotypic impacts of PBAN RNA interference in an ant, Solenopsis invicta, and a moth, Helicoverpa zea. J Insect Physiol. 2012;58:1159–1165. [DOI] [PubMed] [Google Scholar]
- 25. Choi MY, Vander Meer RK. Phenotypic effects of PBAN RNAi using oral delivery of dsRNA to Corn earworm (Lepidoptera: Noctuidae) and Tobacco budworm larvae. J Econ Entomol. 2019;112:434–439. [DOI] [PubMed] [Google Scholar]
- 26. Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. [DOI] [PubMed] [Google Scholar]
- 27. Takiff HE, Chen SM, Court DL. Genetic analysis of the rnc operon of Escherichia coli. J Bacteriol. 1989;171:2581–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoch R, Sethi A, Thakur N, Murdock LL. RNAi for insect control: current perspective and future challenges. Appl Biochem Biotechnol. 2013;171:847–873. [DOI] [PubMed] [Google Scholar]
- 29. Tenllado F, Martinez-Garcia B, Vargas M, Diaz-Ruiz JR. Crude extracts of bacterially expressed dsRNA can be used to protect plants against virus infections. BMC Biotechnol. 2003;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang D, Buchholz F, Huang Z, et al. Short RNA duplexes produced by hydrolysis with Escherichia coli RNase III mediate effective RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:9942–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Figure_S1_R2_xyz1607784970975_(4) for Microbial-Based Double-Stranded RNA Production to Develop Cost-Effective RNA Interference Application for Insect Pest Management by Seung-Joon Ahn, Kelly Donahue, Youngho Koh, Robert R. Martin and Man-Yeon Choi in International Journal of Insect Science